The Antioxidant and Antiproliferative Activities of 1,2,3-Triazolyl-L-Ascorbic Acid Derivatives

 , ,
, ,  and
and
Abstract
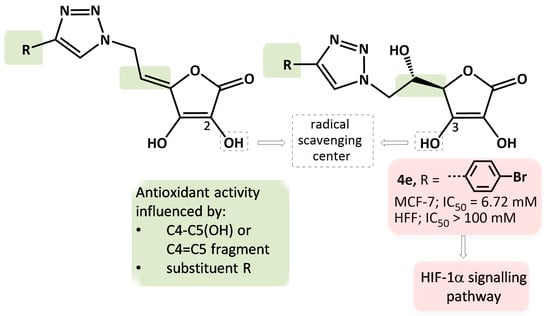
1. Introduction
2. Results and Discussion
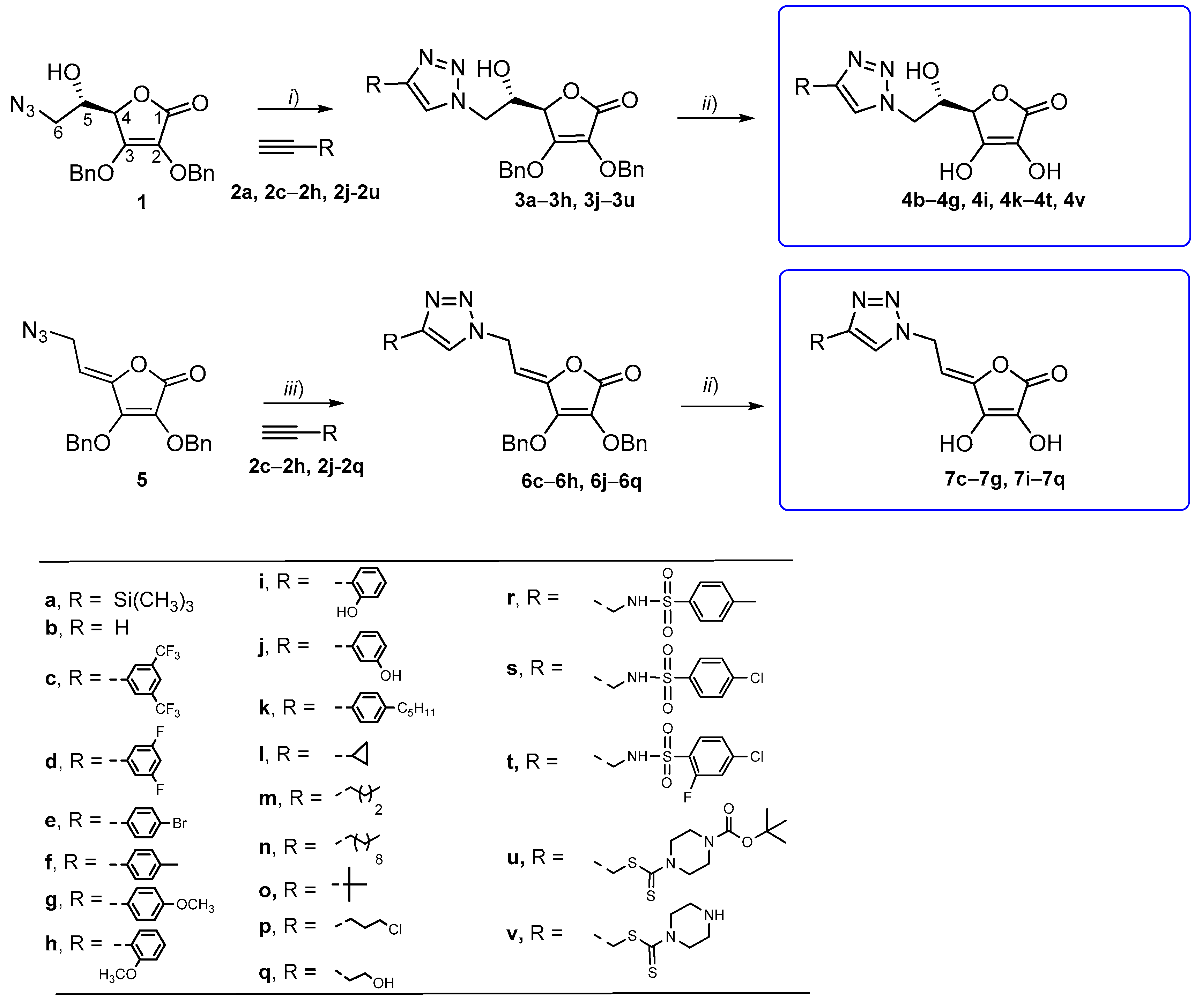
2.1. Synthesis
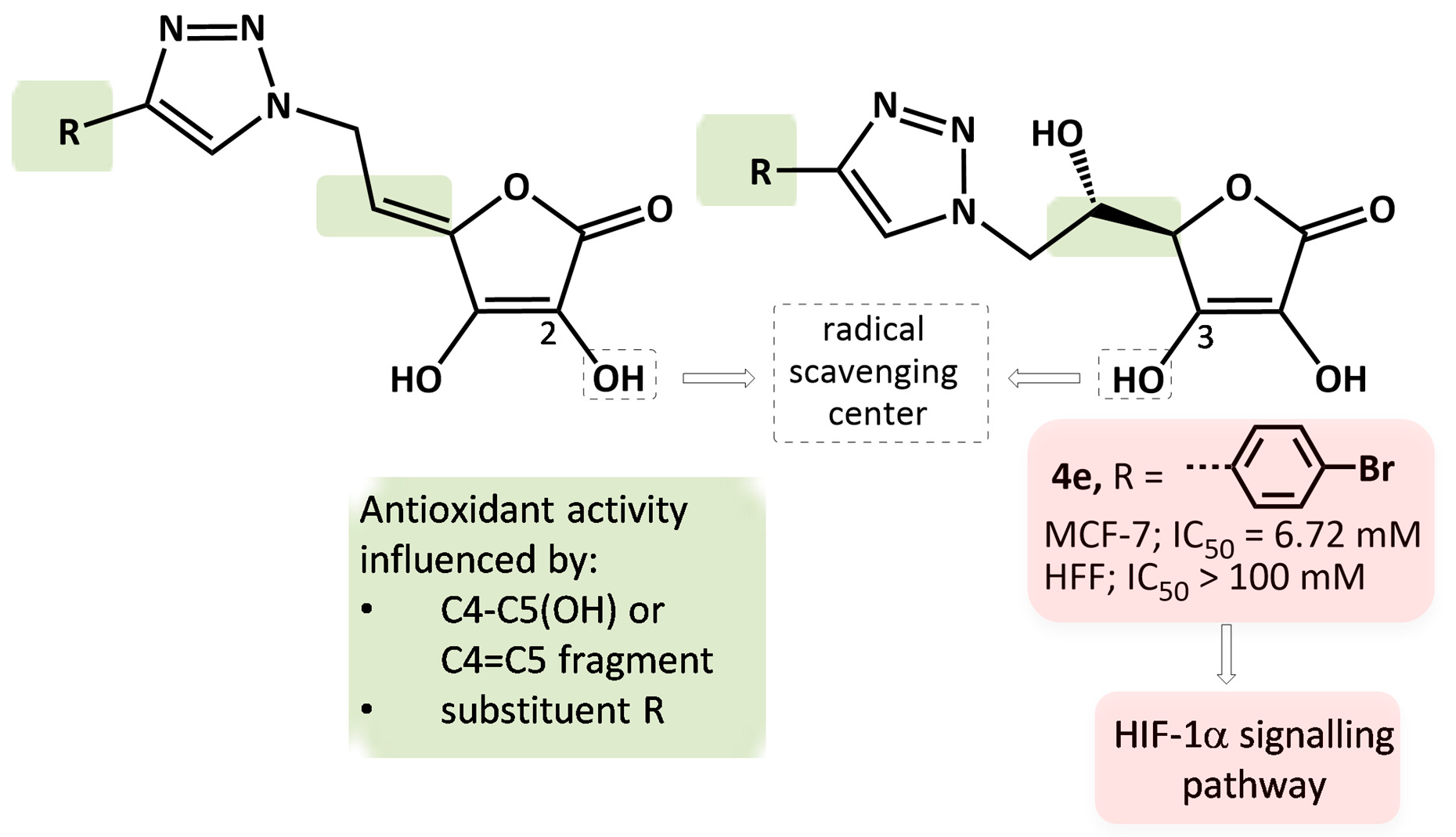
2.2. DPPH• Radical Scavenging Activity in Vitro
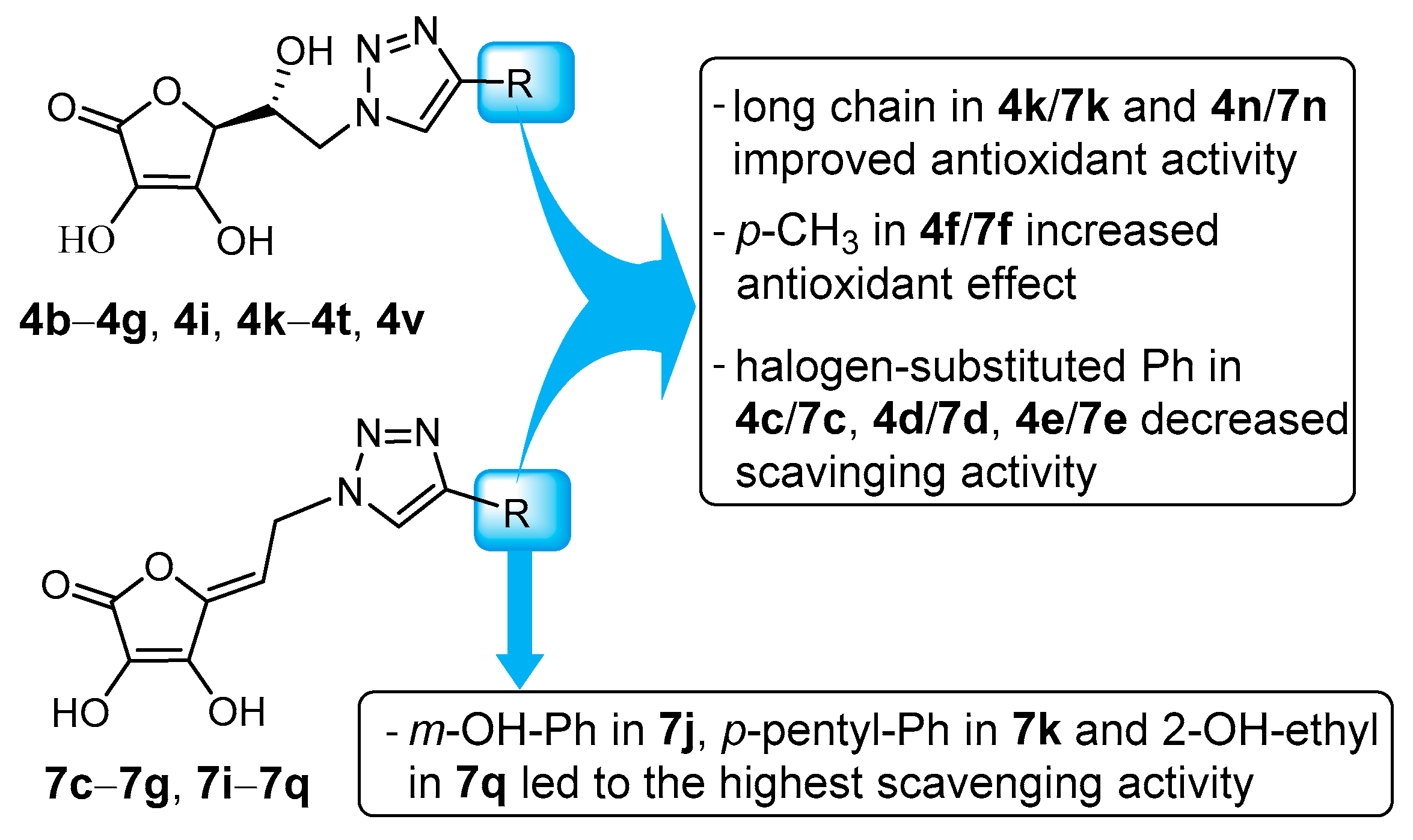
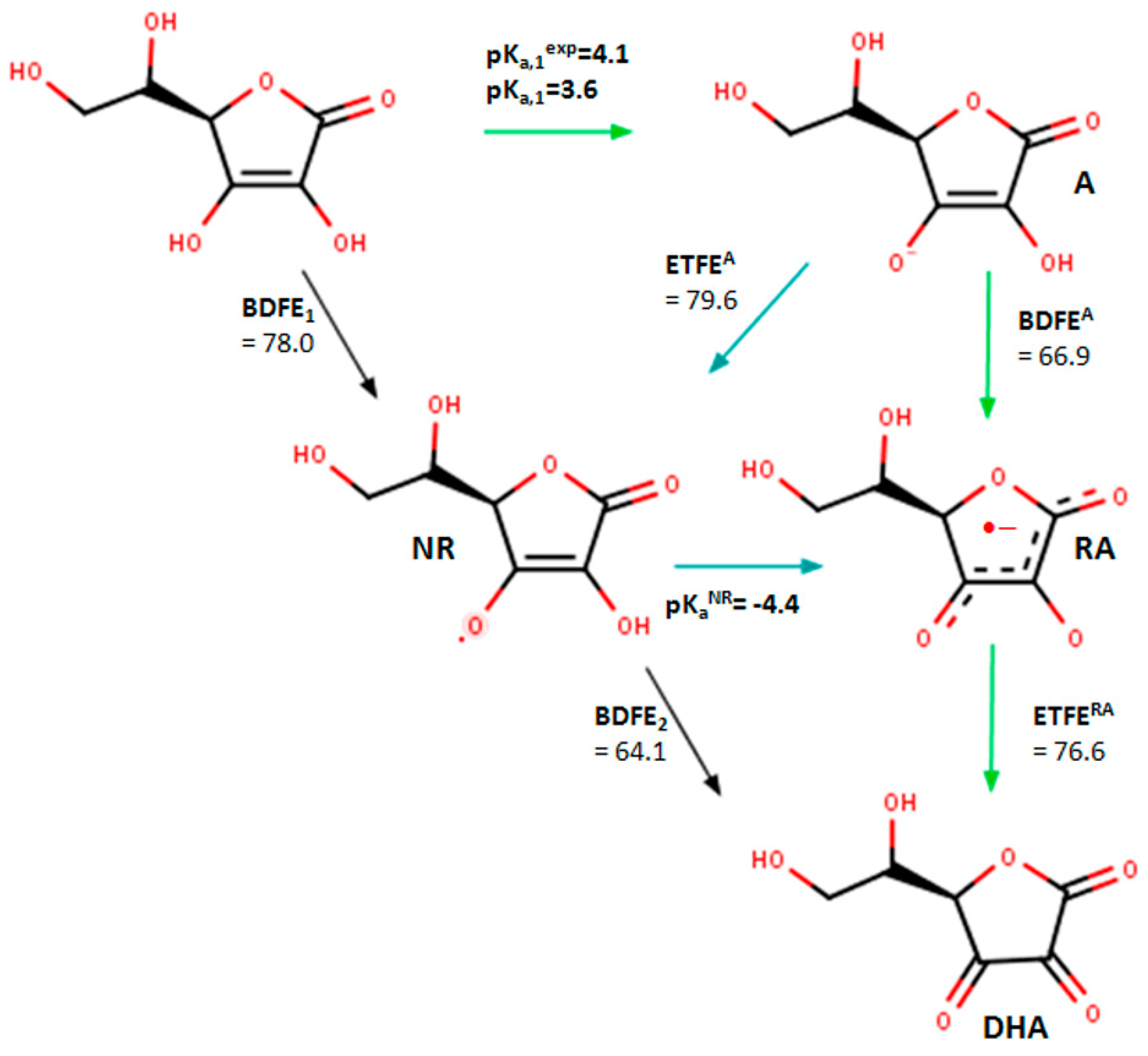
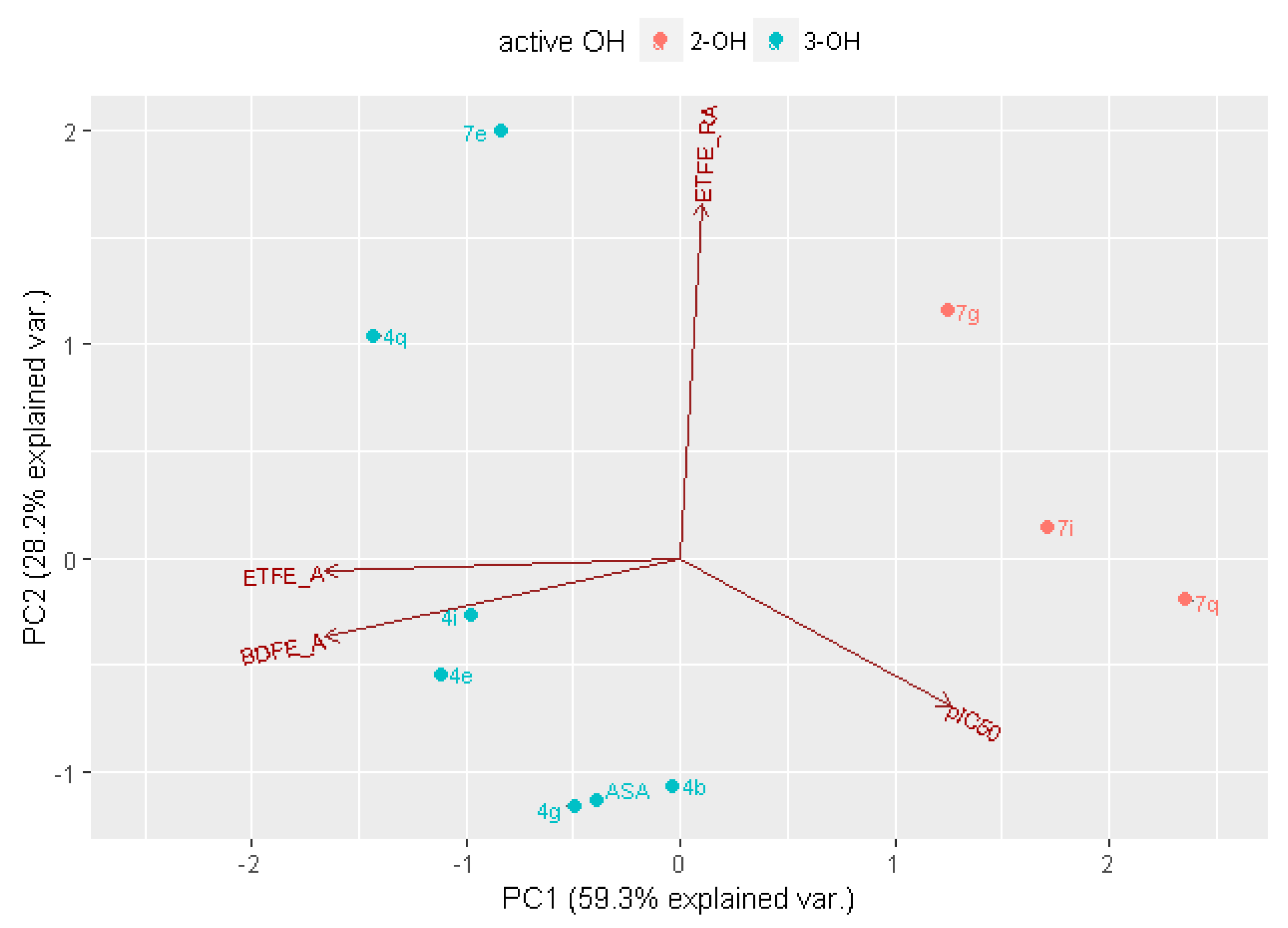
2.3. In Silico Analysis of Radical Scavenging Capacities
2.4. Biological Evaluations
2.4.1. Antiproliferative Activities in Vitro
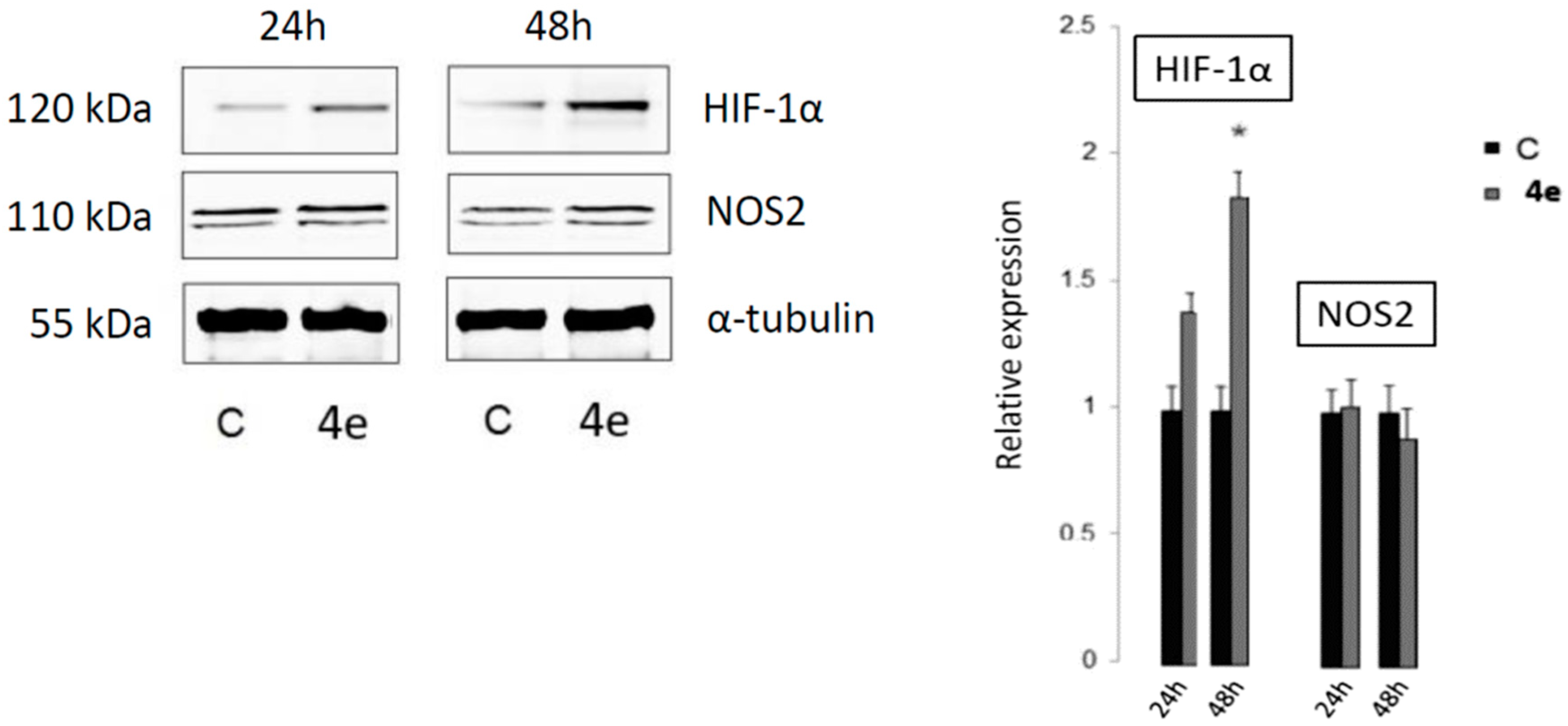
2.4.2. The Detection of Apoptosis and the Validation of HIF-1α and NOS2 Protein Targets for 4e
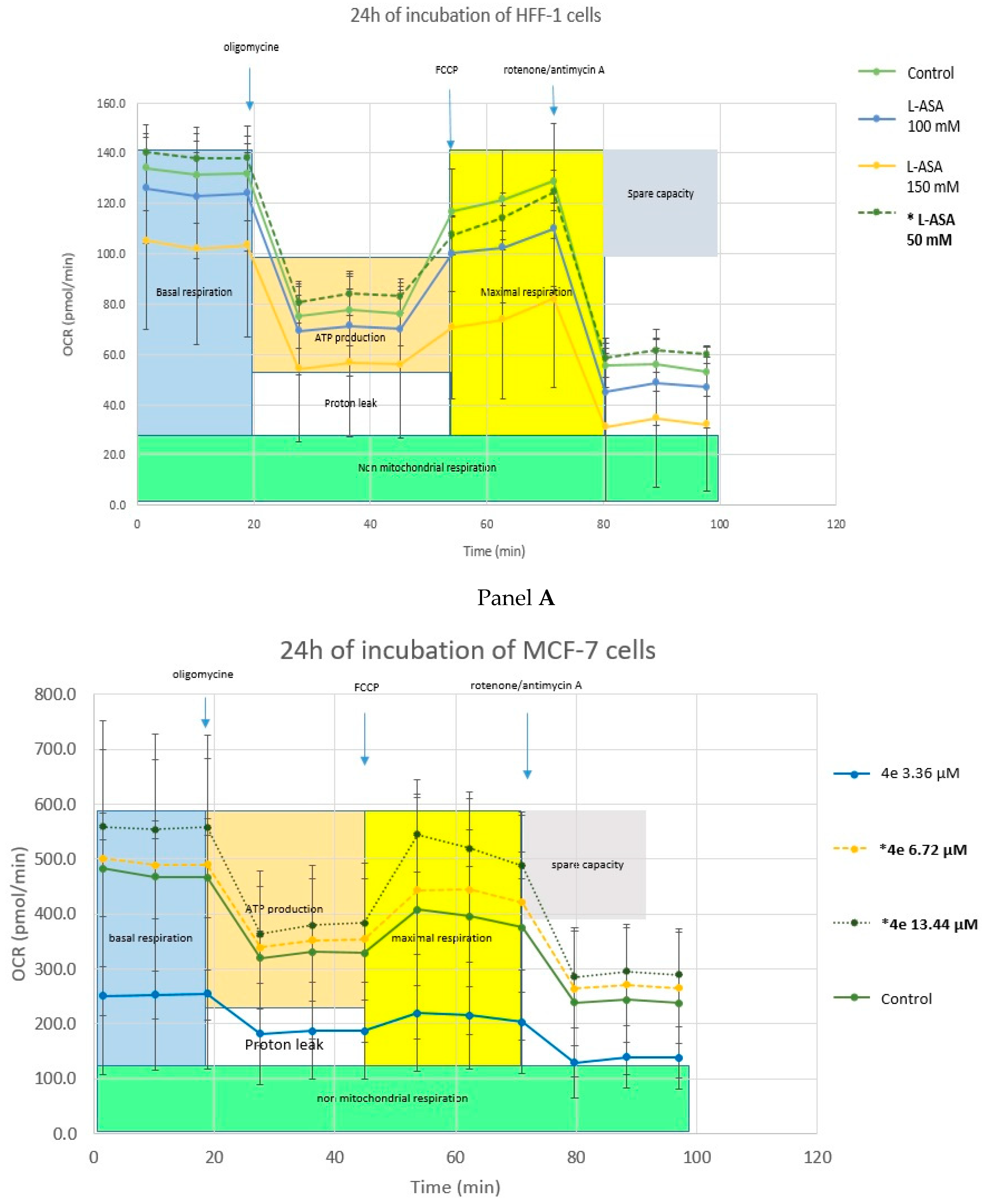
2.4.3. Cell Metabolism Analysis—Mitochondrial Toxicity
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Preparation of Compounds
3.2. Biological Evaluation
3.2.1. DPPH Assay
3.2.2. In Silico Approaches—DFT Calculations and PCA Analysis
3.2.3. Cell Culturing
3.2.4. Proliferation Assay- MTT Assay
3.2.5. Apoptosis Detection—Annexin V Assay
3.2.6. Western Blot Analysis
3.2.7. Seahorse XF Cell Myto Stress Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Salman, K.A.; Ashraf, S. Reactive oxygen species: A link between chronic inflammation and cancer. Asia-Pacific J. Mol. Biol. Biotechnol. 2013, 21, 42–49. [Google Scholar]
- Krishnamurthy, P.; Wadhwani, A. Antioxidant Enzymes and Human Health. In Antioxidant Enzyme; Mohammed Amr El-Missiry, Ed.; IntechOpen: London, UK, 2012. [Google Scholar]
- Adly, A.A.M. Oxidative Stress and Disease: An Updated Review. Res. J. Immunol. 2010, 3, 129–145. [Google Scholar]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; De Ciucis, C.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-Z.; Yang, S.; Wu, G. Free radicals, antioxidants, and nutrition. Nutrition 2002, 18, 872–879. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13–757. [Google Scholar] [CrossRef]
- Ryan, M.J.; Dudash, H.J.; Docherty, M.; Geronilla, K.B.; Baker, B.A.; Haff, G.G.; Cutlip, R.G.; Alway, S.E. Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes and positive muscle work in chronically loaded muscles of aged rats. Exp. Gerontol. 2010, 45, 882–895. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Levine, M.; Dutta, A.; et al. Vitamin C as an Antioxidant: Evaluation of Its Role in Disease Prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta. 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Meščić Macan, A.; Gazivoda Kraljević, T.; Raić-Malić, S. Therapeutic perspectives of vitamin C and its derivatives. Antioxidants 2019, 8, 247. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R.; Jurkiewicz, B.A. Ascorbate free radical as a marker of oxidative stress: An EPR study. Free Radic. Biol. Med. 1993, 14, 49–55. [Google Scholar] [CrossRef]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Weber, V.; Coudert, P.; Rubat, C.; Duroux, E.; Leal, F.; Couqulet, J.; Pharmacologie, L. De Antioxidant Properties of Novel Lipophilic Ascorbic Acid Analogues. J. Pharm. Pharmacol. 2000, 2, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Nihro, Y.; Miyataka, H.; Sudo, T.; Matsumoto, H.; Satoh, T. 3-O-Alkylascorbic acids as free-radical quenchers: Synthesis and inhibitory effect on lipid peroxidation. J. Med. Chem. 1991, 34, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Terao, S.; Shimamoto, N.; Hirata, M. Studies on Scavengers of Active Oxygen Species. 1. Synthesis and Biological Activity of 2 - 0 -Alkylascorbic Acids. J. Med. Chem. 1988, 31, 793–798. [Google Scholar]
- Tao, Z.; Ren, Y.; Tong, W.; Wei, D. Synthesis of 6-O-acyl-L-ascorbic acid-2-O-phosphates and study of their antioxidant effects in 95-D cells. Pharmacol. Rep. 2005, 57, 77–83. [Google Scholar]
- Du, C.B.; Liu, J.W.; Su, W.; Ren, Y.H.; Wei, D.Z. The protective effect of ascorbic acid derivative on PC12 cells: Involvement of its ROS scavenging ability. Life Sci. 2003, 74, 771–780. [Google Scholar] [CrossRef]
- Lopez, E.; del Carmen Ortega-Liébana, M.; Salido, S.; Salido, G.M.; Altarejos, J.; Rosado, J.A.; Redondo, P.C. Evaluation of the antiaggregant activity of ascorbyl phenolic esters with antioxidant properties. J. Physiol. Biochem. 2015, 71, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Fritz, H.; Flower, G.; Weeks, L.; Cooley, K.; Callachan, M.; McGowan, J.; Skidmore, B.; Kirchner, L.; Seely, D. Intravenous vitamin C and cancer: A systematic review. Integr. Cancer Ther. 2014, 13, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C Pharmacokinetics: Implications for Oral and Intravenous Use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, A.Y.; Chen, Q.; Espey, M.G.; Drisko, J.; Levine, M. Vitamin C: Intravenous Use by Complementary and Alternative Medicine Practitioners and Adverse Effects. PLoS ONE 2010, 5, e11414. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic metals, ascorbate and free radicals: Combinations to avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. Revisiting vitamin C and cancer. Science 2015, 350, 1317–1318. [Google Scholar] [CrossRef] [PubMed]
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by Vitamin C-induced oxidative stress. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Byun, J.S.; Kwon, H.S.; Kim, D.Y. Cellular toxicity driven by high-dose vitamin C on normal and cancer stem cells. Biochem. Biophys. Res. Commun. 2018, 497, 347–353. [Google Scholar] [CrossRef]
- Kawada, H.; Kaneko, M.; Sawanobori, M.; Uno, T.; Matsuzawa, H.; Nakamura, Y.; Matsushita, H.; Ando, K. High Concentrations of L-Ascorbic Acid Specifically Inhibit the Growth of Human Leukemic Cells via Downregulation of HIF-1α Transcription. PLoS ONE 2013, 8, e62717. [Google Scholar] [CrossRef]
- Miles, S.L.; Fischer, A.P.; Joshi, S.J.; Niles, R.M. Ascorbic acid and ascorbate-2-phosphate decrease HIF activity and malignant properties of human melanoma cells. BMC Cancer 2015, 15, 867. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J.; Raval, R.R.; Harris, A.L.; Ratcliffe, P.J. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003, 63, 1764–1768. [Google Scholar] [PubMed]
- Fischer, A.P.; Miles, S.L.; Venturelli, S.; Sinnberg, T.W.; Niessner, H.; Busch, C.; Osipyants, A.I.; Poloznikov, A.A.; Smirnova, N.A.; Hushpulian, D.M.; et al. Pharmacologic ascorbate (P-AscH − ) suppresses hypoxia-inducible Factor-1α (HIF-1α) in pancreatic adenocarcinoma. Free Radic. Biol. Med. 2018, 8, 1–10. [Google Scholar]
- Blaszczak, W.; Barczak, W.; Masternak, J.; Kopczyński, P.; Zhitkovich, A.; Rubiś, B. Vitamin C as a Modulator of the Response to Cancer Therapy. Molecules 2019, 24, 453. [Google Scholar] [CrossRef]
- Vissers, M.C.M.; Das, A.B. Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Kuiper, C.; Vissers, M.C.M. Ascorbate as a co-factor for fe- and 2-oxoglutarate dependent dioxygenases: Physiological activity in tumor growth and progression. Front. Oncol. 2014, 4, 359. [Google Scholar] [CrossRef]
- Kuiper, C.; Dachs, G.U.; Currie, M.J.; Vissers, M.C.M. Intracellular ascorbate enhances hypoxia-inducible factor ()-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic. Biol. Med. 2014, 69, 308–317. [Google Scholar] [CrossRef]
- Meščić Macan, A.; Harej, A.; Cazin, I.; Klobučar, M.; Stepanić, V.; Pavelić, K.; Kraljević Pavelić, S.; Schols, D.; Snoeck, R.; Andrei, G.; et al. Antitumor and antiviral activities of 4-substituted 1,2,3-triazolyl-2,3-dibenzyl-L-ascorbic acid derivatives. Eur. J. Med. Chem. (submitted for review).
- Wang, X.; Huang, B.; Liu, X.; Zhan, P. Discovery of bioactive molecules from CuAAC click-chemistry-based combinatorial libraries. Drug Discov. Today 2016, 21, 118–132. [Google Scholar] [CrossRef]
- Stepanić, V.; Gall Trošelj, K.; Lučić, B.; Marković, Z.; Amić, D. Bond dissociation free energy as a general parameter for flavonoid radical scavenging activity. Food Chem. 2013, 141, 1562–1570. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Gregorić, T.; Sedić, M.; Grbčić, P.; Tomljenović Paravić, A.; Kraljević Pavelić, S.; Cetina, M.; Vianello, R.; Raić-Malić, S. Novel pyrimidine-2,4-dione–1,2,3-triazole and furo [2,3-d]pyrimidine-2-one–1,2,3-triazole hybrids as potential anti-cancer agents: Synthesis, computational and X-ray analysis and biological evaluation. Eur. J. Med. Chem. 2017, 125, 1247–1267. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S. Pathophysiological Role of S-Nitrosylation and Transnitrosylation Depending on S-Nitrosoglutathione Levels Regulated by S-Nitrosoglutathione Reductase. Biomol. Ther. (Seoul). 2018, 26, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Poyton, R.; Hendrickson, M. Crosstalk between nitric oxide and hypoxia-inducible factor signaling pathways: An update. Res. Rep. Biochem. 2015, 5, 147–161. [Google Scholar] [CrossRef]
- Olson, N.; van der Vliet, A. Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide 2011, 25, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Ball, K.A.; Nelson, A.W.; Foster, D.G.; Poyton, R.O. Nitric oxide produced by cytochrome c oxidase helps stabilize HIF-1α in hypoxic mammalian cells. Biochem. Biophys. Res. Commun. 2012, 420, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.-Y. Regulation of HIF-1α Stability through S-Nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef]
- Berchner-Pfannschmidt, U.; Tug, S.; Kirsch, M.; Fandrey, J. Oxygen-sensing under the influence of nitric oxide. Cell. Signal. 2010, 22, 349–356. [Google Scholar] [CrossRef]
- Metzen, E.; Zhou, J.; Jelkmann, W.; Fandrey, J.; Brüne, B. Nitric Oxide Impairs Normoxic Degradation of HIF-1α by Inhibition of Prolyl Hydroxylases. Mol. Biol. Cell 2003, 14, 3470–3481. [Google Scholar] [CrossRef]
- Mateo, J.; García-Lecea, M.; Cadenas, S.; Hernández, C.; Moncada, S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Muellner, M.K.; Schreier, S.M.; Schmidbauer, B.; Moser, M.; Quehenberger, P.; Kapiotis, S.; Goldenberg, H.; Laggner, H. Vitamin C inhibits NO-induced stabilization of HIF-1α in HUVECs. Free Radic. Res. 2010, 44, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Pathi, S.S.; Lei, P.; Sreevalsan, S.; Chadalapaka, G.; Jutooru, I.; Safe, S. Pharmacologic doses of ascorbic acid repress specificity protein (Sp) transcription factors and Sp-regulated genes in colon cancer cells. Nutr. Cancer 2011, 63, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Isnaini, I.; Permatasari, N.; Mintaroem, K.; Prihardina, B.; Widodo, M.A. Oxidants-Antioxidants Profile in the Breast Cancer Cell Line MCF-7. Asian Pac. J. Cancer Prev. 2018, 19, 3175–3178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Meščić, A.; Šalić, A.; Gregorić, T.; Zelić, B.; Raić-Malić, S. Continuous flow-ultrasonic synergy in click reactions for the synthesis of novel 1,2,3-triazolyl appended 4,5-unsaturated l -ascorbic acid derivatives. RSC Adv. 2017, 7, 791–800. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Bartmess, J.E. Thermodynamics of the Electron and the Proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Roduner, E. Hydrophobic solvation, quantum nature, and diffusion of atomic hydrogen in liquid water. Radiat. Phys. Chem. 2005, 72, 201–206. [Google Scholar] [CrossRef]
- Alongi, K.S.; Shields, G.C. Theoretical Calculations of Acid Dissociation Constants: A Review Article. Annu. Rep. Comput. Chem. 2010, 6, 113–138. [Google Scholar]
- Zhan, C.-G.; Dixon, D.A. The Nature and Absolute Hydration Free Energy of the Solvated Electron in Water. J. Phys. Chem. 2003, 107, 4403–4417. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 28 January 2019).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | ||||||
|---|---|---|---|---|---|---|---|
| Comp. | R | IC50 (mM)a | Comp. | R | IC50 (mM)a | ||
| Treatm. 5 min | Treatm. 10 min | Treatm. 5 min | Treatm. 10 min | ||||
| L-ASA | 0.18 ± 0.06 | 0.17 ± 0.05 | 4t |  | 0.22 ± 0.08 | 0.12 ± 0.02 | |
| 4b | H | 0.13 ± 0.05 | 0.13 ± 0.05 | 4v |  | 0.27 ± 0.01 | 0.27 |
| 4c |  | 0.26 ± 0.02 | 0.25 | 7c |  | 0.28 | 0.28 |
| 4d |  | 0.27 ± 0.01 | 0.27 | 7d |  | 0.23 | 0.23 |
| 4e |  | 0.20 ± 0.01 | 0.19 ± 0.01 | 7e |  | 0.28 | 0.28 |
| 4f |  | 0.20 ± 0.02 | 0.14 ± 0.01 | 7f |  | 0.10 ± 0.02 | 0.10 ± 0.02 |
| 4g |  | 0.19 ± 0.01 | 0.19 ± 0.01 | 7g |  | > 0.28 | 0.26 |
| 4i |  | >0.28 | >0.28 | 7i |  | 0.10 ± 0.05 | 0.10 ± 0.04 |
| 4k |  | 0.21 ± 0.07 | 0.13 ± 0.04 | 7j |  | 0.06 | 0.06 |
| 4l |  | 0.17 ± 0.16 | 0.16 ± 0.14 | 7k |  | 0.06 | 0.06 |
| 4m |  | >0.28 | >0.28 | 7l |  | 0.20 ± 0.01 | 0.20 |
| 4n |  | 0.15 ± 0.06 | 0.10 ± 0.03 | 7m |  | 0.20 | 0.19 |
| 4o |  | 0.24 ± 0.028 | 0.24 ± 0.04 | 7n |  | 0.14 | 0.10 |
| 4p |  | 0.15 ± 0.06 | 0.15 ± 0.06 | 7o |  | 0.20 | 0.20 |
| 4q |  | 0.24 ± 0.02 | 0.23 | 7p |  | 0.20 | 0.20 |
| 4r |  | 0.25 | 0.27 ± 0.02 | 7q |  | 0.07 | 0.07 |
| 4s |  | 0.27 ± 0.02 | 0.25 | ||||
| Comp. | Active OH | pKa | ETFEA | pKaNR | BDFEA | ETFERA |
|---|---|---|---|---|---|---|
| L-ASA | 2-OH | 10.3 | 72.6 | −6.3 | 57.3 | |
| 3-OH | 3.6 | 79.6 | −4.4 | 66.9 | 76.6 | |
| L-ASA_C4=C5a | 2-OH | 7.8 | 73.9 | −4.4 | 61.3 | |
| 3-OH | 3.6 | 81.3 | −5.6 | 66.9 | 77.9 | |
| 4b | 2-OH | 10.4 | 72.7 | −6.7 | 56.9 | |
| 3-OH | 3.7 | 79.3 | −5.0 | 66.6 | 76.9 | |
| 7b | 2-OH | 7.4 | 75.1 | −5.0 | 61.6 | |
| 3-OH | 3.5 | 82.0 | −5.8 | 67.0 | 79.0 | |
| 4e | 2-OH | 10.4 | 71.0 | −5.3 | 60.4 | |
| 3-OH | 3.5 | 82.0 | −4.1 | 67.9 | 77.3 | |
| 7e | 2-OH | 9.5 | 72.7 | −4.2 | 60.4 | |
| 3-OH | 5.1 | 80.6 | −5.4 | 66.5 | 79.7 | |
| 4g | 2-OH | 10.2 | 79.8 | −11.0 | 58.2 | |
| 3-OH | 3.4 | 79.2 | −3.8 | 67.3 | 76.5 | |
| 7g | 2-OH | 7.6 | 74.7 | −4.6 | 61.8 | |
| 3-OH | 4.3 | 80.3 | −5.4 | 66.3 | 78.3 | |
| 4i | 2-OH | 10.4 | 72.5 | −6.1 | 57.6 | |
| 3-OH | 3.3 | 79.8 | −4.2 | 67.3 | 77.1 | |
| 7i | 2-OH | 7.4 | 76.0 | −5.1 | 62.3 | |
| 3-OH | 4.0 | 80.5 | −5.0 | 66.9 | 78.1 | |
| 4q | 2-OH | 10.7 | 72.8 | −6.5 | 60.4 | |
| 3-OH | 2.4 | 82.3 | −5.1 | 68.6 | 79.0 | |
| 7q | 2-OH | 7.3 | 74.5 | −4.2 | 62.0 | |
| 3-OH | 3.5 | 80.8 | −5.1 | 67.1 | 78.0 |
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | R | IC50 a (µM) | clogPb | ||||||||
| A549 | CFPAC-1 | HCT-116 | HeLa | HepG2 | MCF-7 | SW620 | WI-38 | HFF-1 | |||
| 4b | H | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | NA | −2.89 |
| 4c |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.56 |
| 4d |  | 81.65 | 75.82 | 64.79 | 56.76 | 95.34 | 39.81 | 92.04 | >100 | 0.12 | −0.94 |
| 4e |  | 25.44 | >100 | >100 | >100 | >100 | 6.72 | >100 | 73.93 | >100 | −0.42 |
| 4f |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 69.32 | −0.80 |
| 4g |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | −1.21 |
| 4i |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.92 | −1.49 |
| 4k |  | >100 | >100 | >100 | >100 | >100 | 26.91 | >100 | >100 | 0.21 | 0.98 |
| 4l |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 4.61 | −1.73 |
| 4m |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.12 | −1.17 |
| 4n |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.06 | 1.56 |
| 4o |  | >100 | >100 | >100 | 93.04 | >100 | >100 | >100 | 94.95 | <0.01 | −1.26 |
| 4p |  | >100 | >100 | >100 | 48.01 | >100 | 99.59 | >100 | 52.65 | 6.05 | −1.39 |
| 4q |  | >100 | >100 | 63.34 | >100 | >100 | >100 | >100 | >100 | >100 | −3.00 |
| 4r |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | −1.95 |
| 4s |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.40 | −1.69 |
| 4t |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | −1.59 |
| 4v |  | 97.98 | >100 | >100 | >100 | >100 | 37.33 | >100 | >100 | NA | −2.40 |
| L-ASA | >100 | >100 | >100 | >100 | >100 | >100 | >100 | NA | >100 | −2.46 | |
| Carboxyamidotriazole | 51.61 | 41.55 | 19.88 | 31.84 | 40.09 | 14.69 | 41.33 | / | 32.61 | ||
| 5-FU | 2.80 | 0.14 | / | 8.81 | 9.04 | 0.096 | 0.08 | 0.94 | / | ||
| Cell Line | Duration of Treatment | Comp.Concentration | Early Apoptosis (%) | Late Apoptosis (%) | Necrosis (%) |
|---|---|---|---|---|---|
| MCF-7 | 24 h | Control | 18.2 | 4.4 | 0 |
| 4e (6.72 µM) | 30.6 | 6.4 | 0.8 | ||
| 4e (13.44 µM) | 5.3 | 38.1 | 15.9 | ||
| 48 h | Control | 11.4 | 10.6 | 0 | |
| 4e (6.72 µM) | 46.6 | 4.2 | 0.8 | ||
| 4e (13.44 µM) | 9.1 | 2.3 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harej, A.; Macan, A.M.; Stepanić, V.; Klobučar, M.; Pavelić, K.; Pavelić, S.K.; Raić-Malić, S. The Antioxidant and Antiproliferative Activities of 1,2,3-Triazolyl-L-Ascorbic Acid Derivatives. Int. J. Mol. Sci. 2019, 20, 4735. https://doi.org/10.3390/ijms20194735
Harej A, Macan AM, Stepanić V, Klobučar M, Pavelić K, Pavelić SK, Raić-Malić S. The Antioxidant and Antiproliferative Activities of 1,2,3-Triazolyl-L-Ascorbic Acid Derivatives. International Journal of Molecular Sciences. 2019; 20(19):4735. https://doi.org/10.3390/ijms20194735
Chicago/Turabian StyleHarej, Anja, Andrijana Meščić Macan, Višnja Stepanić, Marko Klobučar, Krešimir Pavelić, Sandra Kraljević Pavelić, and Silvana Raić-Malić. 2019. "The Antioxidant and Antiproliferative Activities of 1,2,3-Triazolyl-L-Ascorbic Acid Derivatives" International Journal of Molecular Sciences 20, no. 19: 4735. https://doi.org/10.3390/ijms20194735
APA StyleHarej, A., Macan, A. M., Stepanić, V., Klobučar, M., Pavelić, K., Pavelić, S. K., & Raić-Malić, S. (2019). The Antioxidant and Antiproliferative Activities of 1,2,3-Triazolyl-L-Ascorbic Acid Derivatives. International Journal of Molecular Sciences, 20(19), 4735. https://doi.org/10.3390/ijms20194735