A New Approach for the Fabrication of Cytocompatible PLLA-Magnetite Nanoparticle Composite Scaffolds

 ,
,
Abstract
1. Introduction
2. Results
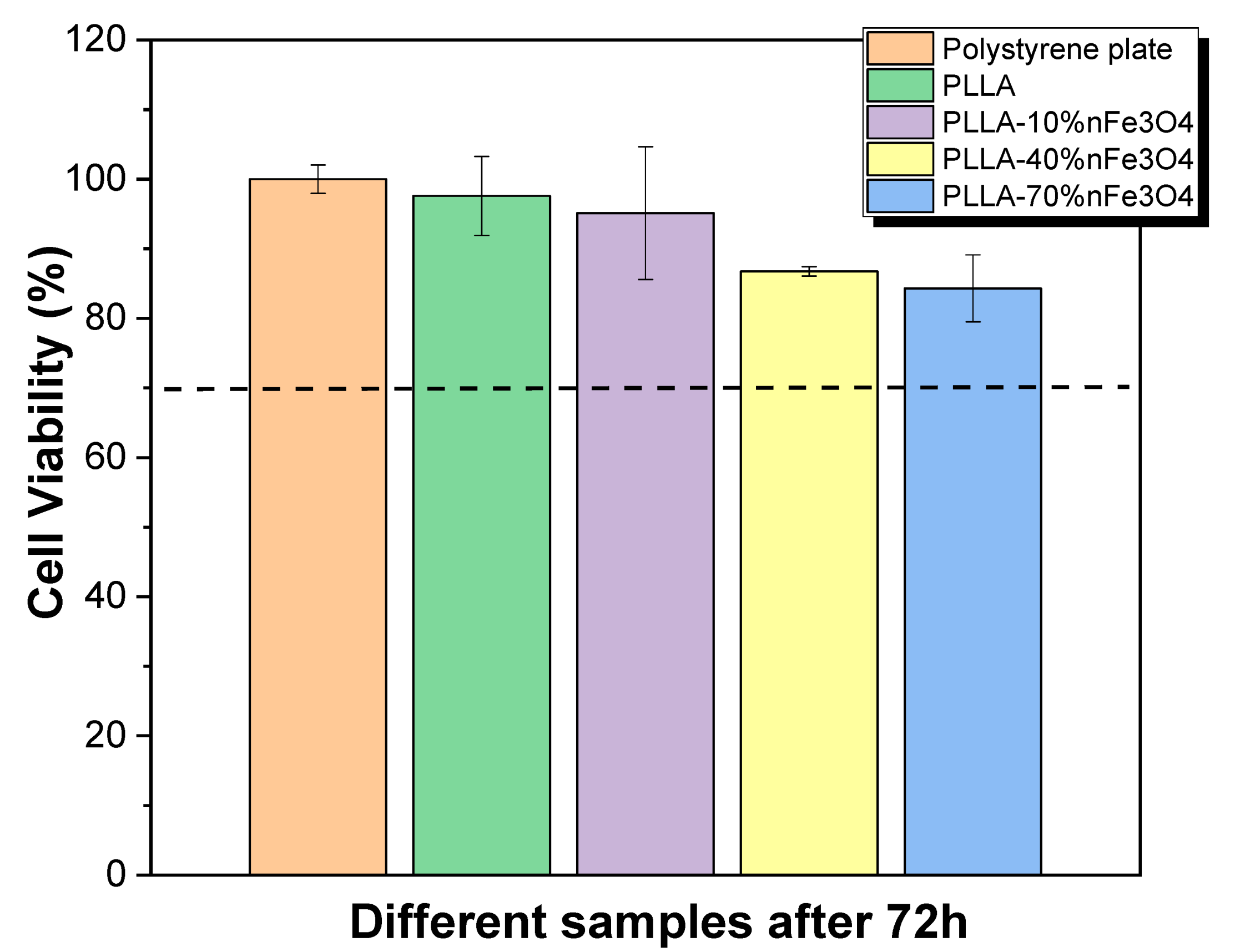
2.1. Cytotoxicity
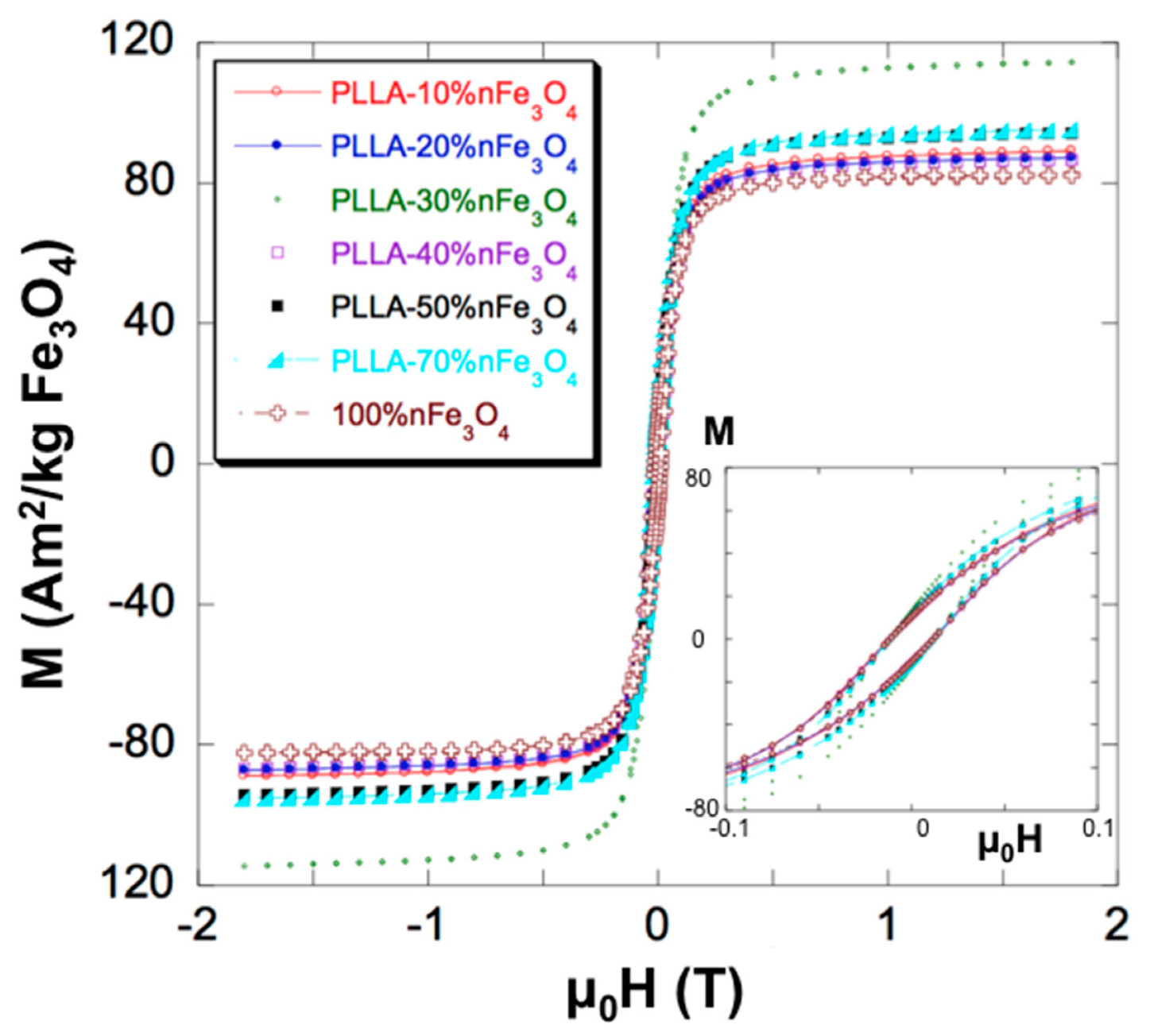
2.2. Magnetic Analysis
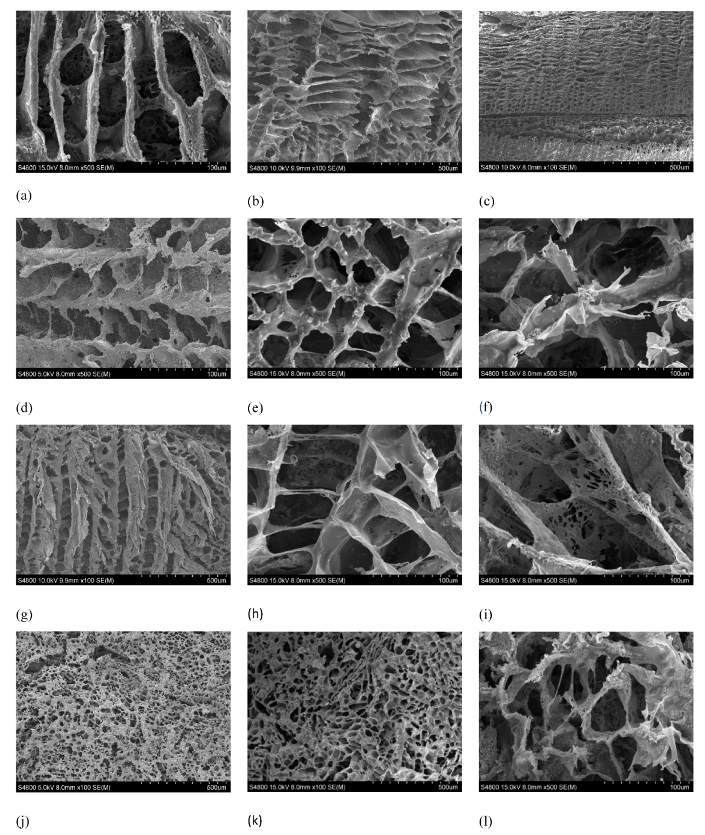
2.3. SEM
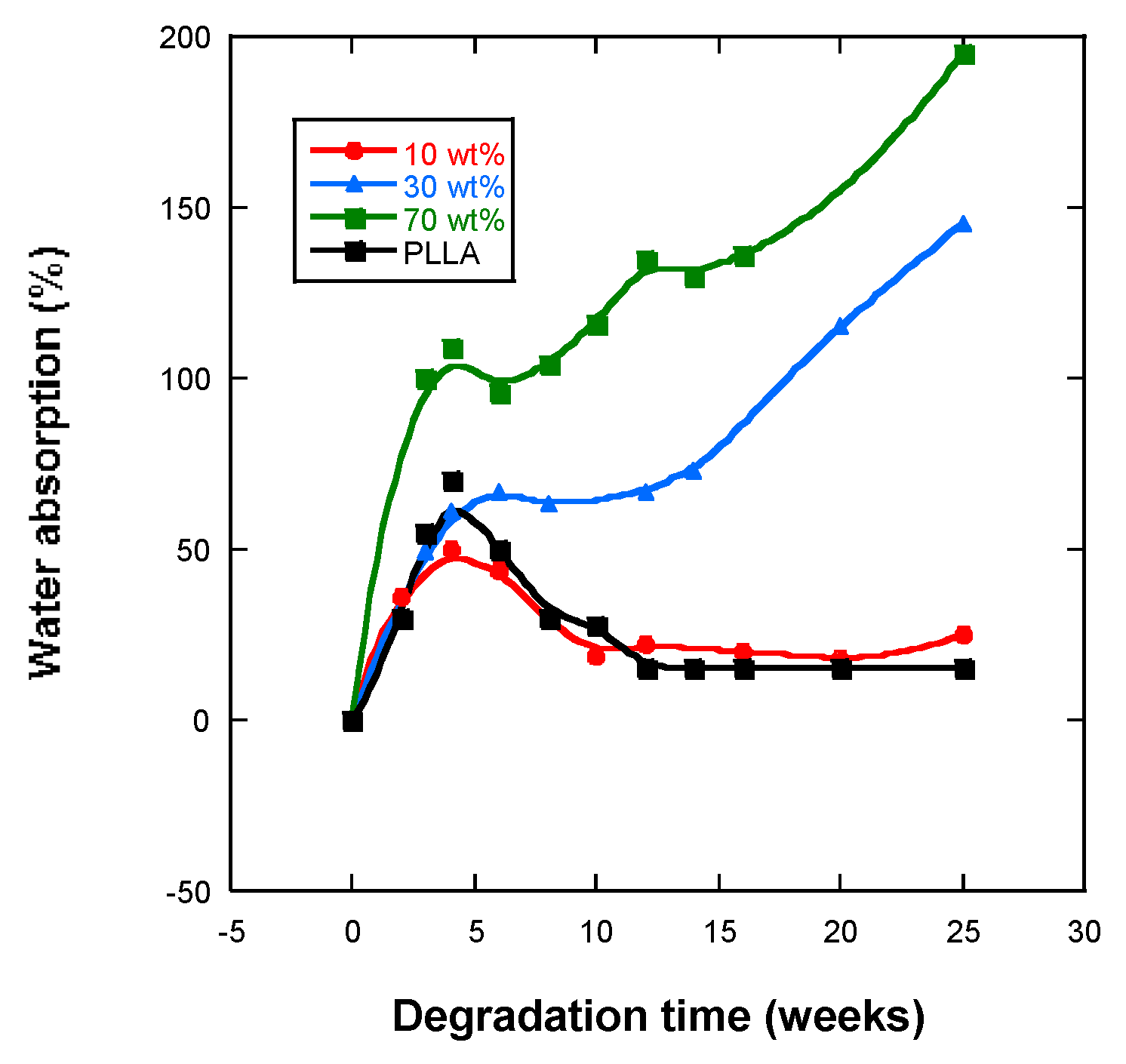
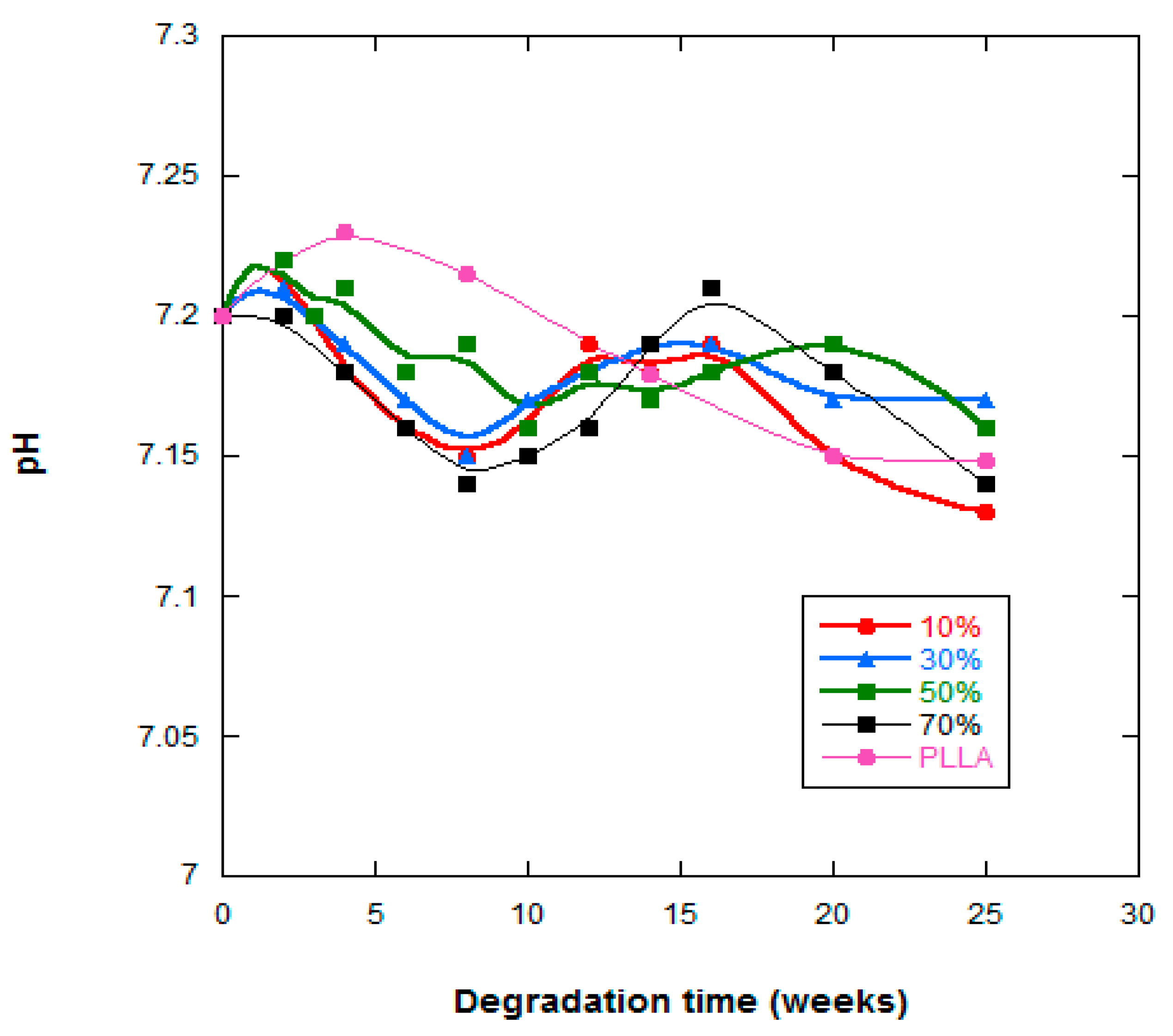
2.4. Water Absorption and pH
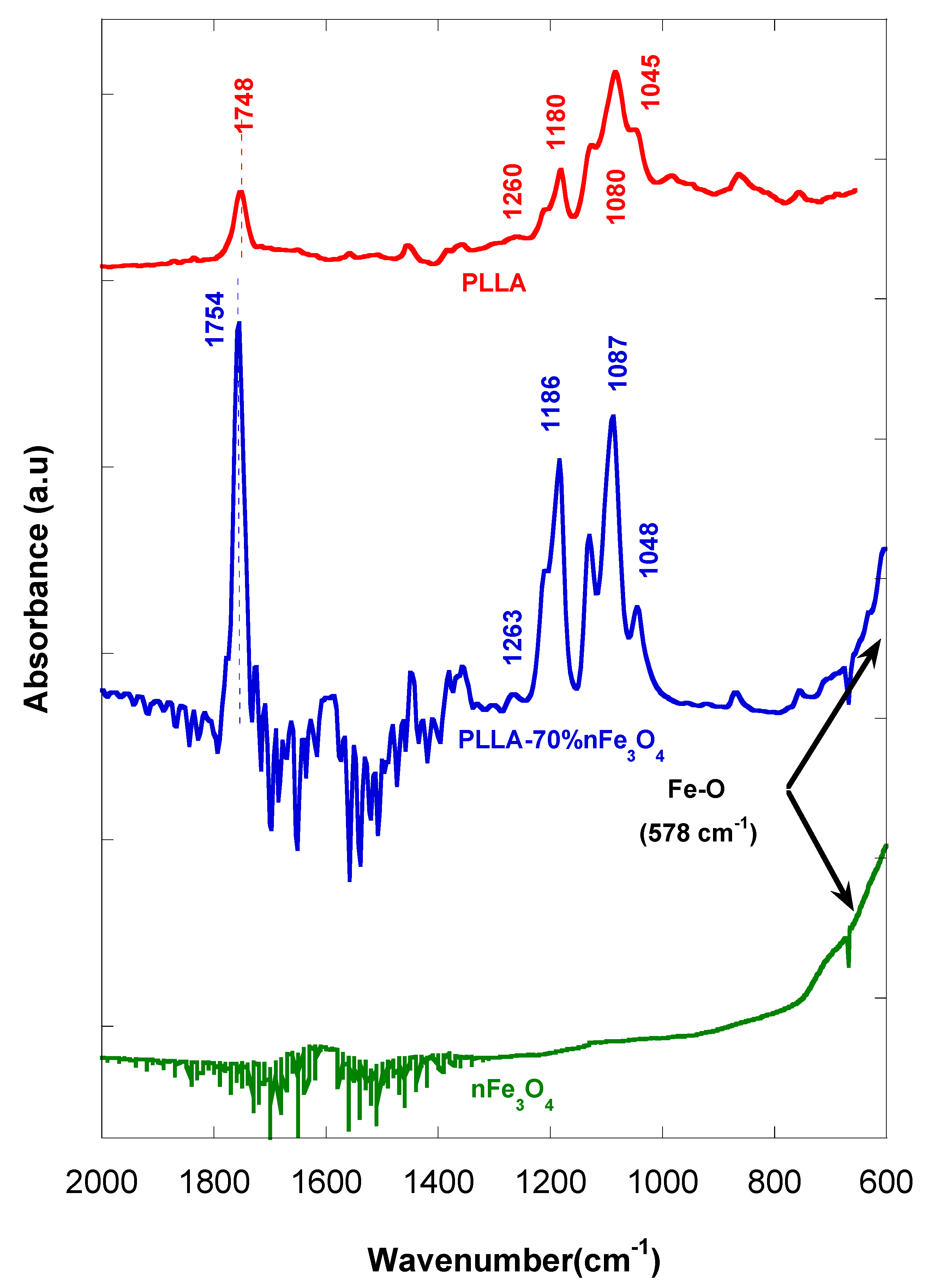
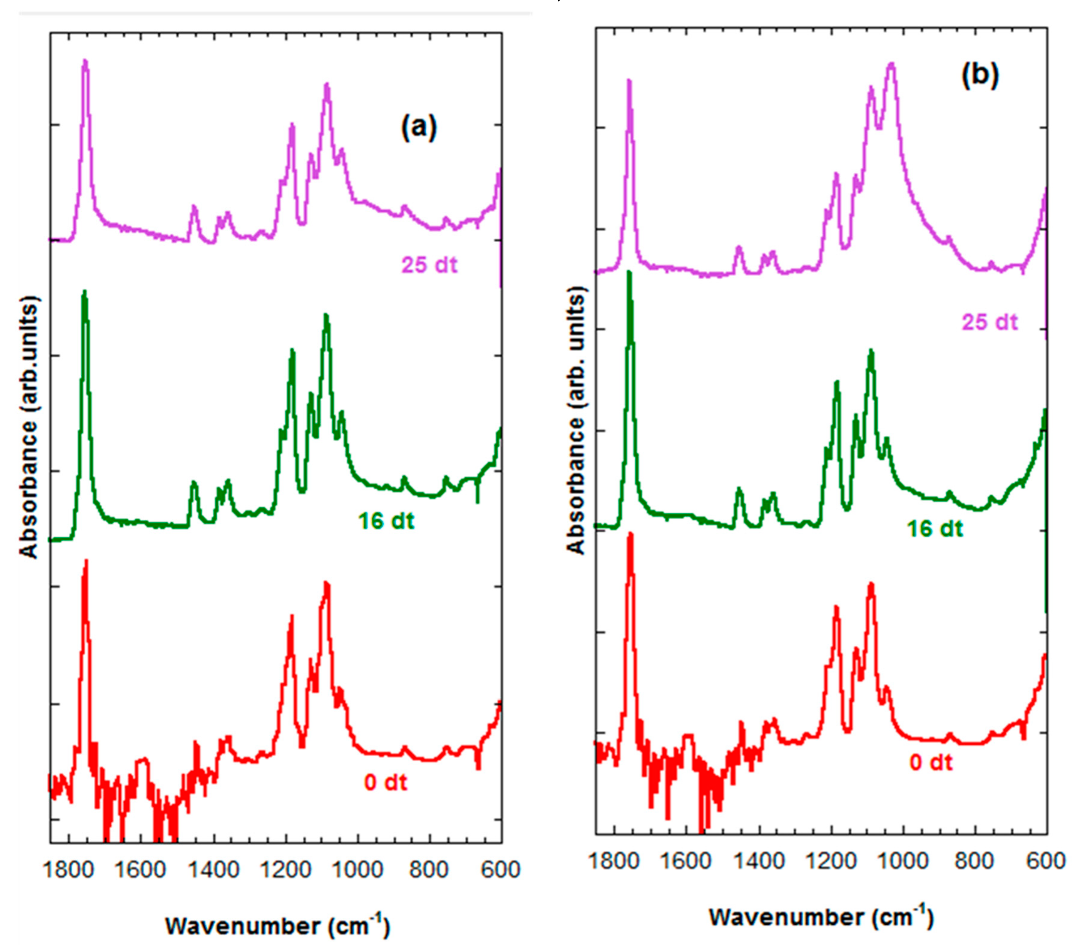
2.5. Infrared Spectroscopy (FTIR)
2.6. Mass Loss and Weight Loss
2.7. Differential Scanning Calorimetry (DSC)
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Fabrication of PLLA/nFe3O4 of Porous Devices
4.3. In Vitro Degradation
4.4. Cytotoxicity
4.5. Magnetic Analysis
5. Differential Scanning Calorimetry (DSC)
6. Infrared Spectroscopy (FTIR)
7. Gel Permeation Chromatography (GPC)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PLLA | poly(L-lactide) |
| Fe3O4 | magnetite |
| PCL | poly(ε-caprolactone) |
| PLCL | poly(ε-caprolactone) |
| PLGA | poly(Lactide-co-Glycolide) |
| MNPs | magnetic nanoparticles |
| MC3T3-E1 | preosteoblast cells |
| PBS | phosphate-buffered saline |
| DMEM | Dulbbecco’s Modified Eagle’s Medium-high glucose (DMEM) |
| SEM | Scanning electron microscopy |
| DSC | Differential scanning calorimetry |
| FTIR | Fourier-Transform Infrared Spectroscopy |
| VSM | Vibrating Sample Magnetometer |
| GPC | Gel permeation chromatography |
| MTT | Colorimetric assay for assessing cell metabolic activity |
References
- Liu, Y.; Lu, Y.; Tian, X.; Cui, G.; Zhao, Y.; Yang, Q.; Yu, S.; Xing, G.; Zhang, B. Segmental bone regeneration using an rhBMP-2-loaded gelatin/nanohydroxyapatite/fibrin scaffold in a rabbit model. Biomaterials 2009, 30, 6276–6285. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, M.J.; Quirk, R.A.; Howdle, S.M.; Shakesheff, K.M. Growth factor release from tissue engineering scaffolds. J. Pharm. Pharmacol. 2001, 53, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Glowacki, J. Angiogenesis in fracture repair. Clin. Orthop. Relat. Res. 1998, 355, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.M.; Zhang, J.; Zhang, X.; Hlavaty, K.A.; Ricci, C.F.; Leonard, J.N.; Shea, L.D.; Gower, R.M. Transforming growth factor-beta 1 delivery from microporous scaffolds decreases inflammation post-implant and enhances function of transplanted islets. Biomaterials 2016, 80, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Milkiewicz, M.; Ispanovic, E.; Doyle, J.L.; Haas, T.L. Regulators of angiogenesis and strategies for their therapeutic manipulation. Int. J. Biochem. Cell. Biol. 2006, 38, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Causa, F.; Netti, P.A.; Ambrosio, L.A. multi-functional scaffold for tissue regeneration: The need to engineer a tissue analogue. Biomaterials 2007, 28, 5093–5099. [Google Scholar] [CrossRef] [PubMed]
- Azeem, A.; Marani, L.; Fuller, K.; Spanoudes, K.; Pandit, A.; Zeugolis, D.I. Influence of Nonsulfated Polysaccharides on the Properties of Electrospun Poly(lactic-co-glycolic acid) Fibers. ACS Biomater. Sci. Eng. 2016, 3, 1304–1312. [Google Scholar]
- Huang, H.M.; Lee, S.Y.; Yao, W.C.; Lin, C.T.; Yeh, C.Y. Static magnetic fields up-regulate osteoblast maturity by affecting local differentiation factors. Clin. Orthop. Relat. Res. 2006, 447, 201–208. [Google Scholar] [CrossRef]
- WãJcik-Piotrowicz, K.; Kaszuba-ZwoiåSka, J.; Rokita, E.; Thor, P. Cell viability modulation through changes of Ca2+-dependent signalling pathways. Prog. Biophys. Mol. Biol. 2016, 121, 45–53. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, S.; Zhou, C.; Cheng, L.; Gao, X.; Xie, X.; Sun, J.; Wang, H.; Weir, M.D.; Reynolds, M.A.; et al. Advanced smart biomaterials and constructs for hard tissue engineering and regeneration. Bone Res. 2018, 6, 31. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, Q.; Yang, M.; Zhan, X.; Lan, F.; He, J.; Gu, Z.; Wu, Y. Protein Corona of Magnetic Hydroxyapatite Scaffold Improves Cell Proliferation via Activation of Mitogen-Activated Protein Kinase Signaling Pathway. ACS Nano. 2017, 11, 3690–3704. [Google Scholar] [CrossRef] [PubMed]
- Arjmand, M.; Ardeshirylajimi, A.; Maghsoudi, H.; Azadian, E. Osteogenic differentiation Potential of Mesenchymal Stem Cells cultured on Nanofibrous Scaffold Improved in the Presence of Pulsed Electromagnetic Field. J. Cell. Physiol. 2017, 233, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Murugan, R.; Ramakrishna, S.; Wang, X.; Ma, Y.X.; Wang, S. Fabrication of nano-estructured porous PLLA scaffolds intended for nerve tissue engineering. Biomaterials 2004, 25, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; Sandonis, I.; Valle, M.B. In vitro degradation of Poly(caprolactone)/nHA composites. J. Nanomater. 2014, 185, 1–8. [Google Scholar] [CrossRef]
- Jeong, S.I.; Kwon, J.H.; Lim, J.I.; Cho, S.W.; Jung, Y.; Sung, W.J. Mechano-active tissue engineering of vascular smooth muscle using pulsatile perfusion bioreactors and elastic PLCL scaffolds. Biomaterials 2005, 26, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; Ibañez, I.; Puerto, I. Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, 11 Volume Set; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar] [CrossRef]
- Baek, C.H.; Ko, Y.J. Characteristics of tissue-engineered cartilage on macroporous biodegradable PLGA scaffold. Laryngoscope 2006, 116, 1829–1834. [Google Scholar] [CrossRef]
- Huang, D.M.; Hsiaon, J.K.; Chen, Y.C.; Chien, L.Y.; Yao, M.; Yin-Kai, C.H.; Bor-Sheng, K.; Szu-Chun, H.; Lin-Ai, T.; Hui-Ying, C.H.; et al. The promotion of human mesenchymal stem cell proliferation by superparamagnetic iron oxide nanoparticles. Biomaterials 2009, 30, 3645–3651. [Google Scholar] [CrossRef]
- Chrissafis, K.; Antoniadis, G.; Parskevopoulos, K.M.; Vassiliou, A.; Bikiaris, D.N. Comparative study of the effect of different nanoparticles on the mechanical properties and thermal degradation mechanism of in situ prepared poly (ε-caprolactone) nanocomposites. Compos. Sci. Technol. 2007, 67, 2165–2174. [Google Scholar] [CrossRef]
- Li, J.L.; Zheng, W.; Li, L.; Zheng, Y.; Lou, X. Thermal degradation kinetics of g-HA/PLA composite. Thermochim. Acta 2009, 493, 90–95. [Google Scholar] [CrossRef]
- Gupta, B.; Revagade, N.; Hilborn, J. Poly(lactic acid) fiber: An overview. Prog. Polym. Sci. 2007, 32, 455–482. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, W.; Wen, X.; He, B.; Zeng, X.; Wang, G.; Gu, Z. A novel calcium phosphate ceramic-magnetic nanoparticle composite as a potential bone substitute. Biomed. Mater. 2010, 5, 015001. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Shi, Y.; Duan, S.; Wei, Y.; Cai, Q.; Yang, X. Electrospun magnetic poly(l-lactide) (PLLA) nanofibers by incorporating PLLA-stabilized Fe3O4 nanoparticles. Mater. Sci. Eng. C 2013, 33, 3498–3505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, J.; Pang, X.; Zhao, M.; Wang, B.; Yang, L.; Wan, H.; Wu, J.; Fu, S. Magnetic nanoparticle-loaded electrospun polymeric nanofibers for tissue engineering. Mater. Sci. Eng. C 2017, 73, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.Y.; Feng, S.W.; Chan, Y.H.; Chang, W.J.; Wang, H.T.; Huang, H.M. In vivo investigation into effectiveness of Fe3O4/PLLA nanofibers for bone tissue engineering applications. Polymers 2018, 10, 804. [Google Scholar] [CrossRef] [PubMed]
- Areias, A.C.; Ribeiro, C.; Sencadas, V.; García-Giralt, N.; Diez-Perez, A.; Ribelles, J.G.; Lanceros-Méndez, S. Influence of crystallinity and fiber orientation on hydrophobicity and biological response of poly(l-lactide) electrospun mats. Soft Matter 2012, 8, 5818–5825. [Google Scholar] [CrossRef]
- Ribeiro, C.; Sencadas, V.; Areias, A.C.; Gama, F.M.; Lanceros-Méndez, S. Surface roughness dependent osteoblast and fibroblast response on poly(L-lactide) films and electrospun membranes. J. Biomed. Mater. Res. A 2015, 103, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Sencadas, V.; Correia, D.M.; Lanceros-Méndez, S. Piezoelectric polymers as biomaterials for tissue engineeting applications. Colloids Surf. B 2015, 136, 46–55. [Google Scholar] [CrossRef]
- Ribeiro, C.; Correia, V.; Martins, P.; Gama, F.M.; Lanceros-Mendez, S. Piezo-and magnetoelectric polymers as biomaterials for novel tissue engineering strategies. MRS Advances. 2016, 140, 430–436. [Google Scholar] [CrossRef]
- Rescignano, N.; Gonzalez-Alfaro, Y.; Fantechi, E.; Mannini, M.; Innocenti, C.; Ruiz-Hitzky, E.; Kenny, J.M.; Armentano, I. Design, development and characterization of a nanomagnetic system based on iron oxide nanoparticles encapsulated in PLLA-nanospheres. Eur. Polym. J. 2015, 62, 145–154. [Google Scholar] [CrossRef]
- Schugens, C.; Maquet, V.; Grandfils, C.; Jerome, R.; Teyssie, P. Biodegradable and macroporous polylactide implants for cell transplantation. 1.Preparation of macroporous polylactide supports by solid-liquid phase separation. Polymer 1996, 37, 1027–1038. [Google Scholar] [CrossRef]
- Zhang, R.; Ma, P.X. Porous poly(l-lactic acid)/apatite composites created by biomimetic process. J. Biomed. Mater. Res. 1999, 45, 285–293. [Google Scholar] [CrossRef]
- Nam, Y.S.; Park, T.G. Porous biodegradable polymeric scaffolds prepared by thermally induced phase separation. J. Biomed. Mater. Res. 1999, 47, 8–17. [Google Scholar] [CrossRef]
- Schugens, C.; Maquet, V.; Grandfils, C.; Jerome, R.; Teyssie, P. Polylactide macroporous biodegradable implants for cell transplantation. II. Preparation of polylactide foams by liquid-liquid phase separation. J. Biomed. Mater. Res. 1996, 30, 449–461. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, Y.; Zhang, Z.; Ye, W.; Lang, M. Effect of porosity on long-term degradation of poly (ε-caprolactone) scaffolds and their celular response. Polym. Degrad. Stab. 2012, 98, 209–218. [Google Scholar] [CrossRef]
- Zuluaga, F. Algunas aplicaciones del ácido poli-L-láctico. Rev. Acad. Colomb. Cienc. 2013, 37, 125–142. [Google Scholar]
- Díaz, E.; Sandonis, I.; Puerto, I.; Ibañez, I. In Vitro Degradation of PLLA/ nHA Composite Scaffolds. Polym. Eng. Sci. 2014, 54, 2571–2578. [Google Scholar] [CrossRef]
- Huang, J.; Xiong, J.; Liu, J.; Zhu, W.; Wang, D. Investigation of the In Vitro Degradation of a Novel Polylactide/Nanohydroxyapatite Composite for Artificial Bone. J. Nanomater. 2013, 2013, 515741. [Google Scholar]
- Lafisco, M.; Palazzo, B.; Ito, T.; Otsuka, M.; Senna, M.; Delgado-Lopez, J.M.; Gomez-Morales, J.; Tampieri, A.; Prat, M.; Rimondini, L. Preparation of core–shell poly(L-lactic) acid-nanocrystalline apatite hollow microspheres for bone repairing applications. J. Mater. Sci. Mater. Med. 2012, 23, 2659–2669. [Google Scholar] [CrossRef] [PubMed]
- Kister, G.; Cassanas, G.; Vert, M. Effects of morphology, conformation and configuration on the IR and Raman spectra of various poly(lactic acid)s. Polymer 1998, 39, 267–273. [Google Scholar] [CrossRef]
- Nan, A.; Turcu, R.; Craciunescu, I.; Pana, O.; Scharf, H.; Liebscher, J. Microwave-assited graft polymerization of ε-caprolactone onto magnetite. Polym. Sci. Part A Polym. Chem. 2009, 47, 5397–5404. [Google Scholar] [CrossRef]
- Yuan, W.; Yuan, J.; Zhou, L.; Wu, S.; Hong, X. Fe3O4@ poly (2-hydroxyethyl methacrylate)-graft-poly (ε-caprolactone) magnetic nanoparticles with branched brush polymeric shell. Polymers 2010, 51, 2540–2547. [Google Scholar] [CrossRef]
- Atkins, P.; de Paula, J. Química-Física; Panamericana: Buenos Aires, Argentina, 2008. [Google Scholar]
- Kurimura, Y.; Tsuchida, E.; Kaneko, M. Preparation and properties of some water-soluble cobalt (III)–poly-4-vinylpyridine complexes. J. Polym. Sci. 1971, 9, 3511–3519. [Google Scholar] [CrossRef]
- Partini, M.; Pantani, R. FTIR analysis of hydrolysis in aliphatic polyesters. Polym. Degrad. Stab. 2007, 92, 1491–1497. [Google Scholar] [CrossRef]
- Lam, C.X.F.; Hutmacher, D.W.; Schantz, J.T.; Woodruff, M.A.; Teoh, S.H. Evaluation of polycaprolactone scaffold degradation for 6 months in vitro and in vivo. J. Biomed. Mater. Res. A 2008, 90, 906–919. [Google Scholar]
- Yeo, A.; Rai, B.; Sju, E.; Cheong, J.; Teoh, S. The degradation profile of novel, bioresorbable PCL-TCP scaffolds: An in vitro and in vivo study. J. Biomed. Mater. Res. A 2008, 84, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; Valenciano, R.; Katime, I.A. Study of complexes of poly(vinyl pyrrolidone) with copper and cobalt on solid state. J. Appl. Polym. Sci. 2004, 93, 1512–1518. [Google Scholar] [CrossRef]
- Deplaine, H.; Acosta-Santamaría, V.A.; Vidaurre, A.; Gómez-Ribelles, J.L.; Doblaré, M.; Ochoa, I.; Gallego-Ferrer, G. Evolution of the properties of a poly(l-lactic acid) scaffold with double porosity during in vitro degradation in a phosphate-buffered saline solution. J. Appl. Polym. Sci. 2014, 131, 40956. [Google Scholar] [CrossRef]
- Shyam-Roy, S. Synthesis of Biodegradable Poly (Lactic Acid) Polymers. Ph.D. Thesis, University of Poona, Poona, India, 2003. [Google Scholar]
- Taddei, P.; Di Foggia, M.; Causa, I.; Ambrosio, C. In vitro bioactivity of poly(caprolactone)-apatite (PCL-AP) scaffolds for bone tissue engineering: The influence of the PCL-AP ratio. Int. J. Artif. Organs 2006, 29, 719–725. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| wt % nFe3O4 (Nominal) | Ms (Am2/kg nFe3O4) | μ0 Hc (mT) | wt % nFe3O4 (Recalculated) |
|---|---|---|---|
| 100 * | 82.05 | 11.87 | - |
| 10 | 88.70 | 12.05 | 10.8 |
| 20 | 86.85 | 11.75 | 21.2 |
| 30 | 114.04 | 11.75 | 41.7 |
| 40 | 86.30 | 11.67 | 42 |
| 50 | 94 | 11.62 | 57 |
| 70 | 94.82 | 11.83 | 80.5 |
| Sample | Degradation Time(Weeks) | Mw | %Mw | Mn | I |
|---|---|---|---|---|---|
| PLLA | 0 | 144,221 | 104,042 | 1.386 | |
| 15 | 50,365 | 65.07 | 27,894 | 1.800 | |
| 25 | 31,420 | 78.21 | 20,123 | 1.958 | |
| PLLA-10%nFe3O4 | 0 | 86,087 | 40.30 | 55,270 | 1.558 |
| 15 | 70,655 | 48.99 | 36,196 | 1.952 | |
| 25 | 51,040 | 64.60 | 28,758 | 1.775 | |
| PLLA-30%nFe3O4 | 0 | 62,280 | 56.81 | 35,695 | 1.745 |
| 15 | 32,867 | 77.21 | 22,501 | 1.461 | |
| 25 | 50,367 | 65.10 | 20,911 | 2.409 | |
| PLLA-50%nFe3O4 | 0 | 61,018 | 57.69 | 33,257 | 1.835 |
| 14 | 39,878 | 72.35 | 24,554 | 1.624 | |
| 25 | 31,721 | 78.00 | 14,722 | 2.155 |
| PLLA/%nFe3O4 tdeg(weeks) | First Scan | Second Scan | Xc % a | CF% b | Xcc% c | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tm1 (°C) | ΔHm1 (J/g) | Tcc (°C) | ΔHcc (J/g) | Tg (°C) | Tc (°C) | ΔHc (J/g) | ||||
| 0%, 0dt | 184 | 41.1 | 76 | 4.7 | 56 | 96 | 2.3 | 39 | 6 | 39 |
| 8dt | 183 | 42 | 76.5 | 4 | 57 | 96 | 2.5 | 41 | 6 | 39 |
| 25dt | 184 | 42.3 | 76.5 | 4 | 56.5 | 96 | 2.8 | 41 | 6.6 | 39.3 |
| 10%, 0dt | 183 | 37.9 | 76 | 4 | 57.5 | 96 | 4.1 | 36 | 11 | 40.5 |
| 4dt | 180 | 38.6 | 85.5 | 4.0 | 57 | 98.5 | 12.5 | 37 | 32 | 37 |
| 8dt | 180 | 43.5 | 85 | 6.9 | 58 | 96 | 4.2 | 39 | 10 | 44 |
| 14dt | 182 | 39.8 | 57 | 96 | 4.6 | 43 | 12 | 47.5 | ||
| 20dt | 180 | 40.3 | 72.5 | 4.1 | 58.5 | 98 | 4.8 | 39 | 12 | 43 |
| 20%, 0dt | 183 | 37.1 | 3.9 | 59 | 102 | 19.7 | 36 | 53 | 45 | |
| 30%, 0dt | 184 | 35.5 | 76.5 | 1.8 | 61 | 103 | 19.3 | 36 | 54 | 52 |
| 4dt | 183 | 35.6 | 76.5 | 2.4 | 59 | 101 | 14.5 | 36 | 41 | 51 |
| 8dt | 183 | 36.5 | 77 | 1.3 | 58.5 | 102 | 17 | 38 | 47 | 54 |
| 12dt | 183 | 37.9 | 58 | 100 | 8.7 | 41 | 23 | 58 | ||
| 16dt | 183 | 37.5 | 59 | 100 | 6.9 | 40 | 18 | 57 | ||
| 20dt | 183 | 37.9 | 60 | 98 | 4.6 | 41 | 12 | 58 | ||
| 25dt | 182 | 40 | 62 | 100 | 7.6 | 43 | 19 | 61 | ||
| 50%, 0dt | 183 | 19.9 | 76.4 | 2.5 | 63 | 118 | 14.6 | 19 | 73 | 37 |
| 4dt | 182 | 21.4 | 59 | 104 | 9.7 | 23 | 45 | 46 | ||
| 8dt | 182 | 19.1 | 58 | 101 | 7.6 | 21 | 40 | 41 | ||
| 25dt | 180 | 16.6 | 80 | 0.7 | 59 | 100 | 5.3 | 17 | 32 | 34 |
| 70%, 0dt | 183 | 11.7 | 72 | 1.2 | 62 | 112 | 6.8 | 11 | 58 | 38 |
| 4dt | 182 | 11.1 | 72 | 0.5 | 58 | 104 | 4.8 | 11 | 43 | 38 |
| 8dt | 183 | 10.2 | 75 | 0.7 | 57 | 112 | 4.3 | 10 | 42 | 34 |
| 12dt | 183 | 9 | 59 | 106 | 3.5 | 10 | 39 | 32 | ||
| 16dt | 182 | 9.1 | 50 | 102 | 2.7 | 10 | 30 | 33 | ||
| 20dt | 182 | 9 | 75 | 1.1 | 58 | 102 | 2.6 | 9 | 29 | 28 |
| 25dt | 181 | 9 | 57 | 103 | 3.2 | 10 | 36 | 32 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, E.; Valle, M.B.; Ribeiro, S.; Lanceros‑Mendez, S.; Barandiarán, J.M. A New Approach for the Fabrication of Cytocompatible PLLA-Magnetite Nanoparticle Composite Scaffolds. Int. J. Mol. Sci. 2019, 20, 4664. https://doi.org/10.3390/ijms20194664
Díaz E, Valle MB, Ribeiro S, Lanceros‑Mendez S, Barandiarán JM. A New Approach for the Fabrication of Cytocompatible PLLA-Magnetite Nanoparticle Composite Scaffolds. International Journal of Molecular Sciences. 2019; 20(19):4664. https://doi.org/10.3390/ijms20194664
Chicago/Turabian StyleDíaz, Esperanza, María Blanca Valle, Sylvie Ribeiro, Senentxu Lanceros‑Mendez, and José Manuel Barandiarán. 2019. "A New Approach for the Fabrication of Cytocompatible PLLA-Magnetite Nanoparticle Composite Scaffolds" International Journal of Molecular Sciences 20, no. 19: 4664. https://doi.org/10.3390/ijms20194664
APA StyleDíaz, E., Valle, M. B., Ribeiro, S., Lanceros‑Mendez, S., & Barandiarán, J. M. (2019). A New Approach for the Fabrication of Cytocompatible PLLA-Magnetite Nanoparticle Composite Scaffolds. International Journal of Molecular Sciences, 20(19), 4664. https://doi.org/10.3390/ijms20194664