Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond

 ,
, 
Abstract

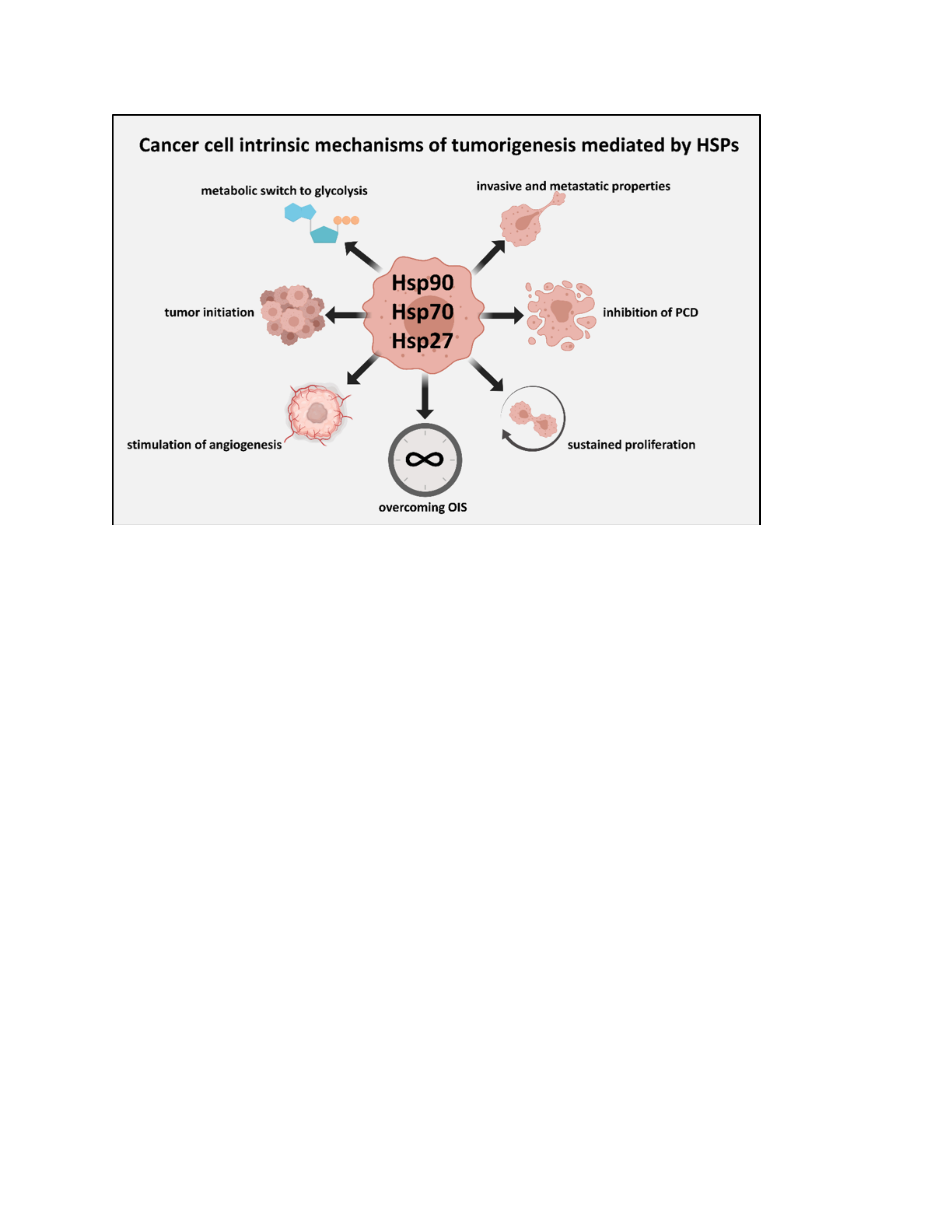{kind=link}
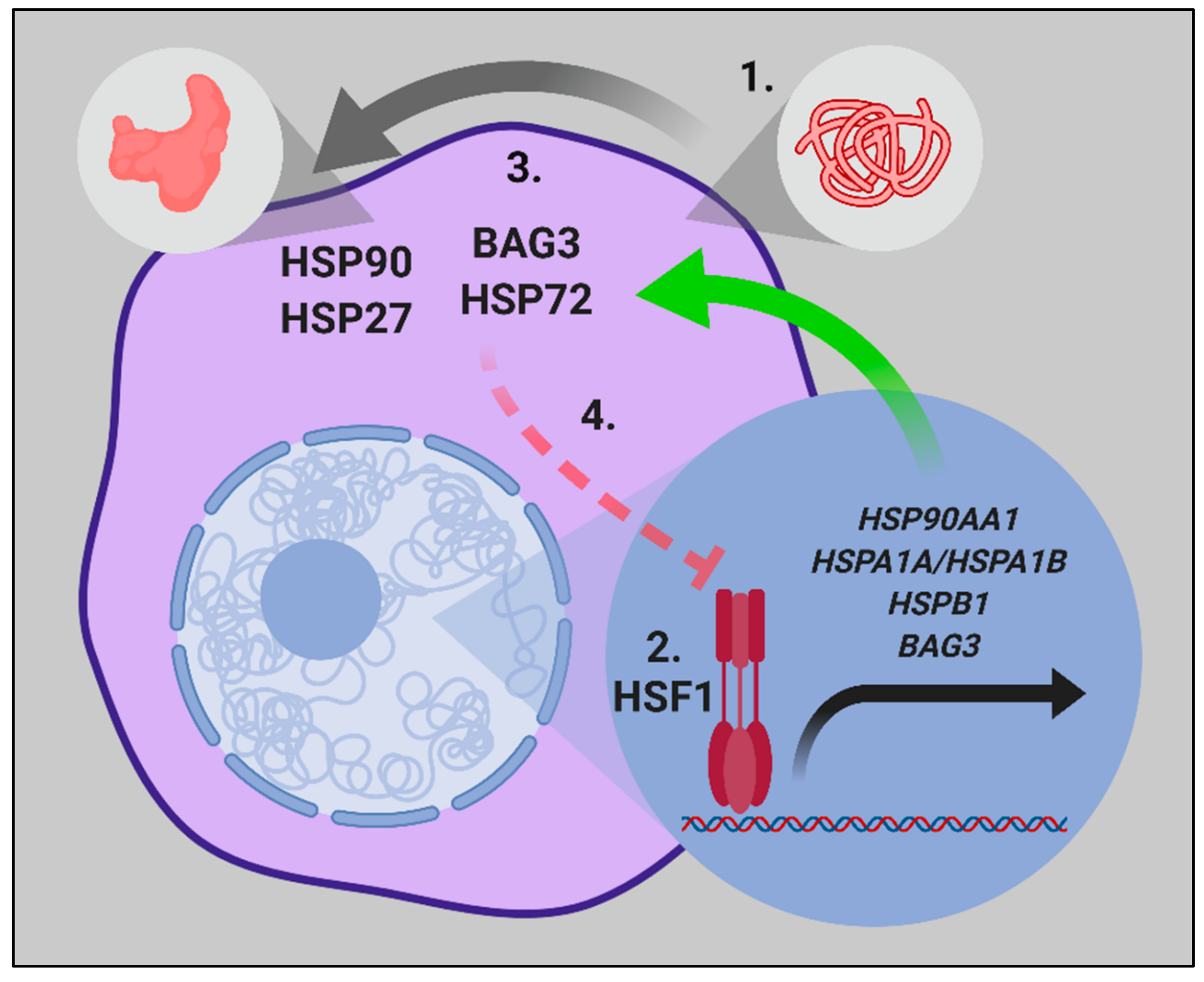{kind=link}
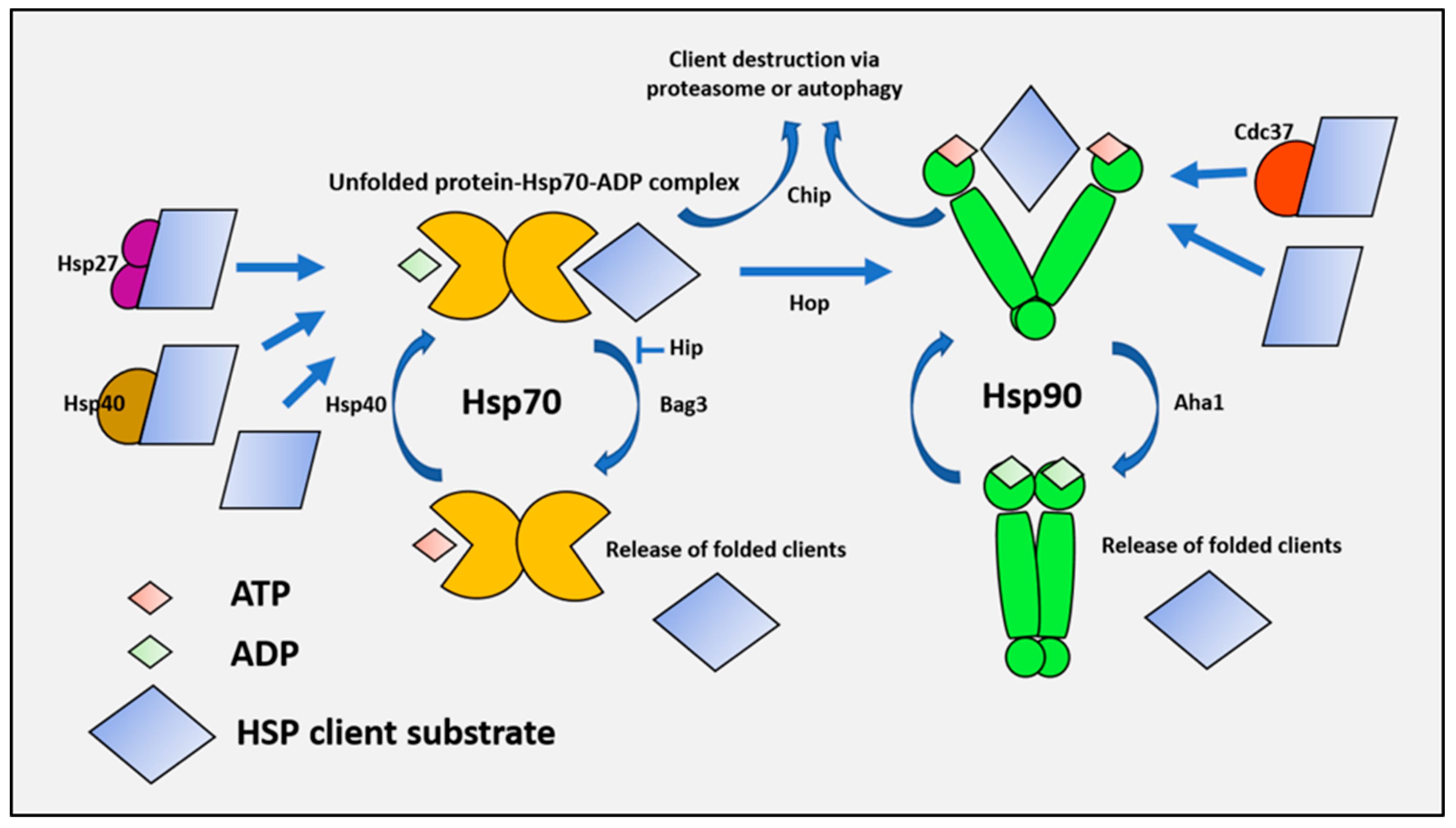{kind=link}
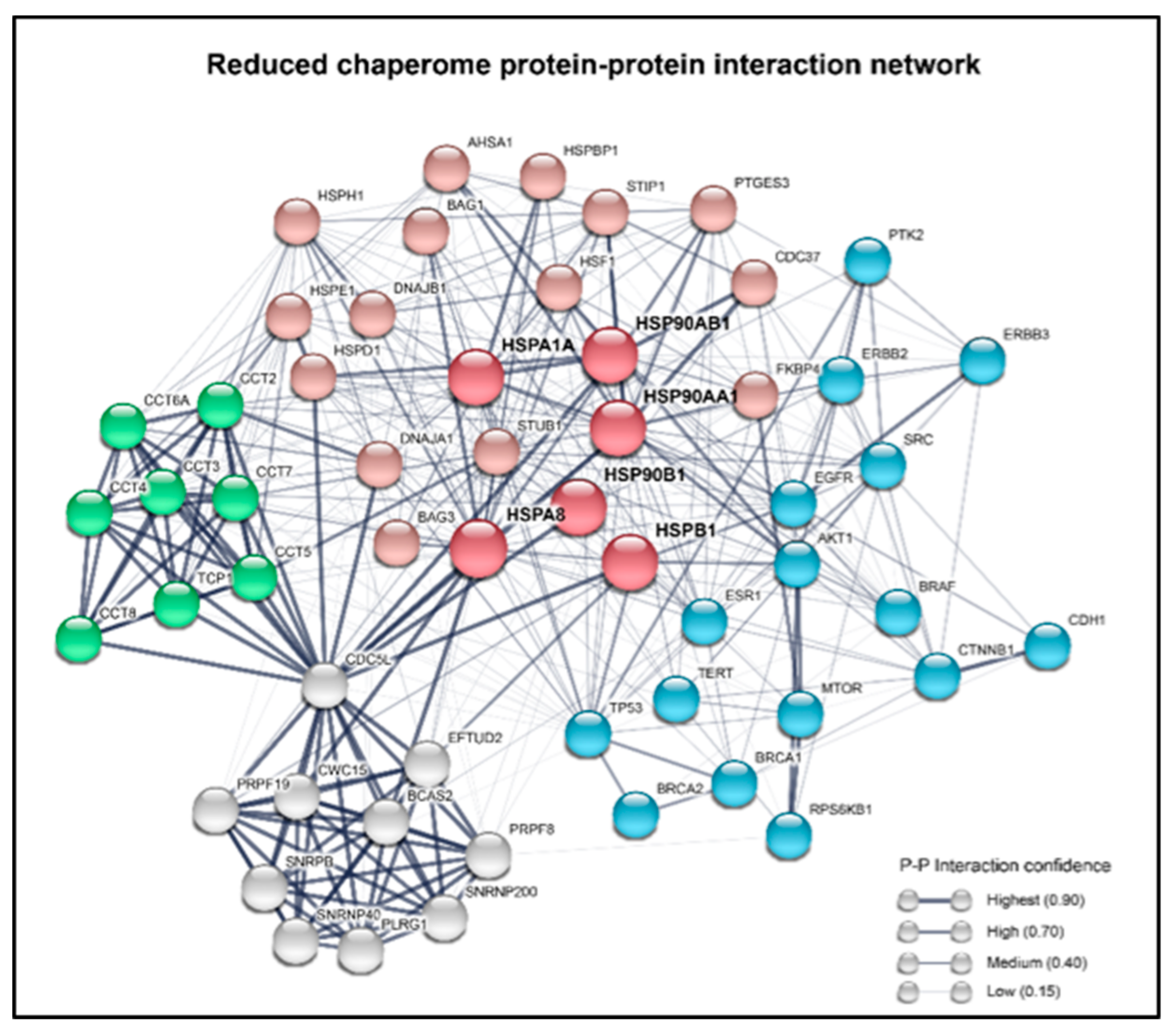{kind=link}
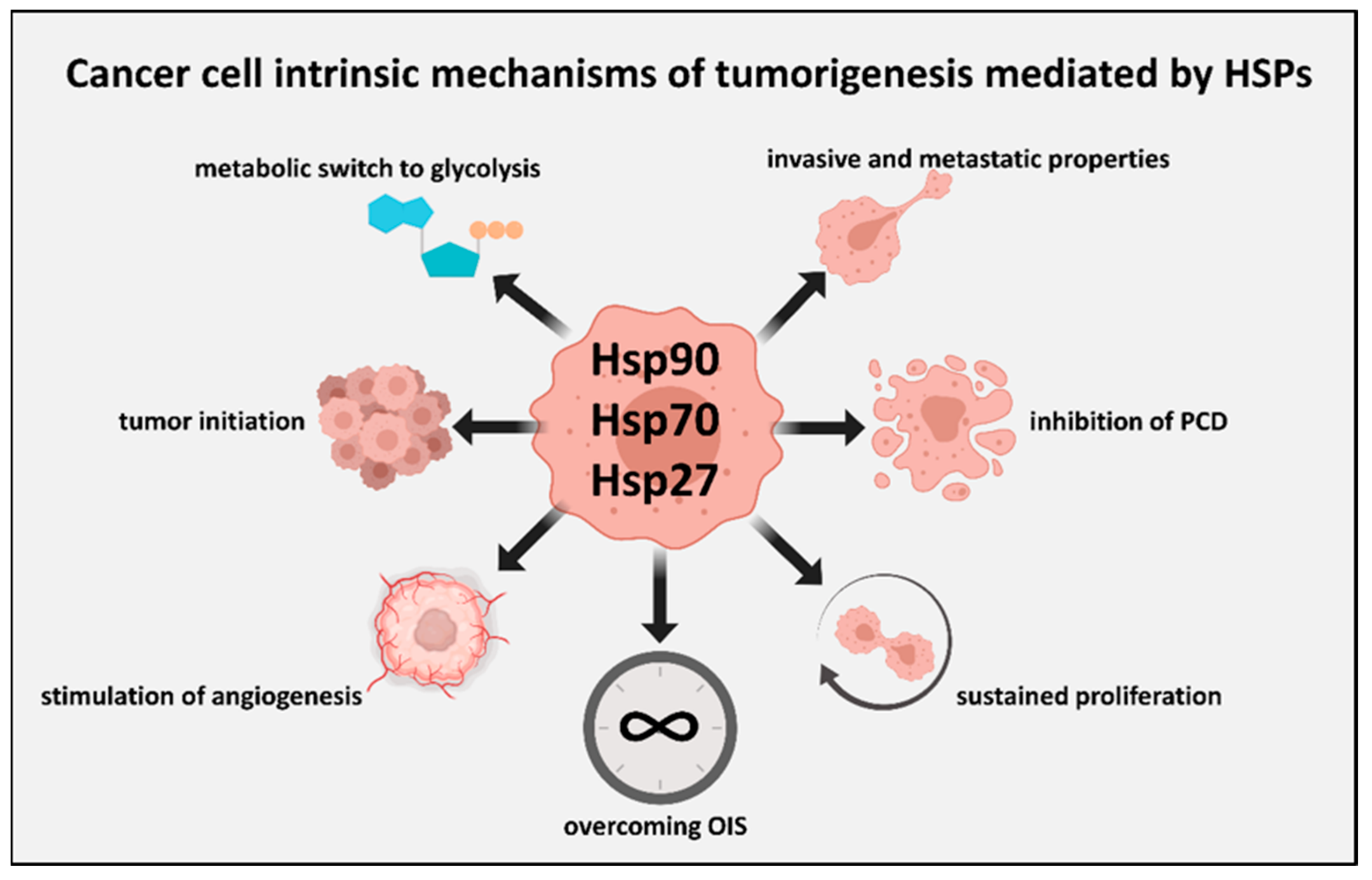{kind=link}
{kind=link}
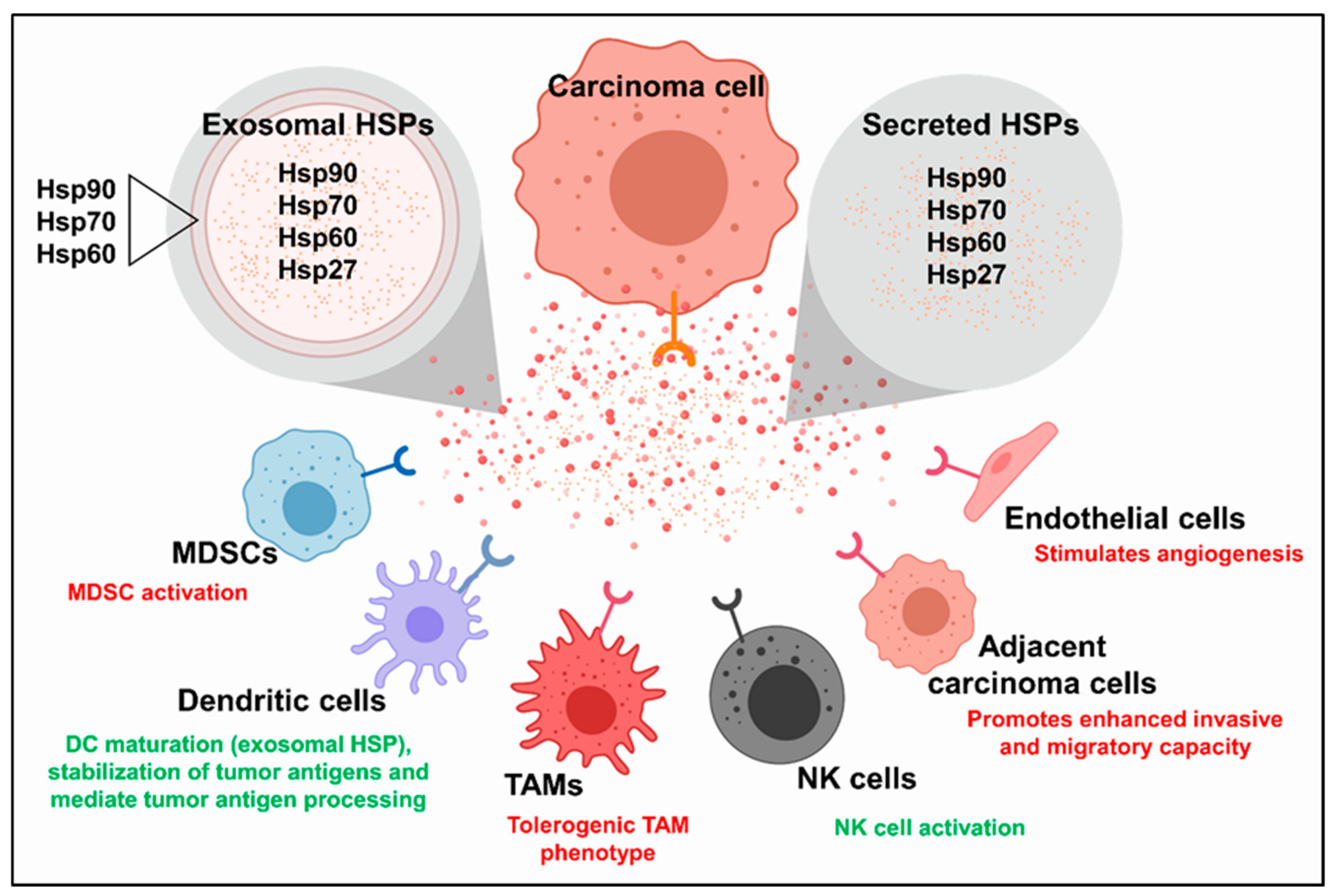{kind=link}
1. Introduction: Molecular Chaperones, Protein Folding, and the Health of the Proteome
1.1. HSF1 and the Heat Shock Response
1.2. Mechanisms of Protein Folding and Chaperone Performance
1.3. Chaperones Work in a Coordinated Manner to Brace Cancer Cell Survival—The Chaperome in Cancer
2. HSPs and the Cell-Intrinsic Properties of Malignant Cells
2.1. The Acquired Properties of Malignant Cells
2.2. HSPs Shore Up the Shared Pro-Malignant Properties of Malignant Cells
2.2.1. Unrestricted Proliferation
2.2.2. Evasion of Programmed Cell Death
2.2.3. Hurdling Replicative Senescence
2.2.4. Angiogenesis and Glucose Metabolism
2.2.5. Invasion and Metastasis
2.2.6. HSPs and Tumor Initiation
3. HSPs and Tumor cell Extrinsic Properties of Tumors
3.1. Influence of the Extracellular Milieu and the Tumor Stroma on the Malignant Phenotype
3.2. Tumor Immunity
4. Extracellular HSPs
5. Interactions between Tumor Cells and HSPs in Treatment of Cancer: Targeting Molecular Chaperone Functions in Cell Survival and Tumor Cell Evolution
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. Heat shock transcription factors: Structure and regulation. Annu. Rev. Cell Dev. Biol. 1995, 11, 441–469. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Murshid, A. Molecular Chaperone Accumulation in Cancer and Decrease in Alzheimer’s Disease: The Potential Roles of HSF1. Front. Neurosci. 2017, 11, 192. [Google Scholar] [CrossRef] [PubMed]
- Voellmy, R. Transduction of the stress signal and mechanisms of transcriptional regulation of heat shock/stress protein gene expression in higher eukaryotes. Crit. Rev. Eukaryot. Gene. Expr. 1994, 4, 357–401. [Google Scholar] [PubMed]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef]
- Bunch, H.; Lawney, B.P.; Lin, Y.F.; Asaithamby, A.; Murshid, A.; Wang, Y.E.; Chen, B.P.; Calderwood, S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015, 6, 10191. [Google Scholar] [CrossRef]
- Zhong, M.; Orosz, A.; Wu, C. Direct sensing of heat and oxidation by Drosophila heat shock transcription factor. Mol. Cell. 1998, 2, 101–108. [Google Scholar] [CrossRef]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar] [CrossRef]
- Kijima, T.; Prince, T.L.; Tigue, M.L.; Yim, K.H.; Schwartz, H.; Beebe, K.; Lee, S.; Budzynski, M.A.; Williams, H.; Trepel, J.B.; et al. HSP90 inhibitors disrupt a transient HSP90-HSF1 interaction and identify a noncanonical model of HSP90-mediated HSF1 regulation. Sci. Rep. 2018, 8, 6976. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Soncin, F.; Price, B.D.; Stevenson, M.A.; Calderwood, S.K. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 1996, 271, 30847–30857. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.D.; Murshid, A.; Eguchi, T.; Gong, J.; Calderwood, S.K. HSF1 regulation of beta-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene 2015, 34, 2178–2188. [Google Scholar] [CrossRef]
- Neef, D.W.; Jaeger, A.M.; Gomez-Pastor, R.; Willmund, F.; Frydman, J.; Thiele, D.J. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 2014, 9, 955–966. [Google Scholar] [CrossRef]
- Tang, D.; Khaleque, M.A.; Jones, E.L.; Theriault, J.R.; Li, C.; Wong, W.H.; Stevenson, M.A.; Calderwood, S.K. Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones 2005, 10, 46–58. [Google Scholar] [CrossRef]
- Khaleque, M.A.; Bharti, A.; Gong, J.; Gray, P.J.; Sachdev, V.; Ciocca, D.R.; Stati, A.; Fanelli, M.; Calderwood, S.K. Heat shock factor 1 represses estrogen-dependent transcription through association with MTA1. Oncogene 2008, 27, 1886–1893. [Google Scholar] [CrossRef]
- Santagata, S.; Hu, R.; Lin, N.U.; Mendillo, M.L.; Collins, L.C.; Hankinson, S.E.; Schnitt, S.J.; Whitesell, L.; Tamimi, R.M.; Lindquist, S.; et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 18378–18383. [Google Scholar] [CrossRef] [PubMed]
- Khaleque, M.A.; Bharti, A.; Sawyer, D.; Gong, J.; Benjamin, I.J.; Stevenson, M.A.; Calderwood, S.K. Induction of heat shock proteins by heregulin beta1 leads to protection from apoptosis and anchorage-independent growth. Oncogene 2005, 24, 6564–6573. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Colvin, T.A.; Gabai, V.L.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Weng, D.; Eguchi, T.; Murshid, A.; Sherman, M.Y.; Song, B.; Calderwood, S.K. Targeting the hsp70 gene delays mammary tumor initiation and inhibits tumor cell metastasis. Oncogene 2015, 34, 5460–5471. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Protein misassembly: Macromolecular crowding and molecular chaperones. Adv. Exp. Med. Biol. 2007, 594, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dinner, A.R.; Sali, A.; Smith, L.J.; Dobson, C.M.; Karplus, M. Understanding protein folding via free-energy surfaces from theory and experiment. Trends Biochem. Sci. 2000, 25, 331–339. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell. Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, S.; Buchberger, A.; Bukau, B. Interaction of Hsp70 chaperones with substrates. Nat. Struct. Biol. 1997, 4, 342–349. [Google Scholar] [CrossRef]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds alpha-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Gibert, B. HspB1 dynamic phospho-oligomeric structure dependent interactome as cancer therapeutic target. Curr. Mol. Med. 2012, 12, 1151–1163. [Google Scholar] [CrossRef]
- Lelj-Garolla, B.; Mauk, A.G. Self-association and chaperone activity of Hsp27 are thermally activated. J. Biol. Chem. 2006, 281, 8169–8174. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.; Napoli, V.; Mazurkie, A.; Stafford, W.F.; Graceffa, P. Phosphorylation dependence of hsp27 multimeric size and molecular chaperone function. J. Biol. Chem. 2009, 284, 18801–18807. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Selig, E.; Griffin, M.D.W.; Carver, J.A.; Ecroyd, H. Small Heat-shock Proteins Prevent a-Synuclein Aggregation via Transient Interactions and Their Efficacy Is Affected by the Rate of Aggregation. J. Biol. Chem. 2016, 291, 22618–22629. [Google Scholar] [CrossRef] [PubMed]
- Ehrnsperger, M.; Graber, S.; Gaestel, M.; Buchner, J. Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 1997, 16, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Patriarca, E.J.; Maresca, B. Acquired Thermotolerance following Heat Shock Protein Synthesis Prevents Impairment of Mitochondrial ATPase Activity at Elevated Temperatures in Saccharomyces cerevisiae. Exp. Cell Res. 1990, 190, 57–64. [Google Scholar] [CrossRef]
- Calderwood, S.K. Molecular cochaperones: Tumor growth and cancer treatment. Scientifica 2013, 2013, 217513. [Google Scholar] [CrossRef] [PubMed]
- Meriin, A.B.; Narayanan, A.; Meng, L.; Alexandrov, I.; Varelas, X.; Cisse, I.I.; Sherman, M.Y. Hsp70-Bag3 complex is a hub for proteotoxicity-induced signaling that controls protein aggregation. Proc. Natl. Acad. Sci. USA 2018, 115, E7043–E7052. [Google Scholar] [CrossRef] [PubMed]
- Antonietti, P.; Linder, B.; Hehlgans, S.; Mildenberger, I.C.; Burger, M.C.; Fulda, S.; Steinbach, J.P.; Gessler, F.; Rodel, F.; Mittelbronn, M.; et al. Interference with the HSF1/HSP70/BAG3 Pathway Primes Glioma Cells to Matrix Detachment and BH3 Mimetic-Induced Apoptosis. Mol. Cancer Ther. 2017, 16, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Tse, E.; Freilich, R.; Mok, S.A.; Makley, L.N.; Southworth, D.R.; Gestwicki, J.E. BAG3 Is a Modular, Scaffolding Protein that physically Links Heat Shock Protein 70 (Hsp70) to the Small Heat Shock Proteins. J. Mol. Biol. 2017, 429, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Workman, P. Altered states: Selectively drugging the Hsp90 cancer chaperone. Trends Mol. Med. 2004, 10, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, J.; Martin-Benito, J.; Scheres, S.H.; Sousa, R.; Moro, F.; Lopez-Vinas, E.; Gomez-Puertas, P.; Muga, A.; Carrascosa, J.L.; Valpuesta, J.M. The structure of CCT-Hsc70 NBD suggests a mechanism for Hsp70 delivery of substrates to the chaperonin. Nat. Struct. Mol. Biol. 2008, 15, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Prince, T.L.; Tran, M.T.; Sogawa, C.; Lang, B.J.; Calderwood, S.K. MZF1 and SCAND1 Reciprocally Regulate CDC37 Gene Expression in Prostate Cancer. Cancers 2019, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.J., Jr.; Prince, T.; Cheng, J.; Stevenson, M.A.; Calderwood, S.K. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer. 2008, 8, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.J., Jr.; Stevenson, M.A.; Calderwood, S.K. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007, 67, 11942–11950. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Ivy, S.P. Heat shock protein 90. Curr. Opin. Oncol. 2003, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef]
- Moran Luengo, T.; Kityk, R.; Mayer, M.P.; Rudiger, S.G.D. Hsp90 Breaks the Deadlock of the Hsp70 Chaperone System. Mol. Cell. 2018, 70, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Moran Luengo, T.; Mayer, M.P.; Rudiger, S.G.D. The Hsp70-Hsp90 Chaperone Cascade in Protein Folding. Trends Cell Biol. 2019, 29, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef]
- Da Silva, V.C.; Ramos, C.H. The network interaction of the human cytosolic 90 kDa heat shock protein Hsp90: A target for cancer therapeutics. J. Proteomics 2012, 75, 2790–2802. [Google Scholar] [CrossRef]
- Brandvold, K.R.; Morimoto, R.I. The Chemical Biology of Molecular Chaperones--Implications for Modulation of Proteostasis. J. Mol. Biol. 2015, 427, 2931–2947. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Wawrzynow, B.; Zylicz, A.; Zylicz, M. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fernandes, N.; Dodge, I.L.; Reddi, A.L.; Rao, N.; Safran, H.; DiPetrillo, T.A.; Wazer, D.E.; Band, V.; Band, H. ErbB2 degradation mediated by the co-chaperone protein CHIP. J. Biol. Chem. 2003, 278, 13829–13837. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Rodina, A.; Dunphy, M.P.; Corben, A.; Modi, S.; Guzman, M.L.; Gewirth, D.T.; Chiosis, G. Chaperome heterogeneity and its implications for cancer study and treatment. J. Biol. Chem. 2019, 294, 2162–2179. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Prince, T.; Ackerman, A.; Cavanaugh, A.; Schreiter, B.; Juengst, B.; Andolino, C.; Danella, J.; Chernin, M.; Williams, H. Dual targeting of HSP70 does not induce the heat shock response and synergistically reduces cell viability in muscle invasive bladder cancer. Oncotarget 2018, 9, 32702–32717. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.L.; Zeisig, A.; Aronson, L.I.; Moore, H.E.; Hockley, S.; Gonzalez, D.; Smith, E.M.; Powers, M.V.; Sharp, S.Y.; Workman, P.; et al. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia 2010, 24, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- Zoppino, F.C.M.; Guerrero-Gimenez, M.E.; Castro, G.N.; Ciocca, D.R. Comprehensive transcriptomic analysis of heat shock proteins in the molecular subtypes of human breast cancer. BMC Cancer 2018, 18, 700. [Google Scholar] [CrossRef] [PubMed]
- Klimczak, M.; Biecek, P.; Zylicz, A.; Zylicz, M. Heat shock proteins create a signature to predict the clinical outcome in breast cancer. Sci. Rep. 2019, 9, 7507. [Google Scholar] [CrossRef] [PubMed]
- Hadizadeh Esfahani, A.; Sverchkova, A.; Saez-Rodriguez, J.; Schuppert, A.A.; Brehme, M. A systematic atlas of chaperome deregulation topologies across the human cancer landscape. PLoS Comput. Biol. 2018, 14, e1005890. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Yokota, J. Tumor progression and metastasis. Carcinogenesis 2000, 21, 497–503. [Google Scholar] [CrossRef]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Ory, B.; Baud’huin, M.; Verrecchia, F.; Royer, B.B.; Quillard, T.; Amiaud, J.; Battaglia, S.; Heymann, D.; Redini, F.; Lamoureux, F. Blocking HSP90 Addiction Inhibits Tumor Cell Proliferation, Metastasis Development, and Synergistically Acts with Zoledronic Acid to Delay Osteosarcoma Progression. Clin. Cancer Res. 2016, 22, 2520–2533. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tu, J.; Dou, C.; Zhang, J.; Yang, L.; Liu, X.; Lei, K.; Liu, Z.; Wang, Y.; Li, L.; et al. HSP90 promotes cell glycolysis, proliferation and inhibits apoptosis by regulating PKM2 abundance via Thr-328 phosphorylation in hepatocellular carcinoma. Mol. Cancer 2017, 16, 178. [Google Scholar] [CrossRef] [PubMed]
- Park, A.M.; Tsunoda, I.; Yoshie, O. Heat shock protein 27 promotes cell cycle progression by down-regulating E2F transcription factor 4 and retinoblastoma family protein p130. J. Biol. Chem. 2018, 293, 15815–15826. [Google Scholar] [CrossRef]
- Budina-Kolomets, A.; Webster, M.R.; Leu, J.I.; Jennis, M.; Krepler, C.; Guerrini, A.; Kossenkov, A.V.; Xu, W.; Karakousis, G.; Schuchter, L.; et al. HSP70 Inhibition Limits FAK-Dependent Invasion and Enhances the Response to Melanoma Treatment with BRAF Inhibitors. Cancer Res. 2016, 76, 2720–2730. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Zylicz, M.; King, F.W.; Wawrzynow, A. Hsp70 interactions with the p53 tumour suppressor protein. EMBO J. 2001, 20, 4634–4638. [Google Scholar] [CrossRef] [PubMed]
- Budina-Kolomets, A.; Balaburski, G.M.; Bondar, A.; Beeharry, N.; Yen, T.; Murphy, M.E. Comparison of the activity of three different HSP70 inhibitors on apoptosis, cell cycle arrest, autophagy inhibition, and HSP90 inhibition. Cancer. Biol. Ther. 2014, 15, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Clarke, P.A.; Workman, P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 2008, 14, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Reed, J.C.; Homma, S. Heat-shock proteins as regulators of apoptosis. Oncogene 2003, 22, 9041–9047. [Google Scholar] [CrossRef] [PubMed]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Lanneau, D.; de Thonel, A.; Maurel, S.; Didelot, C.; Garrido, C. Apoptosis versus cell differentiation: Role of heat shock proteins HSP90, HSP70 and HSP27. Prion 2007, 1, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Simon, S.; Gibert, B.; Virot, S.; Manero, F.; Arrigo, A.P. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp. Cell Res. 2010, 316, 1535–1552. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005, 8, 7–12. [Google Scholar] [CrossRef]
- Campisi, J. Cancer, aging and cellular senescence. In Vivo 2000, 14, 183–188. [Google Scholar]
- Toogun, O.A.; Dezwaan, D.C.; Freeman, B.C. The hsp90 molecular chaperone modulates multiple telomerase activities. Mol. Cell. Biol. 2008, 28, 457–467. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan-Sunol, C.; Gabai, V.L.; Sherman, M.Y. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res. 2007, 67, 11779–11788. [Google Scholar] [CrossRef] [PubMed]
- Yaglom, J.A.; Gabai, V.L.; Sherman, M.Y. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res. 2007, 67, 2373–2381. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Yaglom, J.A.; Waldman, T.; Sherman, M.Y. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol. Cell. Biol. 2009, 29, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Hunt, C.; Yaglom, J.A.; Gabai, V.L.; Sherman, M.Y. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene 2011, 30, 2836–2845. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007, 435, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol. Med. 2002, 8, S62–S67. [Google Scholar] [CrossRef]
- Eichhorn, M.E.; Kleespies, A.; Angele, M.K.; Jauch, K.W.; Bruns, C.J. Angiogenesis in cancer: Molecular mechanisms, clinical impact. Langenbecks Arch. Surg. 2007, 392, 371–379. [Google Scholar] [CrossRef]
- Thuringer, D.; Jego, G.; Wettstein, G.; Terrier, O.; Cronier, L.; Yousfi, N.; Hebrard, S.; Bouchot, A.; Hazoume, A.; Joly, A.L.; et al. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J. 2013, 27, 4169–4183. [Google Scholar] [CrossRef]
- Shiota, M.; Kusakabe, H.; Izumi, Y.; Hikita, Y.; Nakao, T.; Funae, Y.; Miura, K.; Iwao, H. Heat shock cognate protein 70 is essential for Akt signaling in endothelial function. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 491–497. [Google Scholar] [CrossRef]
- Bruns, A.F.; Yuldasheva, N.; Latham, A.M.; Bao, L.; Pellet-Many, C.; Frankel, P.; Stephen, S.L.; Howell, G.J.; Wheatcroft, S.B.; Kearney, M.T.; et al. A heat-shock protein axis regulates VEGFR2 proteolysis, blood vessel development and repair. PLoS ONE 2012, 7, e48539. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Schumann, U.; Liu, Y.; Prokopchuk, O.; Steinacker, J.M. Heat shock protein 70 (Hsp70) inhibits oxidative phosphorylation and compensates ATP balance through enhanced glycolytic activity. J. Appl. Physiol. 2012, 113, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Puliafito, A.; Primo, L.; Celani, A. Cell-size distribution in epithelial tissue formation and homeostasis. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. Epithelial-Mesenchymal Transition (EMT) and Regulation of EMT Factors by Steroid Nuclear Receptors in Breast Cancer: A Review and in Silico Investigation. J. Clin. Med. 2016, 5, 11. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.X.; Wu, H.T.; Li, X.L.; Wen, X.F.; Du, C.W.; Zhang, G.J. Collagen 1A1 (COL1A1) promotes metastasis of breast cancer and is a potential therapeutic target. Discov. Med. 2018, 25, 211–223. [Google Scholar]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Kasioumi, P.; Vrazeli, P.; Vezyraki, P.; Zerikiotis, S.; Katsouras, C.; Damalas, A.; Angelidis, C. Hsp70 (HSP70A1A) downregulation enhances the metastatic ability of cancer cells. Int. J. Oncol. 2019, 54, 821–832. [Google Scholar] [CrossRef]
- Musiani, D.; Konda, J.D.; Pavan, S.; Torchiaro, E.; Sassi, F.; Noghero, A.; Erriquez, J.; Perera, T.; Olivero, M.; Di Renzo, M.F. Heat-shock protein 27 (HSP27, HSPB1) is up-regulated by MET kinase inhibitors and confers resistance to MET-targeted therapy. FASEB J. 2014, 28, 4055–4067. [Google Scholar] [CrossRef] [PubMed]
- Pavan, S.; Musiani, D.; Torchiaro, E.; Migliardi, G.; Gai, M.; Di Cunto, F.; Erriquez, J.; Olivero, M.; Di Renzo, M.F. HSP27 is required for invasion and metastasis triggered by hepatocyte growth factor. Int. J. Cancer 2014, 134, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Gibert, B.; Eckel, B.; Gonin, V.; Goldschneider, D.; Fombonne, J.; Deux, B.; Mehlen, P.; Arrigo, A.P.; Clezardin, P.; Diaz-Latoud, C. Targeting heat shock protein 27 (HspB1) interferes with bone metastasis and tumour formation in vivo. Br. J. Cancer 2012, 107, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Lee, H.J.; Jin, Y.B.; Jang, J.; Kang, G.Y.; Lee, M.; Kim, C.H.; Kim, J.; Yoon, S.S.; Lee, Y.S.; et al. MMP9 processing of HSPB1 regulates tumor progression. PLoS ONE 2014, 9, e85509. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yan, Z.; Huang, L.; Guo, M.; Zhang, Z.; Guo, C. Cell surface heat shock protein 90 modulates prostate cancer cell adhesion and invasion through the integrin-β1/focal adhesion kinase/c-Src signaling pathway. Oncol. Rep. 2011, 25, 1343–1351. [Google Scholar] [PubMed]
- Teng, Y.; Ngoka, L.; Mei, Y.; Lesoon, L.; Cowell, J.K. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J. Biol. Chem. 2012, 287, 10051–10059. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Beebe, K.; Neckers, L. Impact of heat-shock protein 90 on cancer metastasis. Future Oncol. 2009, 5, 679–688. [Google Scholar] [CrossRef]
- Xiong, Y.; He, L.; Shay, C.; Lang, L.; Loveless, J.; Yu, J.; Chemmalakuzhy, R.; Jiang, H.; Liu, M.; Teng, Y. Nck-associated protein 1 associates with HSP90 to drive metastasis in human non-small-cell lung cancer. J. Exp. Clin. Cancer Res. 2019, 38, 122. [Google Scholar] [CrossRef]
- Visvader, J.E.; Stingl, J. Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 2014, 28, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Penzner, J.H.; Song, B.; Koido, S.; Calderwood, S.K.; Gong, J. Metastasis is an early event in mouse mammary carcinomas and is associated with cells bearing stem cell markers. Breast Cancer Res. 2012, 14, R18. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Lang, B.J.; Weng, D.; Eguchi, T.; Murshid, A.; Borges, T.J.; Doshi, S.; Song, B.; Stevenson, M.A.; Calderwood, S.K. Genotoxic stress induces Sca-1-expressing metastatic mammary cancer cells. Mol. Oncol. 2018, 12, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Song, B.; Koido, S.; Calderwood, S.K.; Gong, J. Immunotherapy of radioresistant mammary tumors with early metastasis using molecular chaperone vaccines combined with ionizing radiation. J. Immunol. 2013, 191, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Guyot, C.; Gabbiani, G. The stroma reaction myofibroblast: A key player in the control of tumor cell behavior. Int. J. Dev. Biol. 2004, 48, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumor Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Yaglom, J.A.; Wang, Y.; Meng, L.; Shao, H.; Kim, G.; Colvin, T.; Gestwicki, J.; Sherman, M.Y. Anticancer Effects of Targeting Hsp70 in Tumor Stromal Cells. Cancer Res. 2016, 76, 5926–5932. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.L.; Sholl, L.M.; Ben-Aharon, I.; Beck, A.H.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.M.; Whitesell, L.; et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Dafforn, T.R.; Della, M.; Miller, A.D. The molecular interactions of heat shock protein 47 (Hsp47) and their implications for collagen biosynthesis. J. Biol. Chem. 2001, 276, 49310–49319. [Google Scholar] [CrossRef]
- Ogawa, Y.; Razzaque, M.S.; Kameyama, K.; Hasegawa, G.; Shimmura, S.; Kawai, M.; Okamoto, S.; Ikeda, Y.; Tsubota, K.; Kawakami, Y.; et al. Role of heat shock protein 47, a collagen-binding chaperone, in lacrimal gland pathology in patients with cGVHD. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xiong, G.; Fu, H.; Evers, B.M.; Zhou, B.P.; Xu, R. Chaperone Hsp47 Drives Malignant Growth and Invasion by Modulating an ECM Gene Network. Cancer Res. 2015, 75, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.U.; Lee, W.J.; Tran, T.N.; Jung, I.; Lee, J.H. Hsp70 Knockdown by siRNA Decreased Collagen Production in Keloid Fibroblasts. Yonsei Med. J. 2015, 56, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Bansal, T.; Rana, S.; Datta, K.; Datta Chaudhuri, R.; Chawla-Sarkar, M.; Sarkar, S. Myocyte-Derived Hsp90 Modulates Collagen Upregulation via Biphasic Activation of STAT-3 in Fibroblasts during Cardiac Hypertrophy. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.Y.; Doan, N.D.; DiChiara, A.S.; Papa, L.J.; Cheah, J.H.; Soule, C.K.; Watson, N.; Hulleman, J.D.; Shoulders, M.D. A High-Throughput Assay for Collagen Secretion Suggests an Unanticipated Role for Hsp90 in Collagen Production. Biochemistry 2018, 57, 2814–2827. [Google Scholar] [CrossRef] [PubMed]
- Park, A.M.; Kanai, K.; Itoh, T.; Sato, T.; Tsukui, T.; Inagaki, Y.; Selman, M.; Matsushima, K.; Yoshie, O. Heat Shock Protein 27 Plays a Pivotal Role in Myofibroblast Differentiation and in the Development of Bleomycin-Induced Pulmonary Fibrosis. PLoS ONE 2016, 11, e0148998. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef]
- Murshid, A.; Gong, J.; Calderwood, S.K. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J. Immunol. 2010, 185, 2903–2917. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Sun, X.; Chen, X.; Wang, Y.; Foster, B.A.; Subjeck, J.; Fisher, P.B.; Wang, X.Y. Secretable chaperone Grp170 enhances therapeutic activity of a novel tumor suppressor, mda-7/IL-24. Cancer Res. 2008, 68, 3890–3898. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Sow, H.S.; Ren, J.; Camps, M.; Ossendorp, F.; Ten Dijke, P. Combined Inhibition of TGF-beta Signaling and the PD-L1 Immune Checkpoint Is Differentially Effective in Tumor Models. Cells 2019, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Mambula, S.S.; Calderwood, S.K. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J. Immunol. 2006, 177, 7849–7857. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Sogawa, C.; Okusha, Y.; Uchibe, K.; Iinuma, R.; Ono, K.; Nakano, K.; Murakami, J.; Itoh, M.; Arai, K.; et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. PLoS ONE 2018, 13, e0191109. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Yang, X.; Song, X.; Chen, S.; He, Y.; Wang, Q.; Chen, G.; Luo, C.; Wu, X.; Zhang, Y. Exosomal Hsp70 mediates immunosuppressive activity of the myeloid-derived suppressor cells via phosphorylation of Stat3. Med. Oncol. 2015, 32, 453. [Google Scholar] [CrossRef]
- Kottke, T.; Pulido, J.; Thompson, J.; Sanchez-Perez, L.; Chong, H.; Calderwood, S.K.; Selby, P.; Harrington, K.; Strome, S.E.; Melcher, A.; et al. Antitumor immunity can be uncoupled from autoimmunity following heat shock protein 70-mediated inflammatory killing of normal pancreas. Cancer Res. 2009, 69, 7767–7774. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kottke, T.; Sanchez-Perez, L.; Diaz, R.M.; Thompson, J.; Chong, H.; Harrington, K.; Calderwood, S.K.; Pulido, J.; Georgopoulos, N.; Selby, P.; et al. Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res. 2007, 67, 11970–11979. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring Anticancer Immune Response by Targeting Tumor-Derived Exosomes With a HSP70 Peptide Aptamer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Alzate, O.; Dechkovskaia, A.M.; Keene, J.D.; Sampson, J.H.; Mitchell, D.A.; Bigner, D.D. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009, 23, 1541–1557. [Google Scholar] [CrossRef]
- Campanella, C.; Rappa, F.; Sciume, C.; Marino Gammazza, A.; Barone, R.; Bucchieri, F.; David, S.; Curcuru, G.; Caruso Bavisotto, C.; Pitruzzella, A.; et al. Heat shock protein 60 levels in tissue and circulating exosomes in human large bowel cancer before and after ablative surgery. Cancer 2015, 121, 3230–3239. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, X.; Zhuo, W.; Fu, Y.; Shi, H.; Liang, Y.; Tong, M.; Chang, G.; Luo, Y. The regulatory mechanism of Hsp90 secretion and its function in tumor malignancy. Proc. Natl. Acad. Sci. USA 2009, 106, 21288–21293. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K. Heat shock proteins and cancer: Intracellular chaperones or extracellular signalling ligands? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tsen, F.; Sahu, D.; Bhatia, A.; Chen, M.; Multhoff, G.; Woodley, D.T. Extracellular Hsp90 (eHsp90) as the actual target in clinical trials: Intentionally or unintentionally. Int. Rev. Cell Mol. Biol. 2013, 303, 203–235. [Google Scholar] [CrossRef] [PubMed]
- Tsen, F.; Bhatia, A.; O’Brien, K.; Cheng, C.F.; Chen, M.; Hay, N.; Stiles, B.; Woodley, D.T.; Li, W. Extracellular heat shock protein 90 signals through subdomain II and the NPVY motif of LRP-1 receptor to Akt1 and Akt2: A circuit essential for promoting skin cell migration in vitro and wound healing in vivo. Mol. Cell Biol. 2013, 33, 4947–4959. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.D.; Franco, O.E.; Hance, M.W.; Hayward, S.W.; Isaacs, J.S. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J. Biol. Chem. 2015, 290, 8271–8282. [Google Scholar] [CrossRef]
- Nolan, K.D.; Kaur, J.; Isaacs, J.S. Secreted heat shock protein 90 promotes prostate cancer stem cell heterogeneity. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Eustace, B.K.; Sakurai, T.; Stewart, J.K.; Yimlamai, D.; Unger, C.; Zehetmeier, C.; Lain, B.; Torella, C.; Henning, S.W.; Beste, G.; et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat. Cell Biol. 2004, 6, 507–514. [Google Scholar] [CrossRef]
- Sims, J.D.; McCready, J.; Jay, D.G. Extracellular heat shock protein (Hsp)70 and Hsp90alpha assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE 2011, 6, e18848. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Scroggins, B.; Koga, F.; Lee, M.J.; Trepel, J.; Felts, S.; Carreras, C.; Neckers, L. A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene 2008, 27, 2478–2487. [Google Scholar] [CrossRef]
- Santos, T.G.; Martins, V.R.; Hajj, G.N.M. Unconventional Secretion of Heat Shock Proteins in Cancer. Int. J. Mol. Sci. 2017, 18, 946. [Google Scholar] [CrossRef] [PubMed]
- Cordonnier, M.; Chanteloup, G.; Isambert, N.; Seigneuric, R.; Fumoleau, P.; Garrido, C.; Gobbo, J. Exosomes in cancer theranostic: Diamonds in the rough. Cell Adh. Migr. 2017, 11, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat Shock Protein 70 Surface-Positive Tumor Exosomes Stimulate Migratory and Cytolytic Activity of Natural Killer Cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bai, O.; Zhang, H.; Yuan, J.; Zong, S.; Chibbar, R.; Slattery, K.; Qureshi, M.; Wei, Y.; Deng, Y.; et al. Membrane-bound HSP70-engineered myeloma cell-derived exosomes stimulate more efficient CD8(+) CTL- and NK-mediated antitumour immunity than exosomes released from heat-shocked tumour cells expressing cytoplasmic HSP70. J. Cell Mol. Med. 2010, 14, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Lin, C.F.; Skinner, K.A.; Schiffhauer, L.M.; Peacock, J.; Hicks, D.G.; Redmond, E.M.; Morrow, D.; Huston, A.; Shayne, M.; et al. Heat shock protein 27 differentiates tolerogenic macrophages that may support human breast cancer progression. Cancer Res. 2011, 71, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Salari, S.; Seibert, T.; Chen, Y.X.; Hu, T.; Shi, C.; Zhao, X.; Cuerrier, C.M.; Raizman, J.E.; O’Brien, E.R. Extracellular HSP27 acts as a signaling molecule to activate NF-kappaB in macrophages. Cell Stress Chaperones 2013, 18, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Finka, A.; Goloubinoff, P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 2013, 18, 591–605. [Google Scholar] [CrossRef]
- Schwartz, H.; Scroggins, B.; Zuehlke, A.; Kijima, T.; Beebe, K.; Mishra, A.; Neckers, L.; Prince, T. Combined HSP90 and kinase inhibitor therapy: Insights from The Cancer Genome Atlas. Cell Stress Chaperones 2015, 20, 729–741. [Google Scholar] [CrossRef]
- Pockley, A.G.; Henderson, B. Extracellular cell stress (heat shock) proteins-immune responses and disease: An overview. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Condelli, V.; Crispo, F.; Pietrafesa, M.; Lettini, G.; Matassa, D.S.; Esposito, F.; Landriscina, M.; Maddalena, F. HSP90 Molecular Chaperones, Metabolic Rewiring, and Epigenetics: Impact on Tumor Progression and Perspective for Anticancer Therapy. Cells 2019, 8, 532. [Google Scholar] [CrossRef]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, S.V.; Mollapour, M.; Lee, M.J.; Tsutsumi, S.; Lee, S.; Kim, Y.S.; Prince, T.; Apolo, A.B.; Giaccone, G.; Xu, W.; et al. Tumor-intrinsic and tumor-extrinsic factors impacting hsp90- targeted therapy. Curr. Mol. Med. 2012, 12, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.A.; Ruiz-Gonzalez, M.X.; Moya, A.; Elena, S.F.; Barrio, E. Endosymbiotic bacteria: groEL buffers against deleterious mutations. Nature 2002, 417, 398. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016, 354. [Google Scholar] [CrossRef]
- Shimamura, T.; Li, D.; Ji, H.; Haringsma, H.J.; Liniker, E.; Borgman, C.L.; Lowell, A.M.; Minami, Y.; McNamara, K.; Perera, S.A.; et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008, 68, 5827–5838. [Google Scholar] [CrossRef]
- Pines, G.; Kostler, W.J.; Yarden, Y. Oncogenic mutant forms of EGFR: Lessons in signal transduction and targets for cancer therapy. FEBS Lett. 2010, 584, 2699–2706. [Google Scholar] [CrossRef]
- Miyajima, N.; Tsutsumi, S.; Sourbier, C.; Beebe, K.; Mollapour, M.; Rivas, C.; Yoshida, S.; Trepel, J.B.; Huang, Y.; Tatokoro, M.; et al. The HSP90 inhibitor ganetespib synergizes with the MET kinase inhibitor crizotinib in both crizotinib-sensitive and -resistant MET-driven tumor models. Cancer Res. 2013, 73, 7022–7033. [Google Scholar] [CrossRef]
- Calderwood, S.K. Tumor heterogeneity, clonal evolution, and therapy resistance: An opportunity for multitargeting therapy. Discov. Med. 2013, 15, 188–194. [Google Scholar]
- Chatterjee, S.; Bhattacharya, S.; Socinski, M.A.; Burns, T.F. HSP90 inhibitors in lung cancer: Promise still unfulfilled. Clin. Adv. Hematol. Oncol. 2016, 14, 346–356. [Google Scholar] [PubMed]
- Powers, M.V.; Jones, K.; Barillari, C.; Westwood, I.; van Montfort, R.L.; Workman, P. Targeting HSP70: The second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 2010, 9, 1542–1550. [Google Scholar] [CrossRef]
- Howe, M.K.; Bodoor, K.; Carlson, D.A.; Hughes, P.F.; Alwarawrah, Y.; Loiselle, D.R.; Jaeger, A.M.; Darr, D.B.; Jordan, J.L.; Hunter, L.M.; et al. Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem. Biol. 2014, 21, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Colvin, T.; Rauch, J.N.; Acosta-Alvear, D.; Kampmann, M.; Dunyak, B.; Hann, B.; Aftab, B.T.; Murnane, M.; Cho, M.; et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol. Cancer Ther. 2015, 14, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Frottin, F.; Schueder, F.; Tiwary, S.; Gupta, R.; Korner, R.; Schlichthaerle, T.; Cox, J.; Jungmann, R.; Hartl, F.U.; Hipp, M.S. The nucleolus functions as a phase-separated protein quality control compartment. Science 2019, 365, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Eskew, J.D.; Sadikot, T.; Morales, P.; Duren, A.; Dunwiddie, I.; Swink, M.; Zhang, X.; Hembruff, S.; Donnelly, A.; Rajewski, R.A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. [Google Scholar] [CrossRef]
- Yim, K.H.; Prince, T.L.; Qu, S.; Bai, F.; Jennings, P.A.; Onuchic, J.N.; Theodorakis, E.A.; Neckers, L. Gambogic acid identifies an isoform-specific druggable pocket in the middle domain of Hsp90beta. Proc. Natl. Acad. Sci. USA 2016, 113, E4801–E4809. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Kent, C.N.; Balch, M.; Peng, S.; Mishra, S.J.; Deng, J.; Day, V.W.; Liu, W.; Subramanian, C.; Cohen, M.; et al. Structure-guided design of an Hsp90beta N-terminal isoform-selective inhibitor. Nat. Commun. 2018, 9, 425. [Google Scholar] [CrossRef]
- Kijima, T.; Prince, T.; Neckers, L.; Koga, F.; Fujii, Y. Heat shock factor 1 (HSF1)-targeted anticancer therapeutics: Overview of current preclinical progress. Expert Opin. Ther. Targets 2019, 23, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Noel, P.; Von Hoff, D.D.; Saluja, A.K.; Velagapudi, M.; Borazanci, E.; Han, H. Triptolide and Its Derivatives as Cancer Therapies. Trends Pharmacol. Sci. 2019, 40, 327–341. [Google Scholar] [CrossRef]
- Adams, J.; Kauffman, M. Development of the proteasome inhibitor Velcade (Bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef]
- Arkwright, R.; Pham, T.M.; Zonder, J.A.; Dou, Q.P. The preclinical discovery and development of bortezomib for the treatment of mantle cell lymphoma. Expert Opin. Drug Discov. 2017, 12, 225–235. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Xu, W.; Vos, M.; Yuan, X.; Isaacs, J.S.; Bisht, K.S.; Gius, D.; Neckers, L. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol. Cancer Ther. 2004, 3, 551–566. [Google Scholar] [PubMed]
- Zaarur, N.; Gabai, V.L.; Porco, J.A., Jr.; Calderwood, S.; Sherman, M.Y. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006, 66, 1783–1791. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lang, B.J.; Guerrero-Giménez, M.E.; Prince, T.L.; Ackerman, A.; Bonorino, C.; Calderwood, S.K. Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond. Int. J. Mol. Sci. 2019, 20, 4507. https://doi.org/10.3390/ijms20184507
Lang BJ, Guerrero-Giménez ME, Prince TL, Ackerman A, Bonorino C, Calderwood SK. Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond. International Journal of Molecular Sciences. 2019; 20(18):4507. https://doi.org/10.3390/ijms20184507
Chicago/Turabian StyleLang, Benjamin J., Martín Eduardo Guerrero-Giménez, Thomas L. Prince, Andrew Ackerman, Cristina Bonorino, and Stuart K. Calderwood. 2019. "Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond" International Journal of Molecular Sciences 20, no. 18: 4507. https://doi.org/10.3390/ijms20184507
APA StyleLang, B. J., Guerrero-Giménez, M. E., Prince, T. L., Ackerman, A., Bonorino, C., & Calderwood, S. K. (2019). Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond. International Journal of Molecular Sciences, 20(18), 4507. https://doi.org/10.3390/ijms20184507