Stress-Induced, p53-Mediated Tumor Growth Inhibition of Melanoma by Modulated Electrohyperthermia in Mouse Models without Major Immunogenic Effects

 ,
, 

Abstract
1. Introduction
2. Results
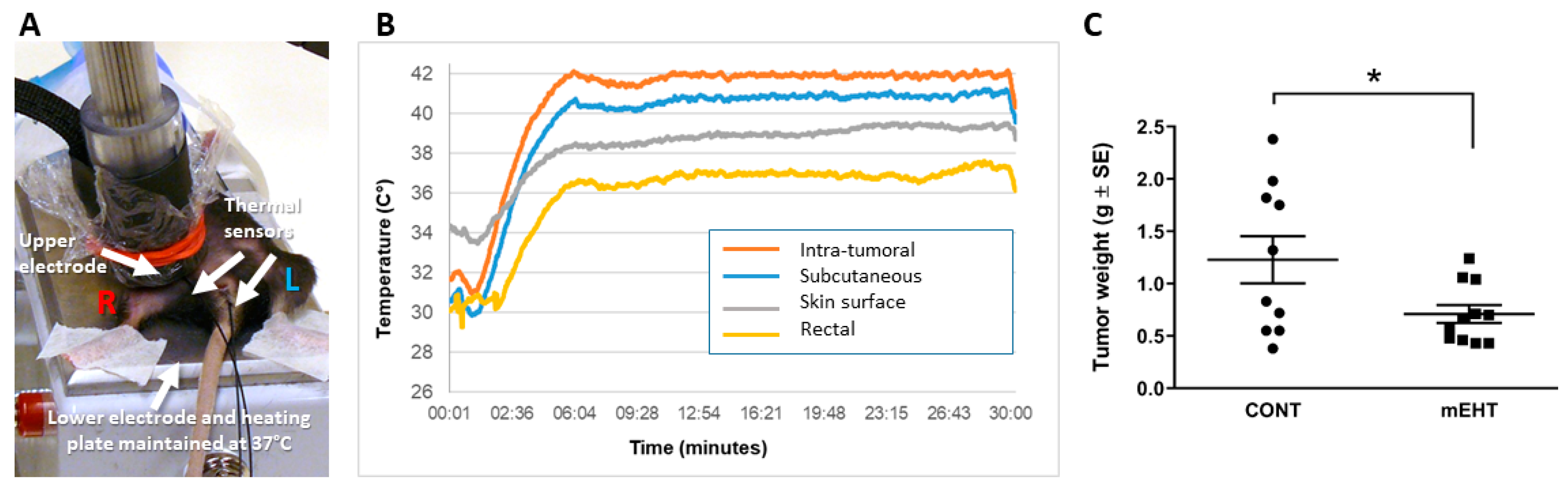
2.1. mEHT Suppressed Melanoma Tumor Growth
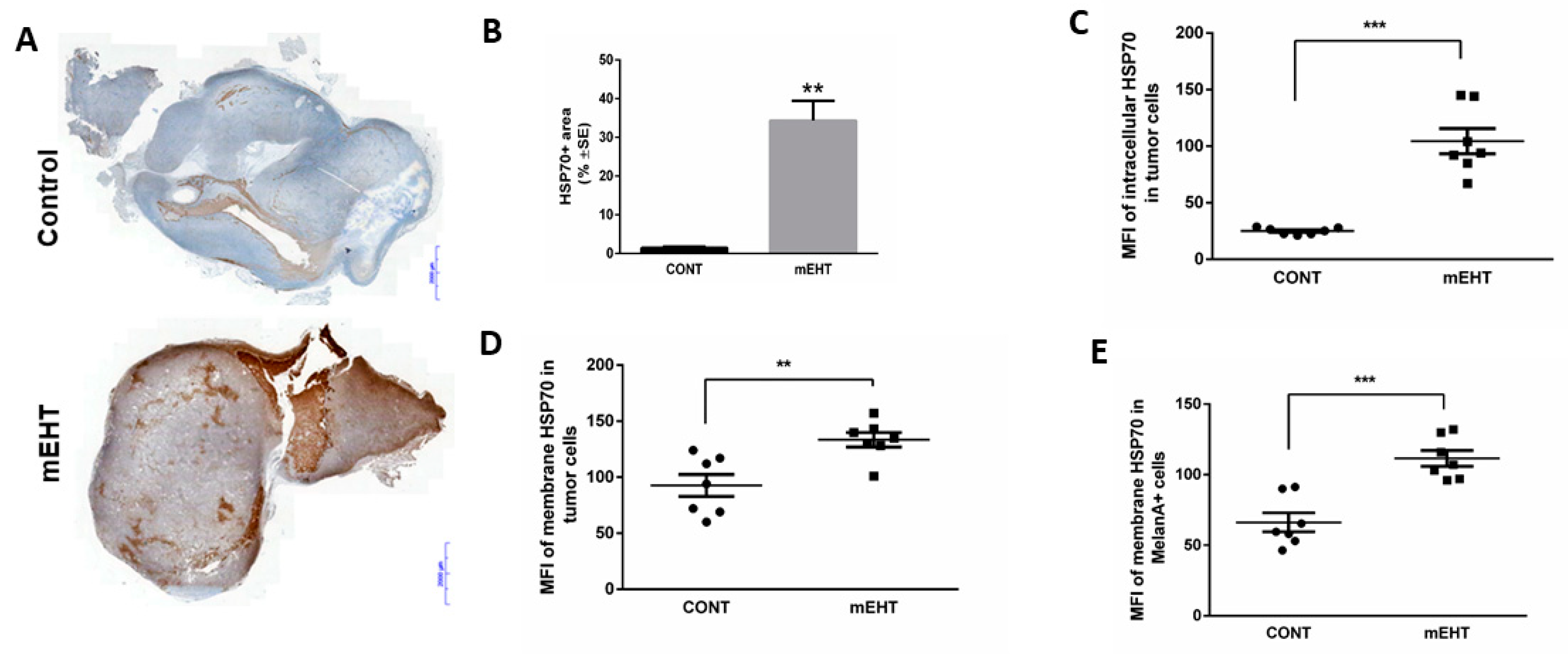
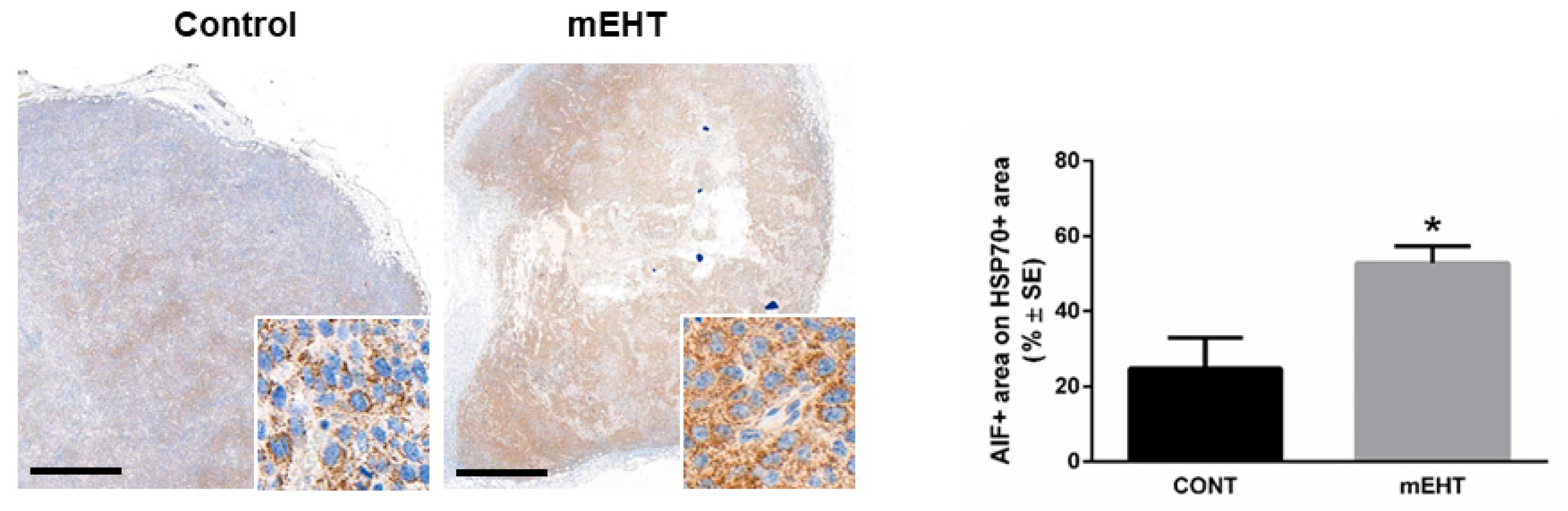
2.2. mEHT Induced the Expression of Different Forms of hsp70
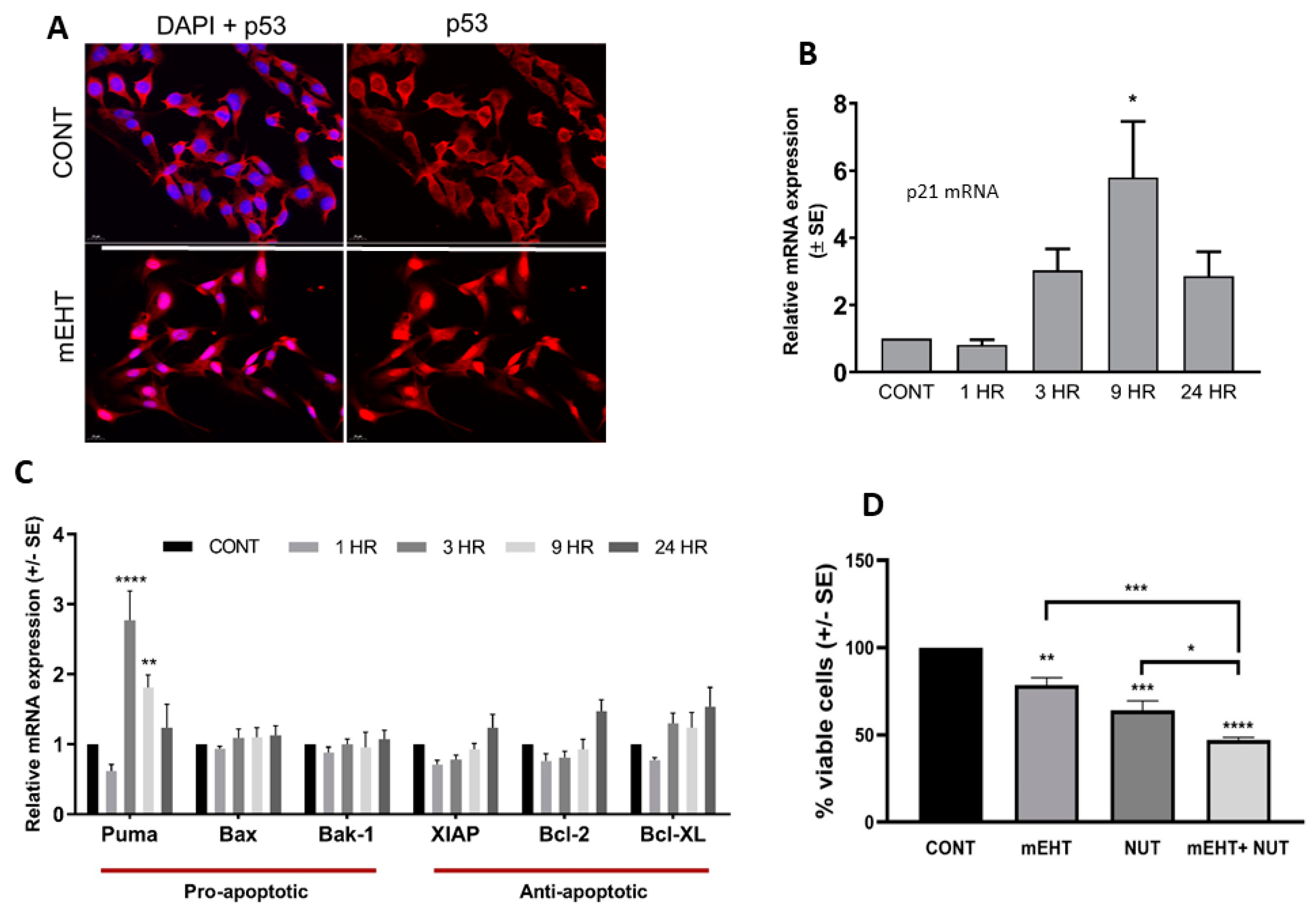
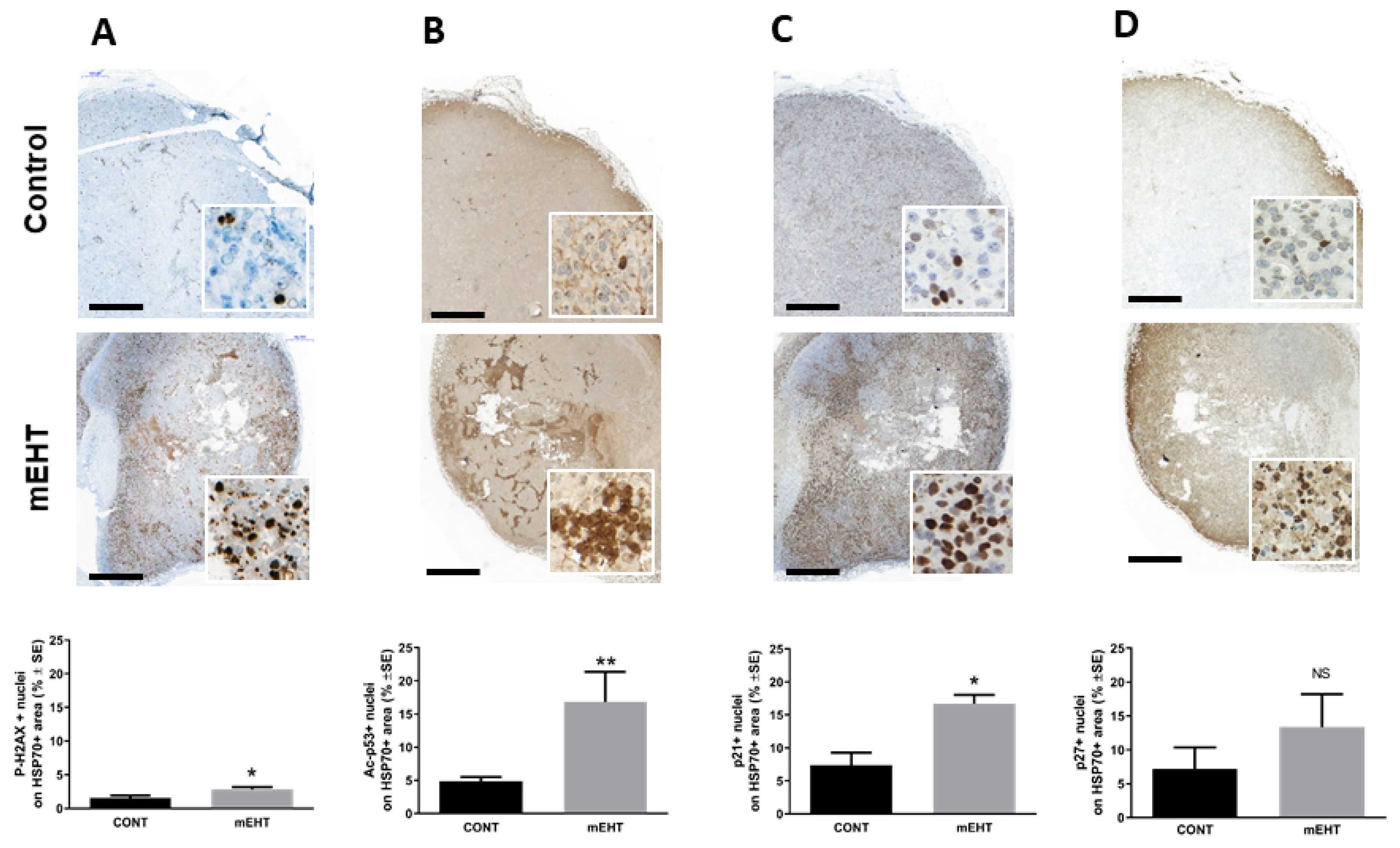
2.3. mEHT Induced p53 Accumulation and Activation In Vitro and In Vivo
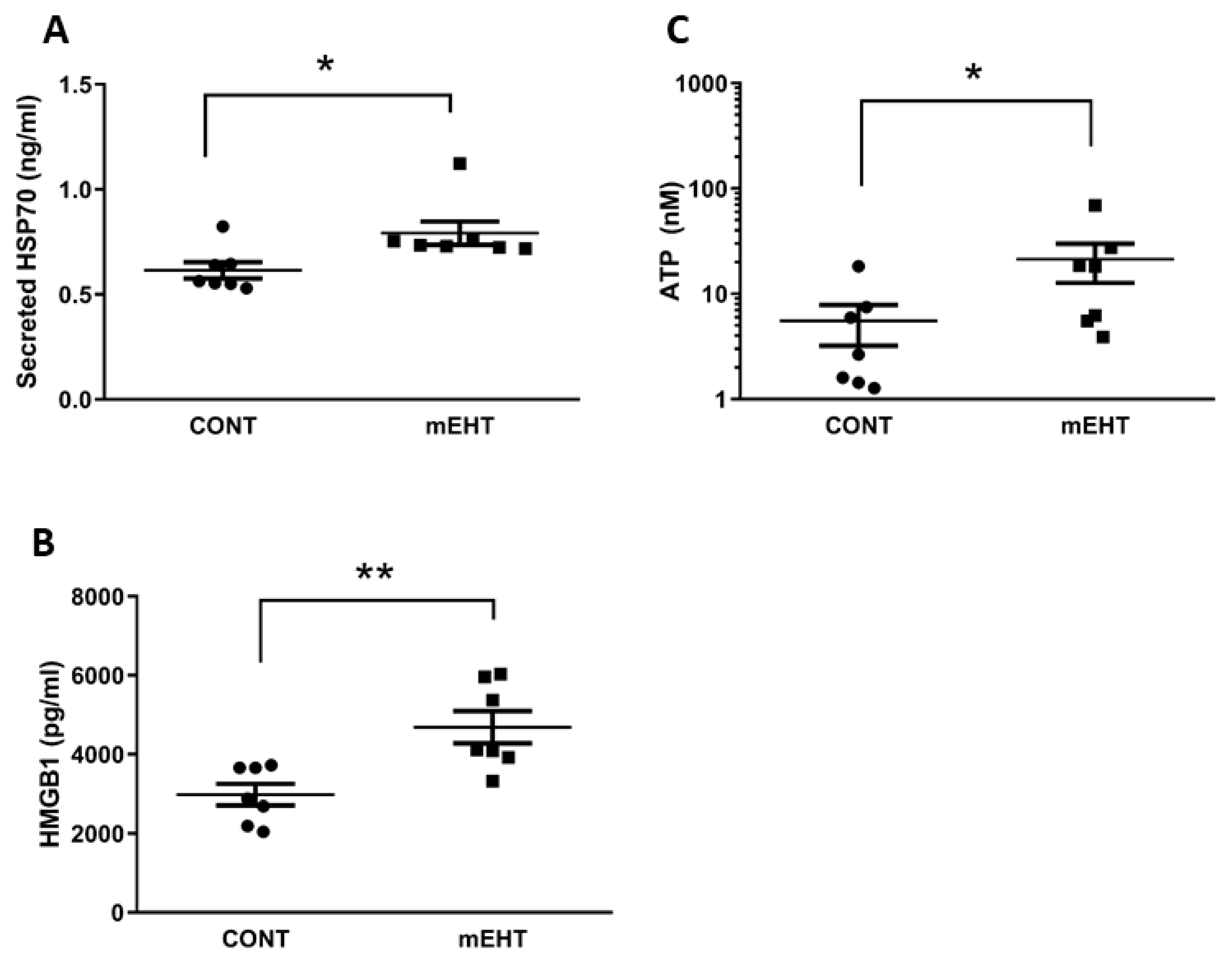
2.4. mEHT Induced DAMP Signal Release
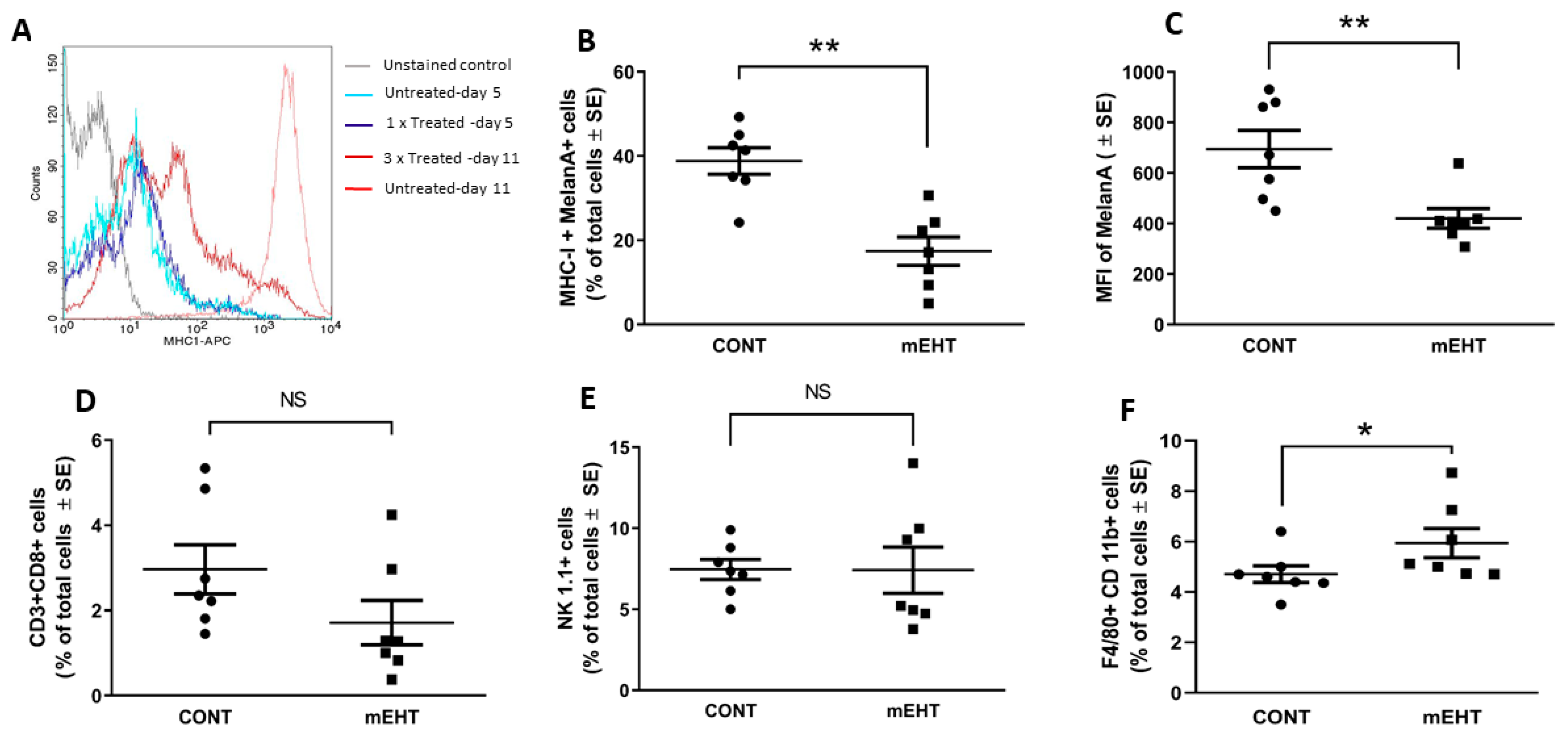
2.5. mEHT-Related Changes of the Anti-Tumor Immune Response
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Quantitative Real-Time PCR
4.3. In Vivo mEHT Treatment
4.4. In Vitro mEHT Treatment
4.5. Immunohisto- and Cytochemistry
4.6. Flow Cytometry
4.7. Measurement of Extracellular hsp70, HMGB1 and ATP
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Roussakow, S.V. Clinical and economic evaluation of modulated electrohyperthermia concurrent to dose-dense temozolomide 21/28 days regimen in the treatment of recurrent glioblastoma: A retrospective analysis of a two-centre German cohort trial with systematic comparison and effect-to-treatment analysis. BMJ Open 2017, 7, e017387. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, F.; Shields, B.; Makhoul, I.; Avaritt, N.; Wong, H.K.; Hutchins, L.F.; Shalin, S.; Tackett, A.J. Immune surveillance in melanoma: From immune attack to melanoma escape and even counterattack. Cancer Biol. Ther. 2017, 18, 451–469. [Google Scholar] [CrossRef] [PubMed]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Invest. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Dang, J.; Ba, D.; Wang, C.; Han, J.; Zheng, F. Potential function of CTLA-4 in the tumourigenic capacity of melanoma stem cells. Oncol. Lett. 2018, 16, 6163–6170. [Google Scholar] [CrossRef]
- Andocs, G.; Szasz, O.; Szasz, A. Oncothermia treatment of cancer: From the laboratory to clinic. Electromagn. Biol. Med. 2009, 28, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, M.S.; Kim, H.J.; Lee, E.; Jeong, J.H.; Park, I.; Jeong, Y.K.; Jang, W.I. Role of HIF-1α in response of tumors to a combination of hyperthermia and radiation in vivo. Int. J. Hyperthermia. 2018, 34, 276–283. [Google Scholar] [CrossRef]
- Kleef, R.; Moss, R.; Szasz, A.M.; Bohdjalian, A.; Bojar, H.; Bakacs, T. Complete Clinical Remission of Stage IV Triple-Negative Breast Cancer Lung Metastasis Administering Low-Dose Immune Checkpoint Blockade in Combination With Hyperthermia and Interleukin-2. Integr. Cancer Ther. 2018, 17, 1297–1303. [Google Scholar] [CrossRef]
- Morimoto, T.; Kimura, S.; Konishi, Y.; Komaki, K.; Uyama, T.; Monden, Y.; Kinouchi, Y.; Iritani, T. A study of the electrical bio-impedance of tumors. J. Invest. Surg. 1993, 6, 25–32. [Google Scholar] [CrossRef]
- Andras, S.; Nora, S.; Oliver, S. Oncothermia: Principles and Practices; Springer Netherlands: Heidelberg, Germany, 2011. [Google Scholar]
- Khramtsov, V.V.; Gillies, R.J. Janus-faced tumor microenvironment and redox. Antioxid. Redox. Sign. 2014, 21, 723–729. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.; Prado-Garcia, H.; Sanchez-Garcia, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Guo, Z. A review of electrical impedance techniques for breast cancer detection. Med. Eng. Phys. 2003, 25, 79–90. [Google Scholar] [CrossRef]
- Pethig, R.; Gascoyne, P.R.; McLaughlin, J.A.; Szent-Gyorgyi, A. Interaction of the 2,6-dimethoxysemiquinone and ascorbyl free radicals with Ehrlich ascites cells: A probe of cell-surface charge. Proc. Natl. Acad. Sci. USA 1984, 81, 2088–2091. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, A.P.; Lazebnik, M.; Bertram, J.M.; Converse, M.C.; Hagness, S.C.; Webster, J.G.; Mahvi, D.M. Dielectric properties of human normal, malignant and cirrhotic liver tissue: In vivo and ex vivo measurements from 0.5 to 20 GHz using a precision open-ended coaxial probe. Phys. Med. Biol. 2007, 52, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Scholoz, B.; Anderson, R. On electrical impedance scanning-principles and simulations. Electromedica 2000, 68, 35–44. [Google Scholar]
- Alon, L.; Sodickson, D.K.; Deniz, C.M. Heat equation inversion framework for average SAR calculation from magnetic resonance thermal imaging. Bioelectromagnetics 2016, 37, 493–503. [Google Scholar] [CrossRef]
- Mátay, G.; Zombory, L. A rádiófrekvenciás sugárzás élettani hatásai és orvosbiológiai alkalmazásai: Egyetemi tankönyv; Budapest: Műegyetem Kiadó, 2000. [Google Scholar]
- Pang, C.L.K.; Zhang, X.; Wang, Z.; Ou, J.; Lu, Y.; Chen, P.; Zhao, C.; Wang, X.; Zhang, H.; Roussakow, S.V. Local modulated electro-hyperthermia in combination with traditional Chinese medicine vs. intraperitoneal chemoinfusion for the treatment of peritoneal carcinomatosis with malignant ascites: A phase II randomized trial. Mol. Clin. Oncol. 2017, 6, 723–732. [Google Scholar] [CrossRef][Green Version]
- Meggyeshazi, N.; Andocs, G.; Balogh, L.; Balla, P.; Kiszner, G.; Teleki, I.; Jeney, A.; Krenacs, T. DNA fragmentation and caspase-independent programmed cell death by modulated electrohyperthermia. Strahlenthe. Onk. 2014, 190, 815–822. [Google Scholar] [CrossRef]
- Vancsik, T.; Kovago, C.; Kiss, E.; Papp, E.; Forika, G.; Benyo, Z.; Meggyeshazi, N.; Krenacs, T. Modulated electro-hyperthermia induced loco-regional and systemic tumor destruction in colorectal cancer allografts. J. Cancer 2018, 9, 41–53. [Google Scholar] [CrossRef]
- Vancsik, T.; Forika, G.; Balogh, A.; Kiss, E.; Krenacs, T. Modulated electro-hyperthermia induced p53 driven apoptosis and cell cycle arrest additively support doxorubicin chemotherapy of colorectal cancer in vitro. Cancer Med. 2019, 8, 4292–4303. [Google Scholar] [CrossRef]
- Cha, J.; Jeon, T.W.; Lee, C.G.; Oh, S.T.; Yang, H.B.; Choi, K.J.; Seo, D.; Yun, I.; Baik, I.H.; Park, K.R.; et al. Electro-hyperthermia inhibits glioma tumorigenicity through the induction of E2F1-mediated apoptosis. Int. J. Hyperth. 2015, 31, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Tsang, Y.W.; Huang, C.C.; Yang, K.L.; Chi, M.S.; Chiang, H.C.; Wang, Y.S.; Andocs, G.; Szasz, A.; Li, W.T.; Chi, K.H. Improving immunological tumor microenvironment using electro-hyperthermia followed by dendritic cell immunotherapy. BMC. Cancer 2015, 15, 708. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qin, W.; Akutsu, Y.; Andocs, G.; Suganami, A.; Hu, X.; Yusup, G.; Komatsu-Akimoto, A.; Hoshino, I.; Hanari, N.; Mori, M.; et al. Modulated electro-hyperthermia enhances dendritic cell therapy through an abscopal effect in mice. Oncol. Rep. 2014, 32, 2373–2379. [Google Scholar] [CrossRef]
- Andocs, G.; Meggyeshazi, N.; Balogh, L.; Spisak, S.; Maros, M.E.; Balla, P.; Kiszner, G.; Teleki, I.; Kovago, C.; Krenacs, T. Upregulation of heat shock proteins and the promotion of damage-associated molecular pattern signals in a colorectal cancer model by modulated electrohyperthermia. Cell. Stress. Chaperon 2015, 20, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Martin, S.; Golab, J.; Agostinis, P. Danger signalling during cancer cell death: Origins, plasticity and regulation. Cell. Death. Differ. 2014, 21, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, J.L.; Prendergast, G.C.; Messerschmidt, G.L. How Cancers Escape Immune Destruction and Mechanisms of Action for the New Significantly Active Immune Therapies: Helping Nonimmunologists Decipher Recent Advances. Oncologist 2016, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Werthmoller, N.; Frey, B.; Ruckert, M.; Lotter, M.; Fietkau, R.; Gaipl, U.S. Combination of ionising radiation with hyperthermia increases the immunogenic potential of B16-F10 melanoma cells in vitro and in vivo. Int. J. Hyperth. 2016, 32, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Tang, Y.P.; Zhao, L.Y.; Geng, C.Y.; Tang, J.T. Antitumor effect and immune response induced by local hyperthermia in B16 murine melanoma: Effect of thermal dose. Oncol. Lett. 2012, 4, 711–718. [Google Scholar] [CrossRef]
- Toraya-Brown, S.; Sheen, M.R.; Zhang, P.; Chen, L.; Baird, J.R.; Demidenko, E.; Turk, M.J.; Hoopes, P.J.; Conejo-Garcia, J.R.; Fiering, S. Local hyperthermia treatment of tumors induces CD8(+) T cell-mediated resistance against distal and secondary tumors. Nanomed. Nanotechnol. 2014, 10, 1273–1285. [Google Scholar] [CrossRef]
- Yang, K.L.; Huang, C.C.; Chi, M.S.; Chiang, H.C.; Wang, Y.S.; Hsia, C.C.; Andocs, G.; Wang, H.E.; Chi, K.H. In vitro comparison of conventional hyperthermia and modulated electro-hyperthermia. Oncotarget 2016, 7, 84082–84092. [Google Scholar] [CrossRef]
- Oei, A.L.; Vriend, L.E.; Crezee, J.; Franken, N.A.; Krawczyk, P.M. Effects of hyperthermia on DNA repair pathways: One treatment to inhibit them all. Raditat. Oncol. 2015, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jaattela, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Djaldetti, M.; Bessler, H. High temperature affects the phagocytic activity of human peripheral blood mononuclear cells. Scand. J. Clin. Lab. Invest. 2015, 75, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Zhang, Q.; Yan, Z.; Chen, R.; Zeh Iii, H.J.; Kang, R.; Lotze, M.T.; Tang, D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death. Dis. 2013, 4, e966. [Google Scholar] [CrossRef] [PubMed]
- Guzhova, I.V.; Shevtsov, M.A.; Abkin, S.V.; Pankratova, K.M.; Margulis, B.A. Intracellular and extracellular Hsp70 chaperone as a target for cancer therapy. Int. J. Hyperth. 2013, 29, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef]
- Shevtsov, M.; Huile, G.; Multhoff, G. Membrane heat shock protein 70: A theranostic target for cancer therapy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 372. [Google Scholar] [CrossRef]
- Tamura, Y.; Torigoe, T.; Kukita, K.; Saito, K.; Okuya, K.; Kutomi, G.; Hirata, K.; Sato, N. Heat-shock proteins as endogenous ligands building a bridge between innate and adaptive immunity. Immunotherapy 2012, 4, 841–852. [Google Scholar] [CrossRef]
- Seliger, B.; Wollscheid, U.; Momburg, F.; Blankenstein, T.; Huber, C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 2001, 61, 1095–1099. [Google Scholar]
- Riond, J.; Rodriguez, S.; Nicolau, M.L.; al Saati, T.; Gairin, J.E. In vivo major histocompatibility complex class I (MHCI) expression on MHCIlow tumor cells is regulated by gammadelta T and NK cells during the early steps of tumor growth. Cancer Immun. 2009, 9, 10. [Google Scholar]
- La Rocca, R.; Tallerico, R.; Talib Hassan, A.; Das, G.; Lakshmikanth, T.; Matteucci, M.; Liberale, C.; Mesuraca, M.; Scumaci, D.; Gentile, F.; et al. Mechanical stress downregulates MHC class I expression on human cancer cell membrane. PLoS ONE 2014, 9, e111758. [Google Scholar] [CrossRef][Green Version]
- Ostberg, J.R.; Dayanc, B.E.; Yuan, M.; Oflazoglu, E.; Repasky, E.A. Enhancement of natural killer (NK) cell cytotoxicity by fever-range thermal stress is dependent on NKG2D function and is associated with plasma membrane NKG2D clustering and increased expression of MICA on target cells. J. Leukoc. Biol. 2007, 82, 1322–1331. [Google Scholar] [CrossRef]
- Karre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef]
- Raulet, D.H. Missing self recognition and self tolerance of natural killer (NK) cells. Semin. Immunol. 2006, 18, 145–150. [Google Scholar] [CrossRef]
- Goldszmid, R.S.; Idoyaga, J.; Bravo, A.I.; Steinman, R.; Mordoh, J.; Wainstok, R. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J. Immunol. 2003, 171, 5940–5947. [Google Scholar] [CrossRef]
- Gordon, S.; Pluddemann, A. Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front. Immunol. 2018, 9, 127. [Google Scholar] [CrossRef]
- Lee, S.; Son, B.; Park, G.; Kim, H.; Kang, H.; Jeon, J.; Youn, H.; Youn, B. Immunogenic Effect of Hyperthermia on Enhancing Radiotherapeutic Efficacy. Int. J. Mol. Sci. 2018, 19, 2795. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| RPLP0 | CTCTCGCTTTCTGGAGGGTG | ACGCGCTTGTACCCATTGAT |
| BCL-2 | CTCGTCGCTACCGTCGTGACTTCG | CAGATGCCGGTTCAGGTACTCAGTC |
| BCL-XL | AACATCCCAGCTTCACATAACCCC | GCGACCCCAGTTTACTCCATCC |
| BAX | AAGCTGAGCGAGTGTCTCCGGCG | GCCACAAAGATGGTCACTGTCTGCC |
| BAK-1 | CAGCTTGCTCTCATCGGAGAT | GGTGAAGAGTTCGTAGGCATTC |
| XIAP | ATGCTTTAGGTGAAGGCGAT | CATGCTGTTCCCAAGGGTCT |
| PUMA | TCTATGGGTGGAGCCTCAGT | GAGGGCTGAGGACCCATTAAA |
| p21 | GCAGAATAAAAGGTGCCACAGG | AAAGTTCCACCGTTCTCGGG |
| Antigen | Type | Reference | Dilution | Vendor |
|---|---|---|---|---|
| AIF | Rabbit, pAb | #4642 | 1:70 | Cell Signaling |
| p-H2AX(Ser139) | Rabbit, mAb | #9718 | 1: 200 | Cell Signaling |
| Cleaved caspase-3 | Rabbit, pAb | #9664 | 1:300 | Cell Signaling |
| Hsp70 | Rabbit, pAb | #4872 | 1:200 | Cell Signaling |
| P21waf1 | Rabbit, mAb | #ab188224 | 1:500 | Abcam |
| P27kip1 | Rabbit, pAb | # RB9019P | 1:50 | Thermo |
| P53 (acetyl K386) | Rabbit, pAb | #ab52172 | 1:200 | Abcam |
| P53 | Goat, pAb | #AF1355 | 1:350 | Bio-Techne |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besztercei, B.; Vancsik, T.; Benedek, A.; Major, E.; Thomas, M.J.; Schvarcz, C.A.; Krenács, T.; Benyó, Z.; Balogh, A. Stress-Induced, p53-Mediated Tumor Growth Inhibition of Melanoma by Modulated Electrohyperthermia in Mouse Models without Major Immunogenic Effects. Int. J. Mol. Sci. 2019, 20, 4019. https://doi.org/10.3390/ijms20164019
Besztercei B, Vancsik T, Benedek A, Major E, Thomas MJ, Schvarcz CA, Krenács T, Benyó Z, Balogh A. Stress-Induced, p53-Mediated Tumor Growth Inhibition of Melanoma by Modulated Electrohyperthermia in Mouse Models without Major Immunogenic Effects. International Journal of Molecular Sciences. 2019; 20(16):4019. https://doi.org/10.3390/ijms20164019
Chicago/Turabian StyleBesztercei, Balázs, Tamás Vancsik, Anett Benedek, Enikő Major, Mbuotidem J. Thomas, Csaba A. Schvarcz, Tibor Krenács, Zoltán Benyó, and Andrea Balogh. 2019. "Stress-Induced, p53-Mediated Tumor Growth Inhibition of Melanoma by Modulated Electrohyperthermia in Mouse Models without Major Immunogenic Effects" International Journal of Molecular Sciences 20, no. 16: 4019. https://doi.org/10.3390/ijms20164019
APA StyleBesztercei, B., Vancsik, T., Benedek, A., Major, E., Thomas, M. J., Schvarcz, C. A., Krenács, T., Benyó, Z., & Balogh, A. (2019). Stress-Induced, p53-Mediated Tumor Growth Inhibition of Melanoma by Modulated Electrohyperthermia in Mouse Models without Major Immunogenic Effects. International Journal of Molecular Sciences, 20(16), 4019. https://doi.org/10.3390/ijms20164019