Protein Kinase CK2—A Putative Target for the Therapy of Diabetes Mellitus?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
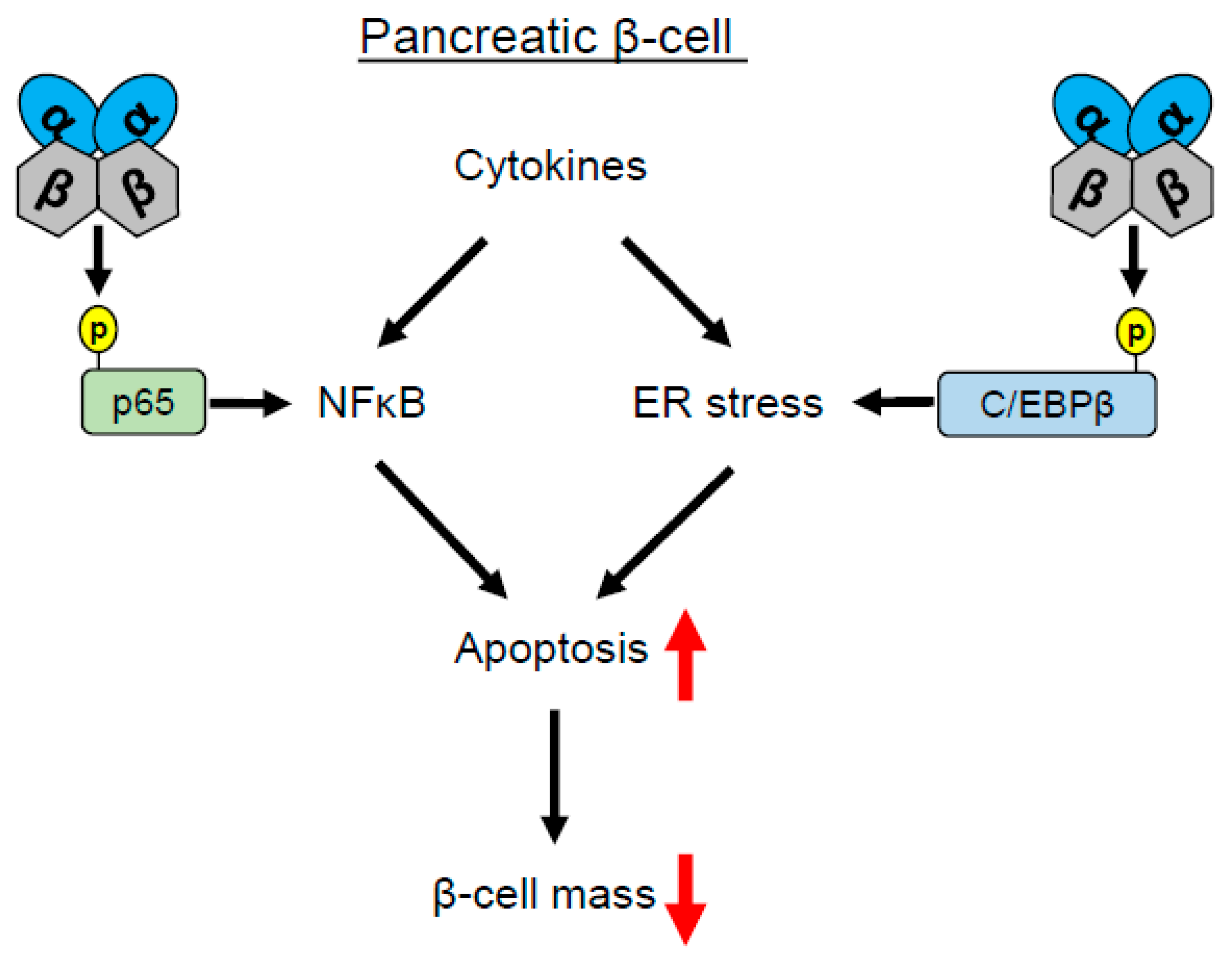
1.1. CK2 Regulates Pancreatic β-Cell Death
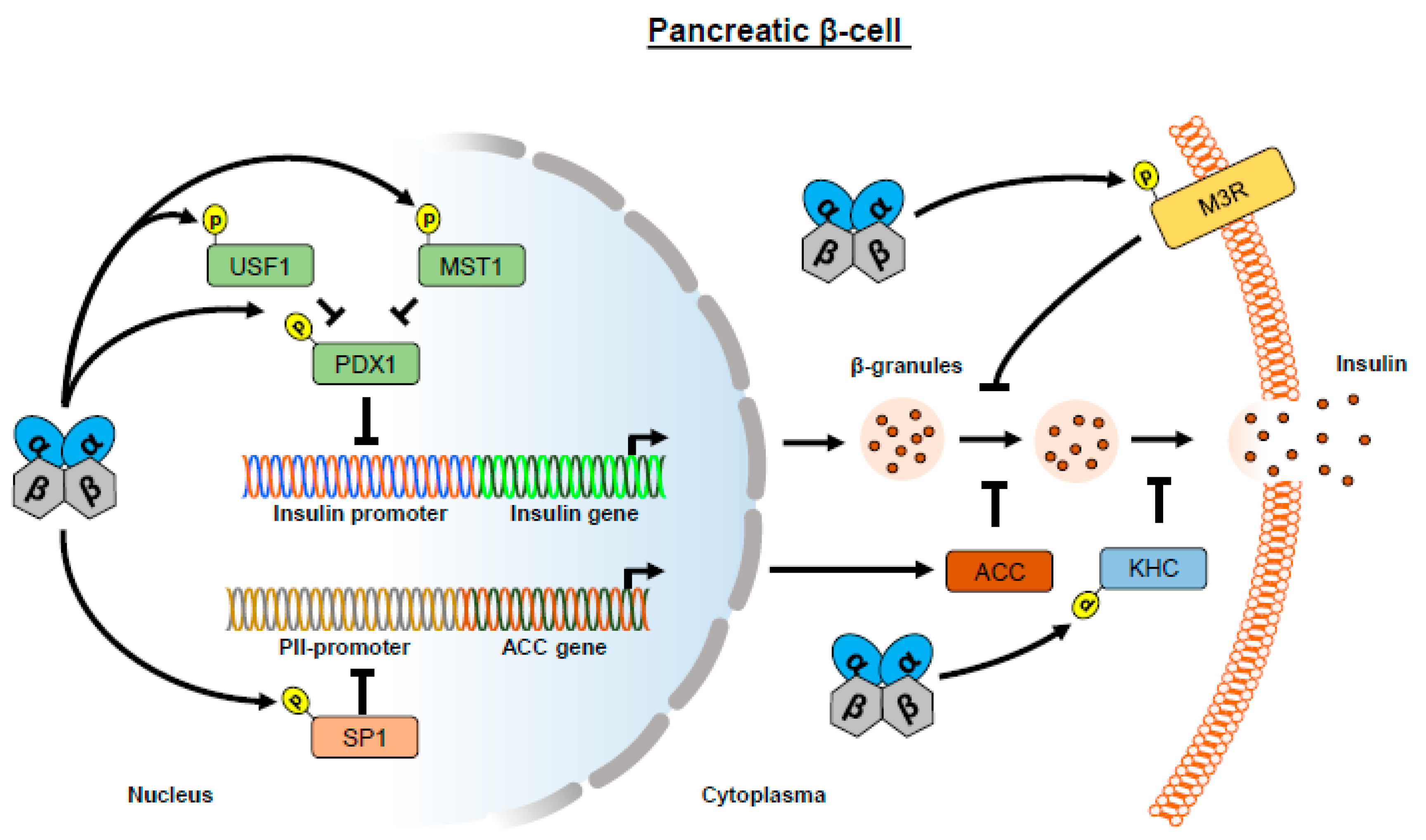
1.2. CK2 Regulates Insulin Expression
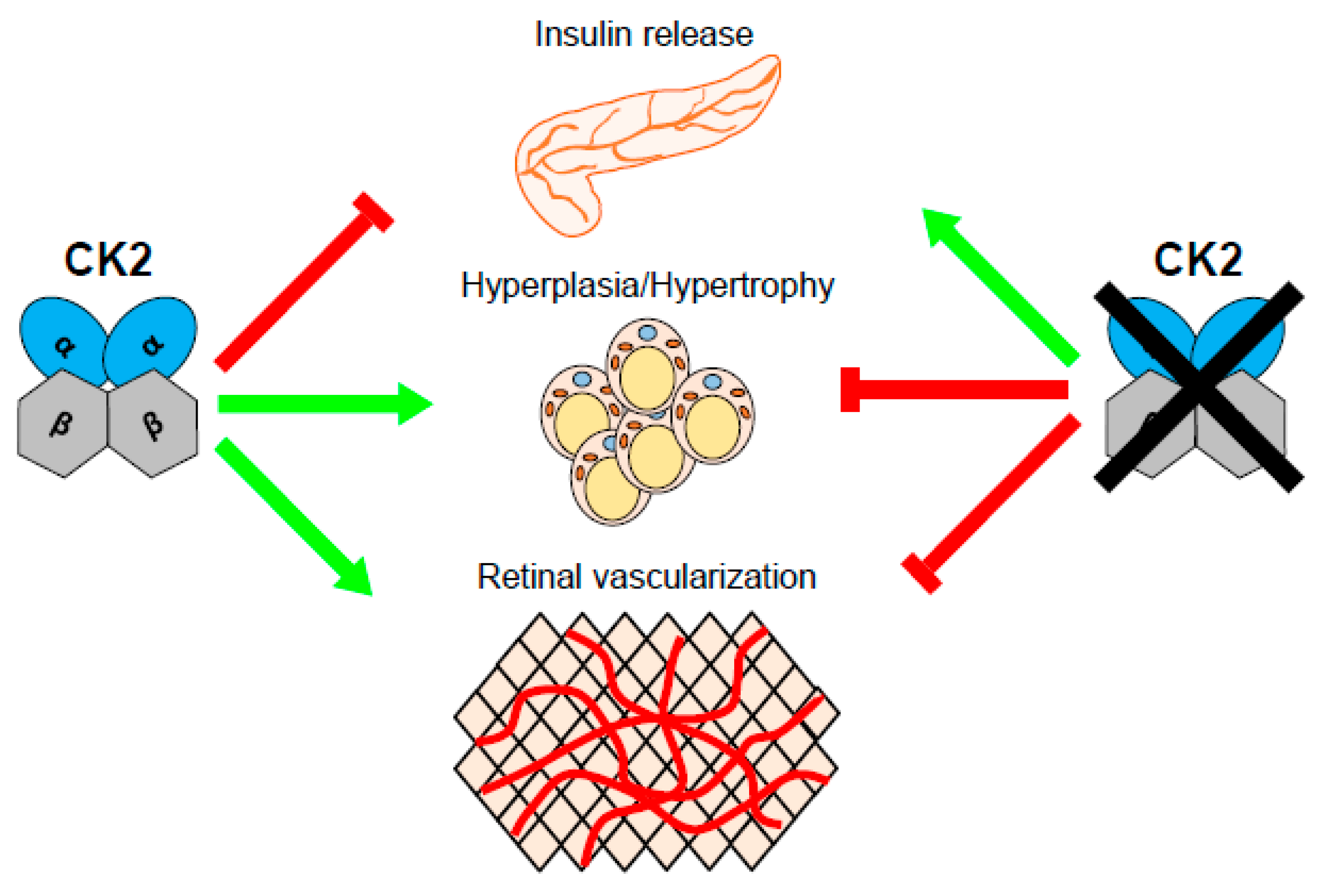
1.3. CK2 Regulates Insulin Release
1.4. CK2 Regulates Insulin Signaling in Adipocytes/Fat Tissue
1.5. CK2 and Diabetic Retinopathy
2. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta 2009, 1793, 847–859. [Google Scholar] [CrossRef]
- Nunez de Villavicencio-Diaz, T.; Rabalski, A.J.; Litchfield, D.W. Protein Kinase CK2: Intricate Relationships within Regulatory Cellular Networks. Pharmaceuticals 2017, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Boldyreff, B.; Meggio, F.; Pinna, L.A.; Issinger, O.G. Protein kinase CK2 structure-function relationship: Effects of the á subunit on reconstitution and activity. Cell. Mol. Biol. Res. 1994, 40, 391–399. [Google Scholar] [PubMed]
- Wirkner, U.; Voss, H.; Lichter, P.; Ansorge, W.; Pyerin, W. The human gene (CSNK2A1) coding for the casein kinase II subunit alpha is located on chromosome 20 and contains tandemly arranged Alu repeats. Genomics 1994, 19, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, K.; Neidhart, T.; Gerber, J.; Waxmann, A.; Pyerin, W. The catalytic subunit alpha’ gene of human protein kinase CK2 (CSNK2A2): genomic organization, promoter identification and determination of Ets1 as a key regulator. Mol. Cell Biochem. 2005, 274, 91–101. [Google Scholar] [CrossRef]
- Albertella, M.R.; Jones, H.; Thomson, W.; Olavesen, M.G.; Neville, M.; Campbell, R.D. Localisation of eight additional genes in the human major histocompatibility complex, including the gene encoding the casein kinase II beta subunit, and DNA sequence analysis of the class III region. DNA Seq. 1996, 7, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Brunstein, E.; Issinger, O.G.; Niefind, K. The interaction of CK2alpha and CK2beta, the subunits of protein kinase CK2, requires CK2beta in a preformed conformation and is enthalpically driven. Protein Sci. 2008, 17, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Boldyreff, B.S.; Marin, O.; Pinna, L.A.; Issinger, O.G. CK2: Role of the á- subunit on the stability and specificity of the recombinant reconstituted holoenzyme. Eur. J. Biochem. 1992, 204, 293–297. [Google Scholar] [CrossRef]
- Boldyreff, B.S.; Meggio, F.; Pinna, L.A.; Issinger, O.G. Casein kinase-2 structure- function relationship: Creation of a set of mutants of the á subunit that variably surrogate the wildtype á subunit function. Biochem. Biophys. Res. Commun. 1992, 188, 228–234. [Google Scholar] [CrossRef]
- Rodriguez, F.A.; Contreras, C.; Bolanos-Garcia, V.; Allende, J.E. Protein kinase CK2 as an ectokinase: The role of the regulatory CK2β subunit. Proc. Natl. Acad. Sci. USA 2008, 105, 5693–5698. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.Y.; Dominguez, I.; Toselli, P.; Landesman-Bollag, E.; O’Brien, C.; Seldin, D.C. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol. Cell Biol. 2008, 28, 131–139. [Google Scholar] [CrossRef]
- Buchou, T.; Vernet, M.; Blond, O.; Jensen, H.H.; Pointu, H.; Olsen, B.B.; Cochet, C.; Issinger, O.G.; Boldyreff, B. Disruption of the regulatory á subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol. Cell. Biol. 2003, 23, 908–915. [Google Scholar] [CrossRef]
- Xu, X.; Toselli, P.A.; Russell, L.D.; Seldin, D.C. Globozoospermia in mice lacking the casein kinase II à’ catalytic subunit. Nat. Genet. 1999, 23, 118–121. [Google Scholar] [CrossRef]
- Intemann, J.; Saidu, N.E.B.; Schwind, L.; Montenarh, M. ER stress signaling in ARPE-19 cells after inhibition of protein kinase CK2 by CX-4945. Cell Signal. 2014, 26, 1567–1575. [Google Scholar] [CrossRef]
- Götz, C.; Gratz, A.; Kucklaender, U.; Jose, J. TF--a novel cell-permeable and selective inhibitor of human protein kinase CK2 induces apoptosis in the prostate cancer cell line LNCaP. Biochim. Biophys. Acta 2012, 1820, 970–977. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, J.J.; Bae, Y.S. Involvement of PI3K-AKT-mTOR pathway in protein kinase CKII inhibition-mediated senescence in human colon cancer cells. Biochem. Biophys. Res. Commun. 2013, 433, 420–425. [Google Scholar] [CrossRef]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Tapia, J.C.; Torres, V.A.; Rodriguez, D.A.; Leyton, L.; Quest, A.F. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef]
- Sarno, S.; Ruzzene, M.; Frascella, P.; Pagano, M.A.; Meggio, F.; Zambon, A.; Mazzorana, M.; Di Maira, G.; Lucchini, V.; Pinna, L.A. Development and exploitation of CK2 inhibitors. Mol. Cell Biochem. 2005, 274, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Cochet, C. New protein kinase CK2 inhibitors: jumping out of the catalytic box. Chem. Biol. 2009, 16, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, N.; Kostelnik, K.; Götz, C.; Montenarh, M. Protein kinase CK2 is implicated in early steps of the differentiation of pre-adipocytes into adipocytes. Mol. Cell Biochem. 2012, 365, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Montenarh, M. Protein kinase CK2 and angiogenesis. Adv. Clin. Exp. Med. 2014, 23, 153–158. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.A.; Litchfield, D.W. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol. Life Sci. 2009, 66, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Al-Quobaili, F.; Montenarh, M. CK2 and the regulation of the carbohydrate metabolism. Metabolism 2012, 61, 1512–1517. [Google Scholar] [CrossRef]
- Welker, S.; Götz, C.; Servas, C.; Laschke, M.W.; Menger, M.D.; Montenarh, M. Glucose regulates protein kinase CK2 in pancreatic beta-cells and its interaction with PDX-1. Int. J. Biochem. Cell Biol. 2013, 45, 2786–2795. [Google Scholar] [CrossRef]
- Wang, K.; Li, F.; Cui, Y.; Cui, C.; Cao, Z.; Xu, K.; Han, S.; Zhu, P.; Sun, Y. The Association between Depression and Type 1 Diabetes Mellitus: Inflammatory Cytokines as Ferrymen in between? Mediat. Inflamm. 2019, 2019, 2987901. [Google Scholar] [CrossRef]
- Andersson, S.A.; Olsson, A.H.; Esguerra, J.L.; Heimann, E.; Ladenvall, C.; Edlund, A.; Salehi, A.; Taneera, J.; Degerman, E.; Groop, L.; et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol. Cell. Endocrinol. 2012, 364, 36–45. [Google Scholar] [CrossRef]
- Donath, M.Y.; Boni-Schnetzler, M.; Ellingsgaard, H.; Halban, P.A.; Ehses, J.A. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab. 2010, 21, 261–267. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: examining the links. Int. J. Physiol. Pathophysiol. Pharm. 2019, 11, 45–63. [Google Scholar]
- Eldor, R.; Yeffet, A.; Baum, K.; Doviner, V.; Amar, D.; Ben-Neriah, Y.; Christofori, G.; Peled, A.; Carel, J.C.; Boitard, C.; et al. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc. Natl. Acad. Sci. USA 2006, 103, 5072–5077. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin Jr, A.S. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Rudzitis-Auth, J.; Dahmke, I.N.; Roessler, O.G.; Thiel, G.; Montenarh, M.; Menger, M.D.; Laschke, M.W. Inhibition of protein kinase CK2 suppresses tumor necrosis factor (TNF)-alpha-induced leukocyte-endothelial cell interaction. Biochim. Biophys. Acta 2015, 1852, 2123–2136. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.A.; Schooley, K.; Dower, S.K.; Hagen, H.; Virca, G.D. Activation of nuclear transcription factor NF-kappaB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J. Biol. Chem. 1997, 272, 32606–32612. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, C.; Thams, P. A critical role for CK2 in cytokine-induced activation of NFkappaB in pancreatic beta cell death. Endocrine 2014, 47, 117–128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duncan, J.S.; Turowec, J.P.; Vilk, G.; Li, S.S.; Gloor, G.B.; Litchfield, D.W. Regulation of cell proliferation and survival: convergence of protein kinases and caspases. Biochim. Biophys. Acta 2010, 1804, 505–510. [Google Scholar] [CrossRef]
- Meng, R.; Götz, C.; Montenarh, M. The role of protein kinase CK2 in the regulation of the insulin production of pancreatic islets. Biochem. Biophys. Res. Commun. 2010, 401, 203–206. [Google Scholar] [CrossRef]
- Spohrer, S.; Gross, R.; Nalbach, L.; Schwind, L.; Stumpf, H.; Menger, M.D.; Ampofo, E.; Montenarh, M.; Götz, C. Functional interplay between the transcription factors USF1 and PDX-1 and protein kinase CK2 in pancreatic beta-cells. Sci. Rep. 2017, 7, 16367. [Google Scholar] [CrossRef]
- Zandomeni, R.; Bunick, D.; Ackerman, S.; Mittleman, B.; Weinmann, R. Mechanism of action of DRB. III. Effect on specific in vitro initiation of transcription. J. Mol. Biol. 1983, 167, 561–574. [Google Scholar] [CrossRef]
- Schneider, C.C.; Hessenauer, A.; Gotz, C.; Montenarh, M. DMAT, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncol. Rep. 2009, 21, 1593–1597. [Google Scholar] [PubMed]
- Eizirik, D.L.; Miani, M.; Cardozo, A.K. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013, 56, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Tyka, K.; Jorns, A.; Turatsinze, J.V.; Eizirik, D.L.; Lenzen, S.; Gurgul-Convey, E. MCPIP1 regulates the sensitivity of pancreatic beta-cells to cytokine toxicity. Cell Death Dis. 2019, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Matsuda, T.; Matsuura, Y.; Inoue, K.; Suzuki, E.; Kanno, A.; Kimura-Koyanagi, M.; Asahara, S.I.; Hatano, N.; Ogawa, W.; et al. Casein kinase 2 phosphorylates and stabilizes C/EBPbeta in pancreatic beta cells. Biochem. Biophys. Res. Commun. 2018, 497, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Sokolowsky, T.; Götz, C.; Montenarh, M. Functional interaction of protein kinase CK2 and activating transcription factor 4 (ATF4), a key player in the cellular stress response. Biochim. Biophys. Acta 2013, 1833, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.C.; Ampofo, E.; Montenarh, M. CK2 regulates ATF4 and CHOP transcription within the cellular stress response signalling pathway. Cell Signal. 2012, 24, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Papa, F.R. Endoplasmic reticulum stress, pancreatic beta-cell degeneration, and diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007666. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Jonsson, J.; Ahlgren, U.; Edlund, T.; Edlund, H. IPF1, a homeodomain protein with a dual function in pancreas development. Int. J. Dev. Biol 1995, 39, 789–798. [Google Scholar]
- McKinnon, C.M.; Docherty, K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001, 44, 1203–1214. [Google Scholar] [CrossRef]
- Meng, R.; Al-Quobaili, F.; Müller, I.; Götz, C.; Thiel, G.; Montenarh, M. CK2 phosphorylation of Pdx-1 regulates its transcription factor activity. Cell Mol. Life Sci. 2010, 67, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Claiborn, K.C.; Sachdeva, M.M.; Cannon, C.E.; Groff, D.N.; Singer, J.D.; Stoffers, D.A. Pcif1 modulates Pdx1 protein stability and pancreatic beta cell function and survival in mice. J. Clin. Investig. 2010, 120, 3713–3721. [Google Scholar] [CrossRef]
- Liu, A.; Oliver-Krasinski, J.; Stoffers, D.A. Two conserved domains in PCIF1 mediate interaction with pancreatic transcription factor PDX-1. FEBS Lett. 2006, 580, 6701–6706. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Meng, R.; Montenarh, M.; Götz, C. The Phosphorylation of PDX-1 by Protein Kinase CK2 Is Crucial for Its Stability. Pharmaceuticals 2016, 10, 2. [Google Scholar] [CrossRef]
- Ostertag, M.S.; Messias, A.C.; Sattler, M.; Popowicz, G.M. The Structure of the SPOP-Pdx1 Interface Reveals Insights into the Phosphorylation-Dependent Binding Regulation. Structure 2019, 27, 327–334 e3. [Google Scholar] [CrossRef] [PubMed]
- Melloul, D.; Tsur, A.; Zangen, D. Pancreatic Duodenal Homeobox (PDX-1) in health and disease. J. Pediatric Endocrinol. Metab. JPEM 2002, 15, 1461–1472. [Google Scholar] [CrossRef]
- Lupp, S.; Götz, C.; Khadouma, S.; Horbach, T.; Dimova, E.Y.; Bohrer, A.M.; Kietzmann, T.; Montenarh, M. The upstream stimulatory factor USF1 is regulated by protein kinase CK2 phosphorylation. Cell Signal. 2014, 26, 2809–2817. [Google Scholar] [CrossRef]
- Frogne, T.; Sylvestersen, K.B.; Kubicek, S.; Nielsen, M.L.; Hecksher-Sorensen, J. Pdx1 is post-translationally modified in vivo and serine 61 is the principal site of phosphorylation. PLoS ONE 2012, 7, e35233. [Google Scholar] [CrossRef]
- An, R.; da Silva Xavier, G.; Semplici, F.; Vakhshouri, S.; Hao, H.X.; Rutter, J.; Pagano, M.A.; Meggio, F.; Pinna, L.A.; Rutter, G.A. Pancreatic and duodenal homeobox 1 (PDX1) phosphorylation at serine-269 is HIPK2-dependent and affects PDX1 subnuclear localization. Biochem. Biophys. Res. Commun. 2010, 399, 155–161. [Google Scholar] [CrossRef]
- Ardestani, A.; Paroni, F.; Azizi, Z.; Kaur, S.; Khobragade, V.; Yuan, T.; Frogne, T.; Tao, W.; Oberholzer, J.; Pattou, F.; et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 2014, 20, 385–397. [Google Scholar] [CrossRef]
- Ardestani, A.; Maedler, K. MST1: a promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia 2016, 59, 1843–1849. [Google Scholar] [CrossRef]
- Servas, C.; Kiehlmeier, S.; Hach, J.; Gross, R.; Gotz, C.; Montenarh, M. The mammalian STE20-like kinase 1 (MST1) is a substrate for the apoptosis inhibiting protein kinase CK2. Cell Signal. 2017, 36, 163–175. [Google Scholar] [CrossRef]
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516. [Google Scholar] [CrossRef]
- Hirokawa, N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 1998, 279, 519–526. [Google Scholar] [CrossRef]
- Donelan, M.J.; Morfini, G.; Julyan, R.; Sommers, S.; Hays, L.; Kajio, H.; Briaud, I.; Easom, R.A.; Molkentin, J.D.; Brady, S.T.; et al. Ca2+-dependent dephosphorylation of kinesin heavy chain on beta-granules in pancreatic beta-cells. Implications for regulated beta-granule transport and insulin exocytosis. J. Biol. Chem. 2002, 277, 24232–24242. [Google Scholar] [CrossRef]
- Gilon, P.; Henquin, J.C. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr. Rev. 2001, 22, 565–604. [Google Scholar]
- Budd, D.C.; McDonald, J.E.; Tobin, A.B. Phosphorylation and regulation of a Gq/11-coupled receptor by casein kinase 1alpha. J. Biol. Chem. 2000, 275, 19667–19675. [Google Scholar] [CrossRef]
- Torrecilla, I.; Spragg, E.J.; Poulin, B.; McWilliams, P.J.; Mistry, S.C.; Blaukat, A.; Tobin, A.B. Phosphorylation and regulation of a G protein-coupled receptor by protein kinase CK2. J. Cell Biol. 2007, 177, 127–137. [Google Scholar] [CrossRef]
- Luo, J.; Busillo, J.M.; Benovic, J.L. M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol. Pharm. 2008, 74, 338–347. [Google Scholar] [CrossRef]
- Willets, J.M.; Mistry, R.; Nahorski, S.R.; Challiss, R.A. Specificity of g protein-coupled receptor kinase 6-mediated phosphorylation and regulation of single-cell m3 muscarinic acetylcholine receptor signaling. Mol. Pharm. 2003, 64, 1059–1068. [Google Scholar] [CrossRef]
- Rossi, M.; Ruiz, d.A.I.; Barella, L.F.; Sakamoto, W.; Zhu, L.; Cui, Y.; Lu, H.; Rebholz, H.; Matschinsky, F.M.; Doliba, N.M.; et al. CK2 acts as a potent negative regulator of receptor-mediated insulin release in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, E6818–E6824. [Google Scholar] [CrossRef] [PubMed]
- Doliba, N.M.; Liu, Q.; Li, C.; Chen, P.; Liu, C.; Naji, A.; Matschinsky, F.M. Inhibition of Cholinergic Potentiation of Insulin Secretion from Pancreatic Islets by Chronic Elevation of Glucose and Fatty Acids: Protection by Casein Kinase 2 Inhibitor. Mol. Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cantley, J.; Davenport, A.; Vetterli, L.; Nemes, N.J.; Whitworth, P.T.; Boslem, E.; Thai, L.M.; Mellett, N.; Meikle, P.J.; Hoehn, K.L.; et al. Disruption of beta cell acetyl-CoA carboxylase-1 in mice impairs insulin secretion and beta cell mass. Diabetologia 2019, 62, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, S.M.; Joseph, J.W.; Ilkayeva, O.; Burgess, S.C.; Lu, D.; Becker, T.C.; Sherry, A.D.; Newgard, C.B. Chronic suppression of acetyl-CoA carboxylase 1 in beta-cells impairs insulin secretion via inhibition of glucose rather than lipid metabolism. J. Biol. Chem. 2008, 283, 14248–14256. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Cousins, R.S.; Liu, S.; Phelps, B.M.; Promes, J.A. Connecting pancreatic islet lipid metabolism with insulin secretion and the development of type 2 diabetes. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kim, K.H. Protein kinase CK2 down-regulates glucose-activated expression of the acetyl-CoA carboxylase gene. Arch. Biochem. Biophys. 1997, 338, 227–232. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Barry, D.A.; Leggett, R.W.; Mueller, C.R. Casein kinase II-mediated phosphorylation of the C terminus of Sp1 decreases its DNA binding activity. J. Biol. Chem. 1997, 272, 13489–13495. [Google Scholar] [CrossRef]
- Schuster, D.P. Obesity and the development of type 2 diabetes: the effects of fatty tissue inflammation. Diabetes Metab. Syndr. Obes. Targets Ther. 2010, 3, 253–262. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, J.; Lee, H.W.; Lee, Y.S.; Kim, J.B. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem. Biophys. Res. Commun. 2004, 321, 942–948. [Google Scholar] [CrossRef]
- Klemm, D.J.; Leitner, J.W.; Watson, P.; Nesterova, A.; Reusch, J.E.; Goalstone, M.L.; Draznin, B. Insulin-induced adipocyte differentiation. Activation of CREB rescues adipogenesis from the arrest caused by inhibition of prenylation. J. Biol. Chem. 2001, 276, 28430–28435. [Google Scholar] [CrossRef]
- Wabitsch, M.; Hauner, H.; Heinze, E.; Teller, W.M. The role of growth hormone/insulin-like growth factors in adipocyte differentiation. Metabolism 1995, 44, 45–49. [Google Scholar] [CrossRef]
- Schwind, L.; Nalbach, L.; Zimmer, A.D.; Kostelnik, K.B.; Menegatti, J.; Grasser, F.; Gotz, C.; Montenarh, M. Quinalizarin inhibits adipogenesis through down-regulation of transcription factors and microRNA modulation. Biochim. Et Biophys. Acta. Gen. Subj. 2017, 1861, 3272–3281. [Google Scholar] [CrossRef]
- Chen, Q.; Hao, W.; Xiao, C.; Wang, R.; Xu, X.; Lu, H.; Chen, W.; Deng, C.X. SIRT6 Is Essential for Adipocyte Differentiation by Regulating Mitotic Clonal Expansion. Cell Rep. 2017, 18, 3155–3166. [Google Scholar] [CrossRef]
- Schäfer, B.; Götz, C.; Dudek, J.; Hessenauer, A.; Matti, U.; Montenarh, M. KIF5C, a new binding partner for protein kinase CK2 with a preference for CK2à’. Cell Mol. Life Sci. 2009, 66, 339–349. [Google Scholar] [CrossRef]
- Schäfer, B.; Götz, C.; Montenarh, M. The kinesin I family member KIF5C is a novel substrate for protein kinase CK2. Biochem. Biophys. Res. Commun. 2008, 375, 179–183. [Google Scholar] [CrossRef]
- Bae, J.S.; Park, S.H.; Jamiyandorj, U.; Kim, K.M.; Noh, S.J.; Kim, J.R.; Park, H.J.; Kwon, K.S.; Jung, S.H.; Park, H.S.; et al. CK2alpha/CSNK2A1 Phosphorylates SIRT6 and Is Involved in the Progression of Breast Carcinoma and Predicts Shorter Survival of Diagnosed Patients. Am. J. Pathol. 2016, 186, 3297–3315. [Google Scholar] [CrossRef]
- Borgo, C.; Milan, G.; Favaretto, F.; Stasi, F.; Fabris, R.; Salizzato, V.; Cesaro, L.; Belligoli, A.; Sanna, M.; Foletto, M.; et al. CK2 modulates adipocyte insulin-signaling and is up-regulated in human obesity. Sci. Rep. 2017, 7, 17569. [Google Scholar] [CrossRef]
- Scherer, P.E. The many secret lives of adipocytes: implications for diabetes. Diabetologia 2019, 62, 223–232. [Google Scholar] [CrossRef]
- Nakamura, A.; Miyoshi, H.; Ukawa, S.; Nakamura, K.; Nakagawa, T.; Terauchi, Y.; Tamakoshi, A.; Atsumi, T. Serum adiponectin and insulin secretion: A direct or inverse association? J. Diabetes Investig. 2018, 9, 1106–1109. [Google Scholar] [CrossRef]
- Okamoto, M.; Ohara-Imaizumi, M.; Kubota, N.; Hashimoto, S.; Eto, K.; Kanno, T.; Kubota, T.; Wakui, M.; Nagai, R.; Noda, M.; et al. Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia 2008, 51, 827–835. [Google Scholar] [CrossRef]
- Heiker, J.T.; Wottawah, C.M.; Juhl, C.; Kosel, D.; Morl, K.; Beck-Sickinger, A.G. Protein kinase CK2 interacts with adiponectin receptor 1 and participates in adiponectin signaling. Cell Signal. 2009, 21, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Juhl, C.; Morl, K.; Beck-Sickinger, A.G. Adiponectin receptor 1 interacts with both subunits of protein kinase CK2. Mol. Cell Biochem. 2011, 356, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Arfken, C.L.; Reno, P.L.; Santiago, J.V.; Klein, R. Development of proliferative diabetic retinopathy in African-Americans and whites with type 1 diabetes. Diabetes Care 1998, 21, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N. Diabetic retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ritt, D.A.; Morrison, D.K.; Der, C.J.; Cox, A.D. Protein Kinase CK2alpha Maintains Extracellular Signal-regulated Kinase (ERK) Activity in a CK2alpha Kinase-independent Manner to Promote Resistance to Inhibitors of RAF and MEK but Not ERK in BRAF Mutant Melanoma. J. Biol. Chem. 2016, 291, 17804–17815. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Bertacchini, J.; Toker, A.; Marmiroli, S. Cross-talk between the CK2 and AKT signaling pathways in cancer. Adv. Biol. Regul. 2017, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Welker, S.; Korbel, C.; Rudzitis-Auth, J.; Menger, M.D.; Montenarh, M.; Laschke, M.W. Protein kinase CK2 is a regulator of angiogenesis in endometriotic lesions. Angiogenesis 2012, 15, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C. Signalling pathways involved in antiproliferative effects of IGFBP-3: a review. Mol. Pathol. MP 2001, 54, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, Q.; Steinle, J.J. Intravitreal injection of IGFBP-3 restores normal insulin signaling in diabetic rat retina. PLoS ONE 2014, 9, e93788. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Doehmen, S.; Le, M.L.; Koizumi, K.; Radetzky, S.; Krohne, T.U.; Poulaki, V.; Semkova, I.; Kociok, N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol. Vis. 2009, 15, 1418. [Google Scholar]
- Zhang, Q.; Soderland, D.; Steinle, J.J. TNFalpha inhibits IGFBP-3 through activation of p38alpha and casein kinase 2 in human retinal endothelial cells. PLoS ONE 2014, 9, e103578. [Google Scholar]
- Ljubimov, A.V.; Caballero, S.; Aoki, A.M.; Pinna, L.A.; Grant, M.B.; Castellon, R. Involvement of protein kinase CK2 in angiogenesis and retinal neovascularization. Invest. Ophthalmol Vis. Sci 2004, 45, 4583–4591. [Google Scholar] [CrossRef] [PubMed]
- Kramerov, A.A.; Saghizadeh, M.; Pan, H.; Kabosova, A.; Montenarh, M.; Ahmed, K.; Penn, J.S.; Chan, C.K.; Hinton, D.R.; Grant, M.B.; et al. Expression of protein kinase CK2 in astroglial cells of normal and neovascularized retina. Am. J. Pathol. 2006, 168, 1722–1736. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ampofo, E.; Schmitt, B.M.; Laschke, M.W.; Menger, M.D. Function of protein kinase CK2 in thrombus formation. Platelets 2018, 1–7. [Google Scholar] [CrossRef]
- Welters, H.J.; Kulkarni, R.N. Wnt signaling: relevance to beta-cell biology and diabetes. Trends Endocrinol. Metab. TEM 2008, 19, 349–355. [Google Scholar] [CrossRef]
- Rulifson, I.C.; Karnik, S.K.; Heiser, P.W.; ten Berge, D.; Chen, H.; Gu, X.; Taketo, M.M.; Nusse, R.; Hebrok, M.; Kim, S.K. Wnt signaling regulates pancreatic beta cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 6247–6252. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Zhou, T.; Zhou, K.K.; Mott, R.; Wu, M.; Boulton, M.; Lyons, T.J.; Gao, G.; Ma, J.X. Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am. J. Pathol. 2009, 175, 2676–2685. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ampofo, E.; Nalbach, L.; Menger, M.D.; Montenarh, M.; Götz, C. Protein Kinase CK2—A Putative Target for the Therapy of Diabetes Mellitus? Int. J. Mol. Sci. 2019, 20, 4398. https://doi.org/10.3390/ijms20184398
Ampofo E, Nalbach L, Menger MD, Montenarh M, Götz C. Protein Kinase CK2—A Putative Target for the Therapy of Diabetes Mellitus? International Journal of Molecular Sciences. 2019; 20(18):4398. https://doi.org/10.3390/ijms20184398
Chicago/Turabian StyleAmpofo, Emmanuel, Lisa Nalbach, Michael D. Menger, Mathias Montenarh, and Claudia Götz. 2019. "Protein Kinase CK2—A Putative Target for the Therapy of Diabetes Mellitus?" International Journal of Molecular Sciences 20, no. 18: 4398. https://doi.org/10.3390/ijms20184398
APA StyleAmpofo, E., Nalbach, L., Menger, M. D., Montenarh, M., & Götz, C. (2019). Protein Kinase CK2—A Putative Target for the Therapy of Diabetes Mellitus? International Journal of Molecular Sciences, 20(18), 4398. https://doi.org/10.3390/ijms20184398