Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy


Abstract
1. Introduction
2. Genetic Basis
3. Clinical Manifestation
4. Pathophysiological Mechanisms of Dystrophic Cardiomyopathy
4.1. Membrane Instability
4.2. Calcium Dysregulation
4.3. Mitochondrial Energetics
4.4. Reactive Oxygen Species Dysregulation
4.5. Nitric Oxide Dysregulation
4.6. Fibrosis
5. Small Molecule Therapies for the Heart
5.1. Angiotensin-Inhibiting Therapies
5.2. Beta-Adrenergic Receptor Blockers
5.3. Mineralocorticoid Receptor Antagonists
5.4. Corticosteroids
6. Gene-Targeted Therapies
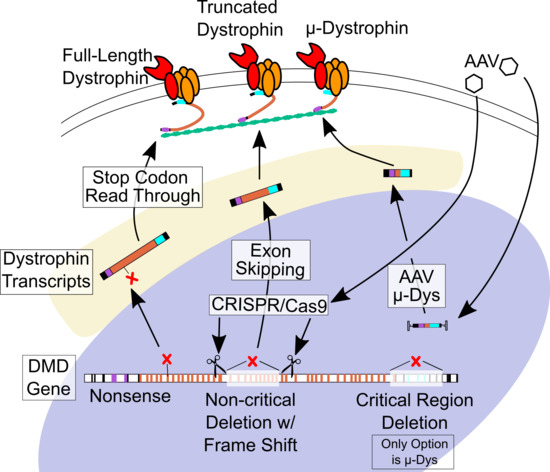
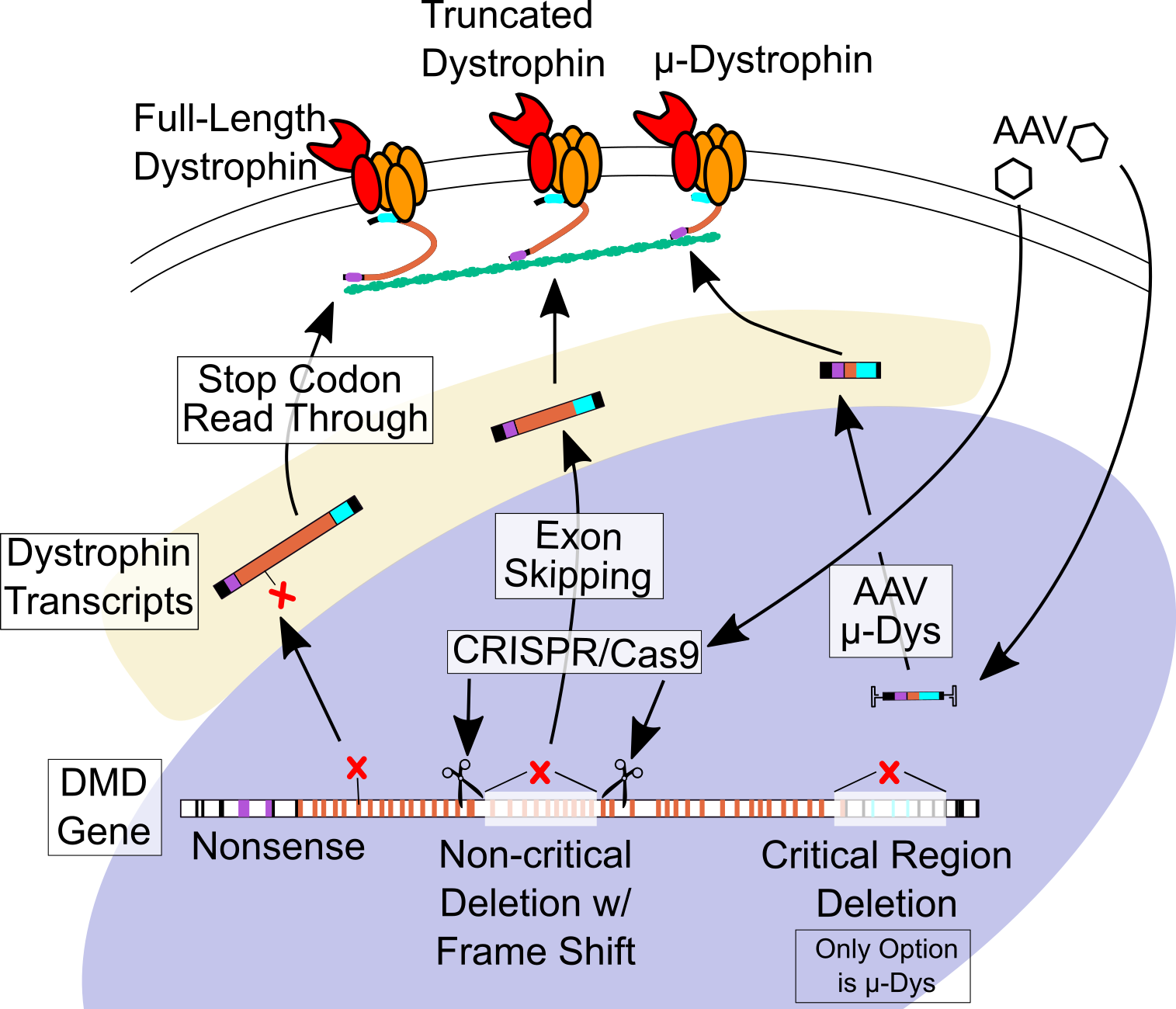
6.1. Stop Codon Readthrough
6.2. Antisense-Mediated Exon Skipping
6.3. Micro-dystrophin Viral Gene Therapy
6.4. CRISPR-Cas9 Gene Editing
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| DMD | Duchenne muscular dystrophy |
| BMD | Becker muscular dystrophy |
| DGC | Dystrophin glycoprotein complex |
| ACEI | Angiotensin converting enzyme inhibitor |
| ARB | Angiotensin receptor blocker |
| RNA | Ribonucleic acid |
| CK | Creatine kinase |
| CMR | Cardiac magnetic resonance |
| LGE | Late gadolinium enhancement |
| MRI | Magnetic resonance imaging |
| cTn | Cardiac troponin |
| ECM | Extracellular matrix |
| IgG | Immunoglobulin |
| LGMD2B | Limb girdle muscular dystrophy 2B |
| LTCC | L-type calcium channel |
| TRP | Transient receptor potential |
| RyR | Ryanodine receptor |
| SR | Sarcoplasmic reticulum |
| SERCA | Sarcoplasmic/endoplasmic reticulum calcium ATP-ase |
| mPTP | Mitochondrial permeability transition pore |
| PCr | Phosphocreatine |
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| ROS | Reactive oxygen species |
| NOX | NADPH oxidase |
| AT1R | Angiotensin II type 1 receptor |
| NOS | Nitric oxide synthase |
| AngII | Angiotensin II |
| AT2R | Angiotensin II type 2 receptor |
| MR | Mineralocorticoid receptor |
| EF | Ejection fraction |
| XLCM | X-linked dilated cardiomyopathy |
| AON | Antisense oligonucleotide |
| PMO | Phosphorodiamidate morpholino oligomer |
| PPMO | Peptide-conjugated PMO |
| AAV | Adeno-associated virus |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
References
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Rafael-Fortney, J.A.; Chadwick, J.A.; Raman, S.V. Duchenne muscular dystrophy mice and men: Can understanding a genetic cardiomyopathy inform treatment of other myocardial diseases? Circ. Res. 2016, 118, 1059–1061. [Google Scholar] [CrossRef]
- 3 Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J.I. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Spurney, C.F.; Shimizu, R.; Morgenroth, L.P.; Kolski, H.; Gordish-Dressman, H.; Clemens, P.R.; Cregan, M.; Goude, E.; Glick, M.; Johnson, L.; et al. CINRG Duchenne Natural History Study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophy. Muscle Nerve 2014, 50, 250–256. [Google Scholar] [CrossRef]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Hoffman, E.P.; Judge, D.P.; Kertesz, N.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.H.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Flanigan, K.M. Duchenne and Becker Muscular Dystrophies. In Swaiman’s Pediatric Neurology, 6th ed.; Elsevier Inc.: Edinburgh, UK, 2017; pp. e2482–e2492. [Google Scholar]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; Hamil, C.; et al. Evidence-Based Path to Newborn Screening for Duchenne Muscular Dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.-J.B.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genomics Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef]
- Nicolas, A.; Raguènès-Nicol, C.; Ben Yaou, R.; Hir, S.A.-L.; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.F.; et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2015, 24, 1267–1279. [Google Scholar] [CrossRef]
- Tyler, K.L. Origins and early descriptions of “Duchenne muscular dystrophy”. Muscle Nerve 2003, 28, 402–422. [Google Scholar] [CrossRef]
- Torriani, M.; Townsend, E.; Thomas, B.J.; Bredella, M.A.; Ghomi, R.H.; Tseng, B.S. Lower leg muscle involvement in Duchenne muscular dystrophy: An MR imaging and spectroscopy study. Skeletal Radiol. 2012, 41, 437–445. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.H.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef]
- Banihani, R.; Smile, S.; Yoon, G.; Dupuis, A.; Mosleh, M.; Snider, A.; McAdam, L. Cognitive and neurobehavioral profile in boys with duchenne muscular dystrophy. J. Child Neurol. 2015, 30, 1472–1482. [Google Scholar] [CrossRef]
- Tandon, A.; Villa, C.R.; Hor, K.N.; Jefferies, J.L.; Gao, Z.; Towbin, J.A.; Wong, B.L.; Mazur, W.; Fleck, R.J.; Sticka, J.J.; et al. Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in Duchenne muscular dystrophy. J. Am. Heart Assoc. 2015, 4, 1–8. [Google Scholar] [CrossRef]
- Hor, K.N.; Wansapura, J.; Markham, L.W.; Mazur, W.; Cripe, L.H.; Fleck, R.; Benson, D.W.; Gottliebson, W.M. Circumferential Strain Analysis Identifies Strata of Cardiomyopathy in Duchenne Muscular Dystrophy. A Cardiac Magnetic Resonance Tagging Study. J. Am. Coll. Cardiol. 2009, 53, 1204–1210. [Google Scholar] [CrossRef]
- Hor, K.N.; Mah, M.L.; Johnston, P.; Cripe, T.P.; Cripe, L.H. Advances in the diagnosis and management of cardiomyopathy in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 711–716. [Google Scholar] [CrossRef]
- Matsumura, T.; Saito, T.; Fujimura, H.; Shinno, S. Cardiac troponin I for accurate evaluation of cardiac status in myopathic patients. Brain Dev. 2007, 29, 496–501. [Google Scholar] [CrossRef]
- Ergul, Y.; Ekici, B.; Nisli, K.; Tatli, B.; Binboga, F.; Acar, G.; Ozmen, M.; Omeroglu, R.E. Evaluation of the North Star Ambulatory Assessment scale and cardiac abnormalities in ambulant boys with Duchenne muscular dystrophy. J. Paediatr. Child Health 2012, 48, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Ramaciotti, C.; Iannaccone, S.T.; Scott, W.A. Myocardial cell damage in Duchenne muscular dystrophy. Pediatr. Cardiol. 2003, 24, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, D.; Khan, S.; Khera, R.; Bhatt, A.; Garg, S.; Grodin, J.L.; Morlend, R.; Araj, F.G.; Amin, A.A.; Thibodeau, J.T.; et al. Predictors of death in adults with Duchenne muscular dystrophy-associated cardiomyopathy. J. Am. Heart Assoc. 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Eagle, M.; Bourke, J.; Bullock, R.; Gibson, M.; Mehta, J.; Giddings, D.; Straub, V.; Bushby, K. Managing Duchenne muscular dystrophy—The additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul. Disord. 2007, 17, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000, 150, 1209–1214. [Google Scholar] [CrossRef]
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, K.M.; Premsukh, M.D.; Townsend, D. Alterations of dystrophin-associated glycoproteins in the heart lacking dystrophin or dystrophin and utrophin. J. Muscle Res. Cell Motil. 2013, 34, 395–405. [Google Scholar] [CrossRef]
- Townsend, D.; Blankinship, M.J.; Allen, J.M.; Gregorevic, P.; Chamberlain, J.S.; Metzger, J.M. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol. Ther. 2007, 15, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Hor, K.N.; Johnston, P.; Kinnett, K.; Mah, M.L.; Stiver, C.; Markham, L.W.; Cripe, L.H. Progression of Duchenne Cardiomyopathy Presenting with Chest Pain and Troponin Elevation. J. Neuromuscul. Dis. 2017, 4, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Meyers, T.A.; Heitzman, J.A.; Krebsbach, A.; Aufdembrink, L.M.; Hughes, R.; Bartolomucci, A.; Townsend, D. Acute AT1R blockade prevents isoproterenol-induced injury in mdx hearts. J. Mol. Cell. Cardiol. 2019, 128, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Skimming, J.W.; Liu, M.; Strawn, T.; Duan, D. Full-length dystrophin expression in half of the heart cells ameliorates β-isoproterenol-induced cardiomyopathy in mdx mice. Hum. Mol. Genet. 2004, 13, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Olthoff, J.T.; Lindsay, A.; Abo-Zahrah, R.; Baltgalvis, K.A.; Patrinostro, X.; Belanto, J.J.; Yu, D.-Y.; Perrin, B.J.; Garry, D.J.; Rodney, G.G.; et al. Loss of peroxiredoxin-2 exacerbates eccentric contraction-induced force loss in dystrophin-deficient muscle. Nat. Commun. 2018, 9, 5104. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Bansal, D.; Miyake, K.; Muniz, V.P.; Weiss, R.M.; McNeil, P.L.; Campbell, K.P. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Invest. 2007, 117, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, B.; Wang, Y.; Guo, A.; Tang, Y.; Khataei, T.; Shi, Y.; Kutschke, W.J.; Zimmerman, K.; Weiss, R.M.; et al. MG53 is dispensable for T-tubule maturation but critical for maintaining T-tubule integrity following cardiac stress. J. Mol. Cell. Cardiol. 2017, 112, 123–130. [Google Scholar] [CrossRef]
- Vanhoutte, D.; Schips, T.G.; Kwong, J.Q.; Davis, J.; Tjondrokoesoemo, A.; Brody, M.J.; Sargent, M.A.; Kanisicak, O.; Yi, H.; Gao, Q.Q.; et al. Thrombospondin expression in myofibers stabilizes muscle membranes. Elife 2016, 5, 1–33. [Google Scholar] [CrossRef]
- Brody, M.J.; Vanhoutte, D.; Schips, T.G.; Boyer, J.G.; Bakshi, C.V.; Sargent, M.A.; York, A.J.; Molkentin, J.D. Defective Flux of Thrombospondin-4 through the Secretory Pathway Impairs Cardiomyocyte Membrane Stability and Causes Cardiomyopathy. Mol. Cell. Biol. 2018, 38, 1–14. [Google Scholar] [CrossRef]
- Harris, E.; Bladen, C.L.; Mayhew, A.; James, M.; Bettinson, K.; Moore, U.; Smith, F.E.; Rufibach, L.; Cnaan, A.; Goebel, D.X.B.; et al. The clinical outcome study for dysferlinopathy: An international multicenter study. Neurol. Genet. 2016, 2, e89. [Google Scholar] [CrossRef]
- He, B.; Tang, R.H.; Weisleder, N.; Xiao, B.; Yuan, Z.; Cai, C.; Zhu, H.; Lin, P.; Qiao, C.; Li, J.; et al. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure inδ-sarcoglycan-deficient hamsters. Mol. Ther. 2012, 20, 727–735. [Google Scholar] [CrossRef]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra85. [Google Scholar] [CrossRef]
- Houang, E.M.; Bates, F.S.; Sham, Y.Y.; Metzger, J.M. All-Atom Molecular Dynamics-Based Analysis of Membrane-Stabilizing Copolymer Interactions with Lipid Bilayers Probed under Constant Surface Tensions. J. Phys. Chem. B 2017, 121, 10657–10664. [Google Scholar] [CrossRef] [PubMed]
- Houang, E.M.; Sham, Y.Y.; Bates, F.S.; Metzger, J.M. Muscle membrane integrity in Duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle 2018, 8, 1–19. [Google Scholar]
- Townsend, D.; Turner, I.; Yasuda, S.; Martindale, J.; Davis, J.; Shillingford, M.; Kornegay, J.N.; Metzger, J.M. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J. Clin. Invest. 2010, 120, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Houang, E.M.; Haman, K.J.; Filareto, A.; Perlingeiro, R.C.; Bates, F.S.; Lowe, D.A.; Metzger, J.M. Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Mol. Ther. Methods Clin. Dev. 2015, 2, 15042. [Google Scholar] [CrossRef]
- Townsend, D.; Yasuda, S.; Metzger, J. Cardiomyopathy of Duchenne muscular dystrophy: Pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert Rev. Cardiovasc. Ther. 2007, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; López de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef]
- Williams, I.A.; Allen, D.G. Intracellular calcium handling in ventricular myocytes from mdx mice. Am. J. Physiol. Circ. Physiol. 2007, 292, H846–H855. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, V.P.A.; Viola, H.M.; Hool, L.C. Dystrophic cardiomyopathy—Potential role of calcium in pathogenesis, treatment and novel therapies. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P. Duchenne muscular dystrophy—What causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 2010, 43, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Lorin, C.; Vögeli, I.; Niggli, E. Dystrophic cardiomyopathy: Role of TRPV2 channels in stretch-induced cell damage. Cardiovasc. Res. 2015, 106, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Yeung, E.W. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: Role of ionic changes. J. Physiol. 2005, 567, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Rubi, L.; Obermair, G.J.; Cervenka, R.; Dang, X.B.; Lukacs, P.; Kummer, S.; Bittner, R.E.; Kubista, H.; Todt, H.; et al. Enhanced currents through L-type calcium channels in cardiomyocytes disturb the electrophysiology of the dystrophic heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H564–H573. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Davies, S.M.K.; Filipovska, A.; Hool, L.C. L-type Ca2+ channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am. J. Physiol. Circ. Physiol. 2013, 304, H767–H775. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.; Doyle, A.D.; Johnson, B.D. Regulation of the cardiac L-type Ca2+ channel by the actin-binding proteins alpha-actinin and dystrophin. Am. J. Physiol. Cell Physiol. 2002, 282, C1502-11. [Google Scholar] [CrossRef]
- Andersson, D.C.; Marks, A.R. Fixing ryanodine receptor Ca2+ leak—A novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov. Today Dis. Mech. 2010, 7, e151–e157. [Google Scholar] [CrossRef] [PubMed]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Sarma, S.; Li, N.; van Oort, R.J.; Reynolds, C.; Skapura, D.G.; Wehrens, X.H.T. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 13165–13170. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, W.; Wang, G.; Rodney, G.G.; Wehrens, X.H.T. Crosstalk between RyR2 oxidation and phosphorylation contributes to cardiac dysfunction in mice with Duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 2015, 89, 177–184. [Google Scholar] [CrossRef]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Rohman, M.S.; Emoto, N.; Takeshima, Y.; Yokoyama, M.; Matsuo, M. Decreased mAKAP, ryanodine receptor, and SERCA2a gene expression in mdx hearts. Biochem. Biophys. Res. Commun. 2003, 310, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Zatz, M.; Starling, A. Calpains and Disease. N. Engl. J. Med. 2005, 352, 2413–2423. [Google Scholar] [CrossRef] [PubMed]
- Letavernier, E.; Zafrani, L.; Perez, J.; Letavernier, B.; Haymann, J.P.; Baud, L. The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res. 2012, 96, 38–45. [Google Scholar] [CrossRef]
- Freitas, A.C.S.; Figueiredo, M.J.; Campos, E.C.; Soave, D.F.; Ramos, S.G.; Tanowitz, H.B.; Celes, M.R.N. Activation of both the calpain and ubiquitin-proteasome systems contributes to septic cardiomyopathy through dystrophin loss/disruption and mTOR inhibition. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–7293. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1395–1401. [Google Scholar] [CrossRef]
- Kyrychenko, V.; Poláková, E.; Janíček, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef]
- Ingwall, J.S. ATP and the Heart, 1st ed.; Springer: Boston, MA, USA, 2002; ISBN 978-1-4613-5391-1. [Google Scholar]
- Cui, W.; Jang, A.; Zhang, P.; Thompson, B.; Townsend, D.; Metzger, J.M.; Zhang, J. Early detection of myocardial bioenergetic deficits: A 9.4 tesla complete non invasive 31P MR spectroscopy study in mice with muscular dystrophy. PLoS ONE 2015, 10, e0135000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; ten Hove, M.; Schneider, J.E.; Stuckey, D.J.; Sebag-Montefiore, L.; Bia, B.L.; Radda, G.K.; Davies, K.E.; Neubauer, S.; Clarke, K. Abnormal cardiac morphology, function and energy metabolism in the dystrophic mdx mouse: An MRI and MRS study. J. Mol. Cell. Cardiol. 2008, 45, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Stelter, Z.; Strakova, J.; Yellamilli, A.; Fischer, K.; Sharpe, K.; Townsend, D. Hypoxia-induced cardiac injury in dystrophic mice. Am. J. Physiol. Hear. Circ. Physiol. 2016, 310, H938–H948. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kang, C.; Badr, M.A.; Kyrychenko, V.; Eskelinen, E.L.; Shirokova, N. Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc. Res. 2018, 58, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Crilley, J.G.; Boehm, E.A.; Rajagopalan, B.; Blamire, A.M.; Styles, P.; Muntoni, F.; Hilton-Jones, D.; Clarke, K. Magnetic resonance spectroscopy evidence of abnormal cardiac energetics in Xp21 muscular dystrophy. J. Am. Coll. Cardiol. 2000, 36, 1953–1958. [Google Scholar] [CrossRef]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Des Rosiers, C.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Saotome, M.; Katoh, H.; Yaguchi, Y.; Tanaka, T.; Urushida, T.; Satoh, H.; Hayashi, H. Transient opening of mitochondrial permeability transition pore by reactive oxygen species protects myocardium from ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2009, 296, H1125–H1132. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 76, 725–734. [Google Scholar] [CrossRef]
- Hausenloy, D.; Wynne, A.; Duchen, M.; Yellon, D. Transient Mitochondrial Permeability Transition Pore Opening Mediates Preconditioning-Induced Protection. Circulation 2004, 109, 1714–1717. [Google Scholar] [CrossRef]
- Kanai, A.J.; Pearce, L.L.; Clemens, P.R.; Birder, L.A.; VanBibber, M.M.; Choi, S.-Y.; de Groat, W.C.; Peterson, J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. USA 2002, 98, 14126–14131. [Google Scholar] [CrossRef]
- Strakova, J.; Dean, J.D.; Sharpe, K.M.; Meyers, T.A.; Odom, G.L.; Townsend, D. Dystrobrevin increases dystrophin’s binding to the dystrophin-glycoprotein complex and provides protection during cardiac stress. J. Mol. Cell. Cardiol. 2014, 76, 106–115. [Google Scholar] [CrossRef][Green Version]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.R.; Treuer, A.V.; Lamirault, G.; Mayo, V.; Cao, Y.; Dulce, R.A.; Hare, J.M. NADPH oxidase-2 inhibition restores contractility and intracellular calcium handling and reduces arrhythmogenicity in dystrophic cardiomyopathy. Am. J. Physiol. Hear. Circ. Physiol. 2014, 307, H710–H721. [Google Scholar] [CrossRef] [PubMed]
- Loehr, J.A.; Wang, S.; Cully, T.R.; Pal, R.; Larina, I.V.; Larin, K.V.; Rodney, G.G. NADPH oxidase mediates microtubule alterations and diaphragm dysfunction in dystrophic mice. Elife 2018, 7, 1–19. [Google Scholar] [CrossRef]
- Belanto, J.J.; Olthoff, J.T.; Mader, T.L.; Chamberlain, C.M.; Nelson, D.M.; McCourt, P.M.; Talsness, D.M.; Gundersen, G.G.; Lowe, D.A.; Ervasti, J.M. Independent variability of microtubule perturbations associated with dystrophinopathy. Hum. Mol. Genet. 2016, 25, 4951–4961. [Google Scholar] [CrossRef]
- Matecki, S.; Fauconnier, J.; Lacampagne, A. Reactive Oxygen Species and Muscular Dystrophy. Syst. Biol. Free Radicals Antioxid. 2014, 3055–3072. [Google Scholar]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mechanisms and Relevance for Cardiovascular Disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2015, 96, 253–305. [Google Scholar] [CrossRef]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 2013, 58, 172–181. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T.; Keaney, J.F.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef]
- Choi, H.; Leto, T.L.; Hunyady, L.; Catt, K.J.; Yun, S.B.; Sue, G.R. Mechanism of angiotensin II-induced superoxide production in cells reconstituted with angiotensin type 1 receptor and the components of NADPH oxidase. J. Biol. Chem. 2008, 283, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F.; Sali, A.; Guerron, A.D.; Iantorno, M.; Yu, Q.; Gordish-Dressman, H.; Rayavarapu, S.; Van Der Meulen, J.; Hoffman, E.P.; Nagaraju, K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-M.; Kim, D.-Y.; Kim, A.-Y.; Lee, E.-J.; Kim, S.-H.; Lee, M.-M.; Sung, S.-E.; Park, J.-K.; Jeong, K.-S. Chronic effects of losartan on the muscles and the serologic profiles of mdx mice. Life Sci. 2015, 143, 35–42. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Martin, E.A.; Barresi, R.; Byrne, B.J.; Tsimerinov, E.I.; Scott, B.L.; Walker, A.E.; Gurudevan, S.V.; Anene, F.; Elashoff, R.M.; Thomas, G.D.; et al. Tadalafil alleviates muscle ischemia in patients with becker muscular dystrophy. Sci. Transl. Med. 2012, 4, 162ra155. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Sander, M.; Lau, K.S.; Huang, P.L.; Stull, J.T.; Victor, R.G. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 15090–15095. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Zhang, L.; Adams, M.E.; Phillips, A.; Freitas, M.A.; Froehner, S.C.; Green-Church, K.B.; Montanaro, F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE 2012, 7, e43515. [Google Scholar] [CrossRef]
- Bia, B.L.; Cassidy, P.J.; Young, M.E.; Rafael-Fortney, J.A.; Leighton, B.; Davies, K.E.; Radda, G.K.; Clarke, K. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 1999, 31, 1857–1862. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, J.; Schneider, J.S.; Crassous, P.-A.; Zheng, R.; Gonzalez, J.P.; Xie, L.-H.; Beuve, A.; Fraidenraich, D.; Peluffo, R.D. Nitric oxide signalling pathway in Duchenne muscular dystrophy mice: Up-regulation of L-arginine transporters. Biochem. J. 2012, 449, 133–142. [Google Scholar] [CrossRef]
- Garbincius, J.F.; Michele, D.E. Dystrophin–glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 13663–13668. [Google Scholar] [CrossRef]
- Balligand, J.L.; Ungureanu-Longrois, D.; Simmons, W.W.; Pimental, D.; Malinski, T.A.; Kapturczak, M.; Taha, Z.; Lowenstein, C.J.; Davidoff, A.J.; Kelly, R.A.; et al. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J. Biol. Chem. 1994, 269, 27580–27588. [Google Scholar] [PubMed]
- Altamirano, F.; López, J.R.; Henríquez, C.; Molinski, T.; Allen, P.D.; Jaimovich, E. Increased resting intracellular calcium modulates NF-κB-dependent inducible nitric-oxide synthase gene expression in dystrophic mdx skeletal myotubes. J. Biol. Chem. 2012, 287, 20876–20887. [Google Scholar] [CrossRef]
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.-L. Nitric oxide and cardiac function: Ten years after, and continuing. Circ. Res. 2003, 93, 388–398. [Google Scholar] [CrossRef]
- Adamo, C.M.; Dai, D.-F.F.; Percival, J.M.; Minami, E.; Willis, M.S.; Patrucco, E.; Froehner, S.C.; Beavo, J.A. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 19079–19083. [Google Scholar] [CrossRef]
- Khairallah, M.; Khairallah, R.J.; Young, M.E.; Allen, B.G.; Gillis, M.A.; Danialou, G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc. Natl. Acad. Sci. USA 2008, 105, 7028–7033. [Google Scholar] [CrossRef]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.G.; Herzka, D.A.; Thompson, W.R.; He, B.; Bibat, G.; Tennekoon, G.; Russell, S.D.; Schuleri, K.H.; Lardo, A.C.; Kass, D.A.; et al. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann. Neurol. 2014, 76, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Kim, G.E.; Holewinski, R.J.; Venkatraman, V.; Zhu, G.; Bedja, D.; Kass, D.A.; Van Eyk, J.E. Transient receptor potential channel 6 regulates abnormal cardiac S-nitrosylation in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E10763–E10771. [Google Scholar] [CrossRef]
- Lai, Y.; Zhao, J.; Yue, Y.; Wasala, N.B.; Duan, D. Partial restoration of cardiac function with ΔPDZ nNOS in aged mdx model of Duchenne cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3189–3199. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Jordan, M.C.; Roos, K.P.; Deng, B.; Tidball, J.G. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet. 2005, 14, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Tidball, J.G. Neuronal nitric oxide synthase-rescue of dystrophin/utrophin double knockout mice does not require nNOS localization to the cell membrane. PLoS ONE 2011, 6, e25071. [Google Scholar] [CrossRef] [PubMed]
- Hor, K.N.; Taylor, M.D.; Al-Khalidi, H.R.; Cripe, L.H.; Raman, S.V.; Jefferies, J.L.; O’Donnell, R.; Benson, D.W.; Mazur, W. Prevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: Effect of age and left ventricular systolic function. J. Cardiovasc. Magn. Reson. 2013, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Heras-Bautista, C.O.; Mikhael, N.; Lam, J.; Shinde, V.; Katsen-Globa, A.; Dieluweit, S.; Molcanyi, M.; Uvarov, V.; Jütten, P.; Sahito, R.G.A.; et al. Cardiomyocytes facing fibrotic conditions re-express extracellular matrix transcripts. Acta Biomater. 2019, 89, 180–192. [Google Scholar] [CrossRef]
- Soslow, J.H.; Xu, M.; Slaughter, J.C.; Crum, K.; Chew, J.D.; Burnette, W.B.; Su, Y.R.; Tomasek, K.; Parra, D.A.; Markham, L.W. The Role of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Duchenne Muscular Dystrophy Cardiomyopathy. J. Card. Fail. 2019, 25, 259–267. [Google Scholar] [CrossRef]
- Smith, L.R.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68–69, 602–615. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef]
- Chadwick, J.A.; Swager, S.A.; Lowe, J.; Welc, S.S.; Tidball, J.G.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Rafael-Fortney, J.A. Myeloid cells are capable of synthesizing aldosterone to exacerbate damage in muscular dystrophy. Hum. Mol. Genet. 2016, 25, 5167–5177. [Google Scholar] [CrossRef]
- Rafael-Fortney, J.A.; Chimanji, N.S.; Schill, K.E.; Martin, C.D.; Murray, J.D.; Ganguly, R.; Stangland, J.E.; Tran, T.; Xu, Y.; Canan, B.D.; et al. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation 2011, 124, 582–588. [Google Scholar] [CrossRef]
- Rodriguez, E.K.; Hunter, W.C.; Royce, M.J.; Leppo, M.K.; Douglas, A.S.; Weisman, H.F. A method to reconstruct myocardial sarcomere lengths and orientations at transmural sites in beating canine hearts. Am. J. Physiol. Circ. Physiol. 2017, 263, H293–H306. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Papa, A.A.; Williams, E.A.; Rago, A.; Palladino, A.; Politano, L.; Nigro, G. ACE inhibition to slow progression of myocardial fibrosis in muscular dystrophies. Trends Cardiovasc. Med. 2018, 28, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Yu, Q.; Fiorillo, A.A.; Tully, C.B.; Tucker, A.; Mazala, D.A.; Uaesoontrachoon, K.; Srinivassane, S.; Damsker, J.M.; Hoffman, E.P.; et al. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci. Alliance 2019, 2, e201800186. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bible, K.L.; Regnier, M.; Adams, M.E.; Froehner, S.C.; Whitehead, N.P. Simvastatin provides long-term improvement of left ventricular function and prevents cardiac fibrosis in muscular dystrophy. Physiol. Rep. 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, C.; Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov. Today 2011, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Iwanami, J.; Mogi, M.; Iwai, M.; Horiuchi, M. Inhibition of the renin–angiotensin system and target organ protection. Hypertens. Res. 2009, 32, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Forrester, S.J.; O’Brien, S.; Baggett, A.; Rizzo, V.; Eguchi, S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017, 125, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Duboc, D.; Meune, C.; Lerebours, G.; Devaux, J.Y.; Vaksmann, G.; Bécane, H.M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2005, 45, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Duboc, D.; Meune, C.; Pierre, B.; Wahbi, K.; Eymard, B.; Toutain, A.; Berard, C.; Vaksmann, G.; Weber, S.; Bécane, H.M. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am. Heart J. 2007, 154, 596–602. [Google Scholar] [CrossRef]
- Eichhorn, E.; Domanski, M.; Krause-Steinrauf, H.; Anderson, J. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. ACC Curr. J. Rev. 2001, 344, 1659–1667. [Google Scholar] [CrossRef]
- Meyers, T.A.; Heitzman, J.A.; Townsend, D. Acute myocardial injury in mdx hearts ameliorated by ARB but not ACE inhibitor treatment. 2019; Submitted. [Google Scholar]
- Bangalore, S.; Fakheri, R.; Toklu, B.; Ogedegbe, G.; Weintraub, H.; Messerli, F.H. Angiotensin-Converting Enzyme Inhibitors or Angiotensin Receptor Blockers in Patients Without Heart Failure? Insights from 254,301 Patients from Randomized Trials. Mayo Clin. Proc. 2016, 91, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Poole-Wilson, P.A.; Segal, R.; Martinez, F.A.; Dickstein, K.; Camm, A.J.; Konstam, M.A.; Riegger, G.; Klinger, G.H.; Neaton, J.; et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: Randomised trial—The Losartan Heart Failure Survival Study ELITE II. Lancet 2000, 355, 1582–1587. [Google Scholar] [CrossRef]
- Allen, H.D.; Flanigan, K.M.; Thrush, P.T.; Viollet-Callendret, L.; Dvorchik, I.; Yin, H.; Canter, C.E.; Connolly, A.M.; Parrish, M.; McDonald, C.M.; et al. A Randomized, Double-Blind Trial of Lisinopril and Losartan for the Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy. PLoS Curr. Muscular Dystrophy 2013, 5. [Google Scholar] [CrossRef]
- Hollenberg, N.K.; Fisher, N.D.L.; Price, D.A. Pathways for angiotensin II generation in intact human tissue: Evidence from comparative pharmacological interruption of the renin system. Hypertension 1998, 32, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Miura, S.; Yahiro, E.; Saku, K. Non-ACE Pathway-induced Angiotensin II Production. Curr. Pharm. Des. 2013, 19, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of Angiotensin Peptides and Recombinant Human ACE2 in Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef]
- Zou, Y.; Akazawa, H.; Qin, Y.; Sano, M.; Takano, H.; Minamino, T.; Makita, N.; Iwanaga, K.; Zhu, W.; Kudoh, S.; et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 2004, 6, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Structure-Function Basis of Attenuated Inverse Agonism of Angiotensin II Type 1 Receptor Blockers for Active-State Angiotensin II Type 1 Receptors. Mol. Pharmacol. 2015, 88, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.S.M.; Allen, T.J. Angiotensin II type 2 receptor (AT2R) in renal and cardiovascular disease. Clin. Sci. 2016, 130, 1307–1326. [Google Scholar] [CrossRef]
- Pavo, N.; Goliasch, G.; Wurm, R.; Novak, J.; Strunk, G.; Gyöngyösi, M.; Poglitsch, M.; Säemann, M.D.; Hülsmann, M. Low- and high-renin heart failure phenotypes with clinical implications. Clin. Chem. 2018, 64, 597–608. [Google Scholar] [CrossRef]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of Sodium and Calcium Dysregulation in Tachyarrhythmias in Sudden Cardiac Death. Circ. Res. 2015, 16, 1956–1970. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The Sympathetic Nervous System in Heart Failure. Physiology, Pathophysiology, and Clinical Implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef]
- Kajimoto, H.; Ishigaki, K.; Okumura, K.; Tomimatsu, H.; Nakazawa, M.; Saito, K.; Osawa, M.; Nakanishi, T. Beta-Blocker Therapy for Cardiac Dysfunction in Patients With Muscular Dystrophy. Circ. J. 2006, 70, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Tamura, T.; Kuru, S.; Kikuchi, Y.; Kawai, M. Carvedilol can Prevent Cardiac Events in Duchenne Muscular Dystrophy. Intern. Med. 2010, 49, 1357–1363. [Google Scholar] [CrossRef]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Halnon, N.J.; Kissel, J.T.; He, X.; Tran, T.; Smart, S.; McCarthy, B.; Taylor, M.D.; et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: A randomised, double-blind, placebo-controlled trial. Lancet. Neurol. 2015, 14, 153–161. [Google Scholar] [CrossRef]
- Janssen, P.M.L.; Murray, J.D.; Schill, K.E.; Rastogi, N.; Schultz, E.J.; Tran, T.; Raman, S.V.; Rafael-Fortney, J.A. Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of duchenne muscular dystrophy. PLoS ONE 2014, 9, e88360. [Google Scholar] [CrossRef]
- Raman, S.V.; Hor, K.N.; Mazur, W.; He, X.; Kissel, J.T.; Smart, S.; McCarthy, B.; Roble, S.L.; Cripe, L.H. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: Results of a two-year open-label extension trial. Orphanet J. Rare Dis. 2017, 12, 1–5. [Google Scholar] [CrossRef]
- Griggs, R.C.; Herr, B.E.; Reha, A.; Elfring, G.; Atkinson, L.; Cwik, V.; Mccoll, E.; Tawil, R.; Pandya, S.; Mcdermott, M.P.; et al. Corticosteroids in Duchenne muscular dystrophy: Major variations in practice. Muscle Nerve 2013, 48, 27–31. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Reeves, E.; Damsker, J.; Nagaraju, K.; McCall, J.M.; Connor, E.M.; Bushby, K. Novel Approaches to Corticosteroid Treatment in Duchenne Muscular Dystrophy. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.; Straub, V.; Blain, A.; Bushby, K.; MacGowan, G.A. Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Schram, G.; Fournier, A.; Leduc, H.; Dahdah, N.; Therien, J.; Vanasse, M.; Khairy, P. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2013, 61, 948–954. [Google Scholar] [CrossRef]
- Markham, L.W.; Spicer, R.L.; Khoury, P.R.; Wong, B.L.; Mathews, K.D.; Cripe, L.H. Steroid therapy and cardiac function in duchenne muscular dystrophy. Pediatr. Cardiol. 2005, 26, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Barber, B.J.; Andrews, J.G.; Lu, Z.; West, N.A.; Meaney, F.J.; Price, E.T.; Gray, A.; Sheehan, D.W.; Pandya, S.; Yang, M.; et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J. Pediatr. 2013, 163, 1080–1084.e1. [Google Scholar] [CrossRef] [PubMed]
- Silversides, C.K.; Webb, G.D.; Harris, V.A.; Biggar, D.W. Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am. J. Cardiol. 2003, 91, 769–772. [Google Scholar] [CrossRef]
- Raman, S.V.; Cripe, L.H. Glucocorticoid Therapy for Duchenne Cardiomyopathy: A Hobson’s Choice? J. Am. Heart Assoc. 2015, 4, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F. Cardiomyopathy of Duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve 2011, 44, 8–19. [Google Scholar] [CrossRef]
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; Van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Scheffer, L.; et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013, 5, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Echigoya, Y.; Fukada, S.; Yokota, T. Current Translational Research and Murine Models For Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2016, 3, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Berko, B.A.; Swift, M. X-Linked dilated cardiomyopathy. N. Engl. J. Med. 1985, 316, 1186–1191. [Google Scholar] [CrossRef]
- Ferlini, A.; Sewry, C.; Melis, M.A.; Mateddu, A.; Muntoni, F. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromuscul. Disord. 1999, 9, 339–346. [Google Scholar] [CrossRef]
- Nakamura, A. X-linked dilated cardiomyopathy: A cardiospecific phenotype of dystrophinopathy. Pharmaceuticals 2015, 8, 303–320. [Google Scholar] [CrossRef]
- Kaspar, R.W.; Allen, H.D.; Ray, W.C.; Alvarez, C.E.; Kissel, J.T.; Pestronk, A.; Weiss, R.B.; Flanigan, K.M.; Mendell, J.R.; Montanaro, F. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ. Cardiovasc. Genet. 2009, 2, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Namgoong, J.H.; Bertoni, C. Clinical potential of ataluren in the treatment of Duchenne muscular dystrophy. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 37–48. [Google Scholar]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Dillmann, U.; Flotats-Bastardas, M.; Poryo, M.; Abdul-Khaliq, H.; Shamdeen, M.G.; Mischo, B.; Zemlin, M.; Meyer, S. Off-Label Use of Ataluren in Four Non-ambulatory Patients With Nonsense Mutation Duchenne Muscular Dystrophy: Effects on Cardiac and Pulmonary Function and Muscle Strength. Front. Pediatr. 2018, 6, 316. [Google Scholar] [CrossRef]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.; Yokota, T. Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy. Am. J. Transl. Res. 2019, 11, 1202–1218. [Google Scholar]
- Wu, B.; Lu, P.; Benrashid, E.; Malik, S.; Ashar, J.; Doran, T.J.; Lu, Q.L. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010, 17, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.Q.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 4213–4218. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Echevarría, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Wyatt, E.J. Mutation-based therapy for duchenne muscular dystrophy: Antisense treatment arrives in the clinic. Circulation 2017, 136, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.R.; Chamberlain, J.S. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2017, 25, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef]
- Wang, B.; Li, J.; Fu, F.H.; Chen, C.; Zhu, X.; Zhou, L.; Jiang, X.; Xiao, X. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther. 2008, 15, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Salva, M.Z.; Himeda, C.L.; Tai, P.W.L.; Nishiuchi, E.; Gregorevic, P.; Allen, J.M.; Finn, E.E.; Nguyen, Q.G.; Blankinship, M.J.; Meuse, L.; et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007, 15, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Hakim, C.H.; Wasala, N.B.; Pan, X.; Kodippili, K.; Yue, Y.; Zhang, K.; Yao, G.; Haffner, B.; Duan, S.X.; Ramos, J.; et al. A Five-Repeat Micro-Dystrophin Gene Ameliorated Dystrophic Phenotype in the Severe DBA/2J-mdx Model of Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2017, 6, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing Anti–Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Chicoine, L.G.; Montgomery, C.L.; Bremer, W.G.; Shontz, K.M.; Griffin, D.A.; Heller, K.N.; Lewis, S.; Malik, V.; Grose, W.E.; Shilling, C.J.; et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol. Ther. 2014, 22, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.-L.; Bassel-Duby, R.; Olson, E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2018, 70, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Yoon, C.; Yokota, T. Applications of CRISPR/Cas9 for the treatment of Duchenne muscular dystrophy. J. Pers. Med. 2018, 8, 38. [Google Scholar] [CrossRef]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef]
- Amoasii, L.; Li, H.; Sanchez-Ortiz, E.; Caballero, D.; Harron, R.; Massey, C.; Shelton, J.; Piercy, R.; Olson, E.N. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 1–9. [Google Scholar]
- Hakim, C.H.; Wasala, N.B.; Nelson, C.E.; Wasala, L.P.; Yue, Y.; Louderman, J.A.; Lessa, T.B.; Dai, A.; Zhang, K.; Jenkins, G.J.; et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight 2018, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lau, Y.S.; Gao, Y.; Li, H.; Han, R. Life-Long AAV-Mediated CRISPR Genome Editing in Dystrophic Heart Improves Cardiomyopathy without Causing Serious Lesions in mdx Mice. Mol. Ther. 2019, 27, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Approach | Target Mutation Type | Dystrophin Product | Strengths | Challenges |
|---|---|---|---|---|
| Stop codon readthrough | Nonsense point mutations | Complete | • Well-tolerated (ataluren) | • Low efficiency in the heart • Low prevalence of amenable mutations • Frequent re-dosing |
| AON-mediated exon skipping 1 | Frameshift mutations (each AON drug targets one exon) | Lacking existing deletion and additional exon(s) | • Well-tolerated • Effective at cell level | • Poor cardiac uptake of PMO3 • Frequent re-dosing • Low number of amenable mutations for each AON drug |
| AAV micro-dystrophin 2 | Any (does not interact with endogenous gene) | Extensively truncated but functional | • High efficacy in heart • High efficacy in skeletal muscle • Lasting (multiple years) | • Potentially immunogenic • Potential for null effect with pre-existing immunity |
| CRISPR-Cas9 | Frameshift, insertion, and nonsense mutations (each sgRNA targets one exon) | Depends on editing strategy (ranging from complete to lacking deletion and additional exon(s)) | • High efficacy in heart and skeletal muscle • Versatile • Genomic correction is life-long (theoretically) | • Potentially immunogenic • Risk of off-target editing • Low number of amenable mutations for each CRISPR drug |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. https://doi.org/10.3390/ijms20174098
Meyers TA, Townsend D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2019; 20(17):4098. https://doi.org/10.3390/ijms20174098
Chicago/Turabian StyleMeyers, Tatyana A., and DeWayne Townsend. 2019. "Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 20, no. 17: 4098. https://doi.org/10.3390/ijms20174098
APA StyleMeyers, T. A., & Townsend, D. (2019). Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 20(17), 4098. https://doi.org/10.3390/ijms20174098