Lipolytic Effects of 3-Iodothyronamine (T1AM) and a Novel Thyronamine-Like Analog SG-2 through the AMPK Pathway



 , and
, and
Abstract
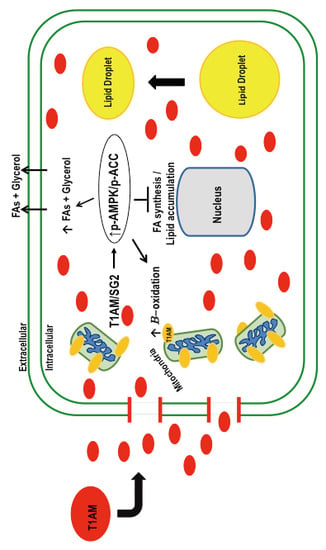
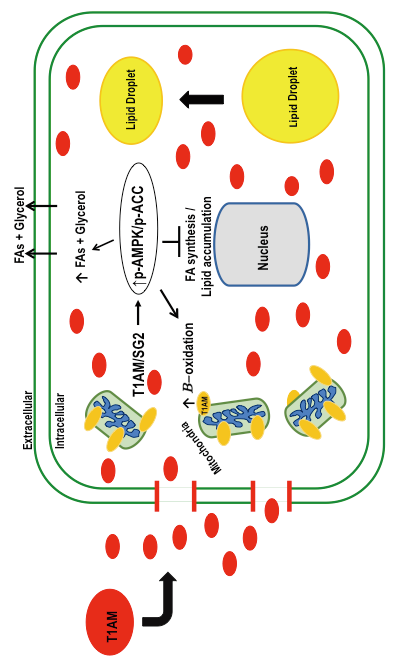{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
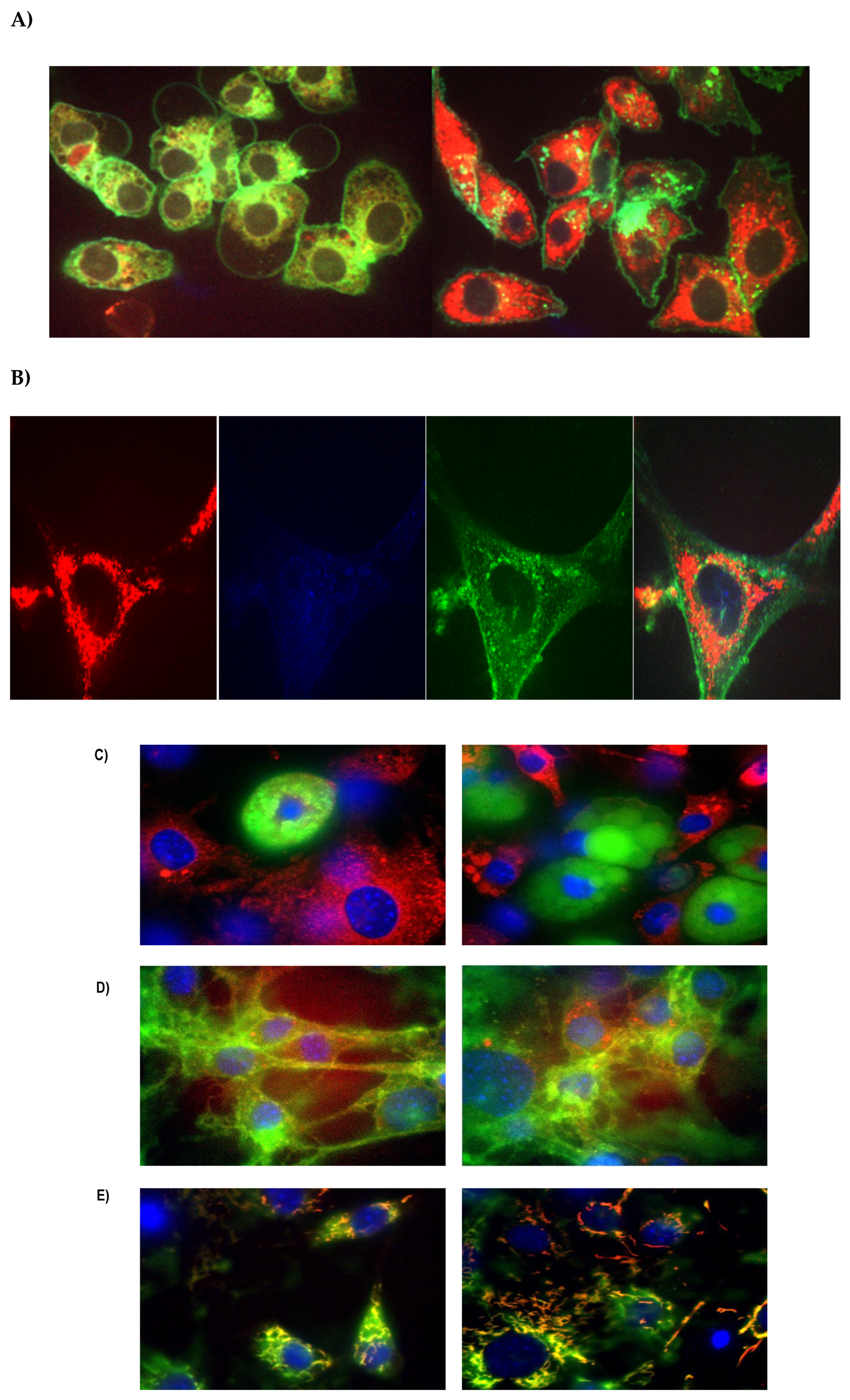
2.1. T1AM Localizes to Mitochondria of Mature 3T3-L1 Cells
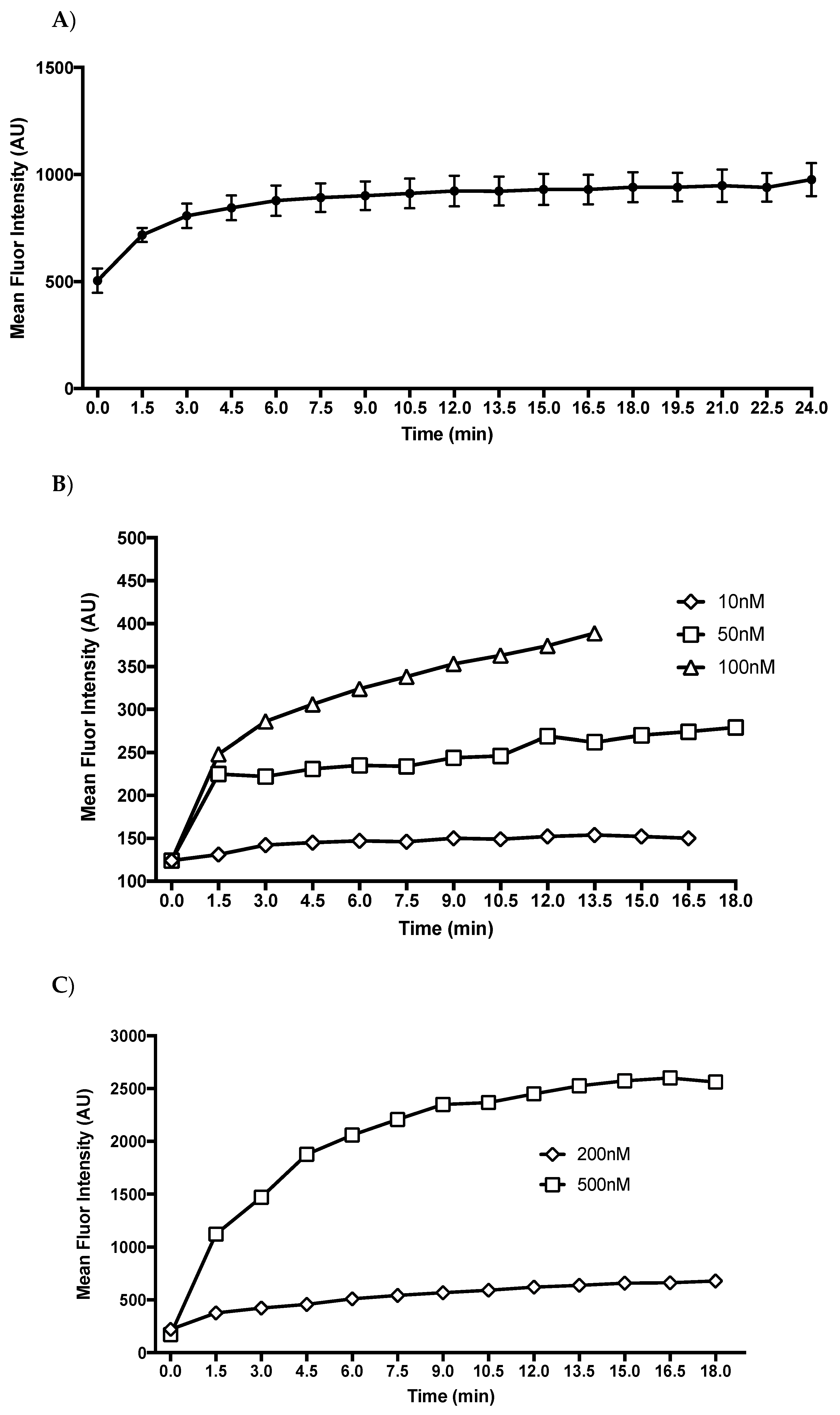
2.2. T1AM Shows a Rapid Cellular Uptake into Adipocytes
2.3. SG-2 and T1AM Show a Different Efficacy in Decreasing Cellular Lipogenesis
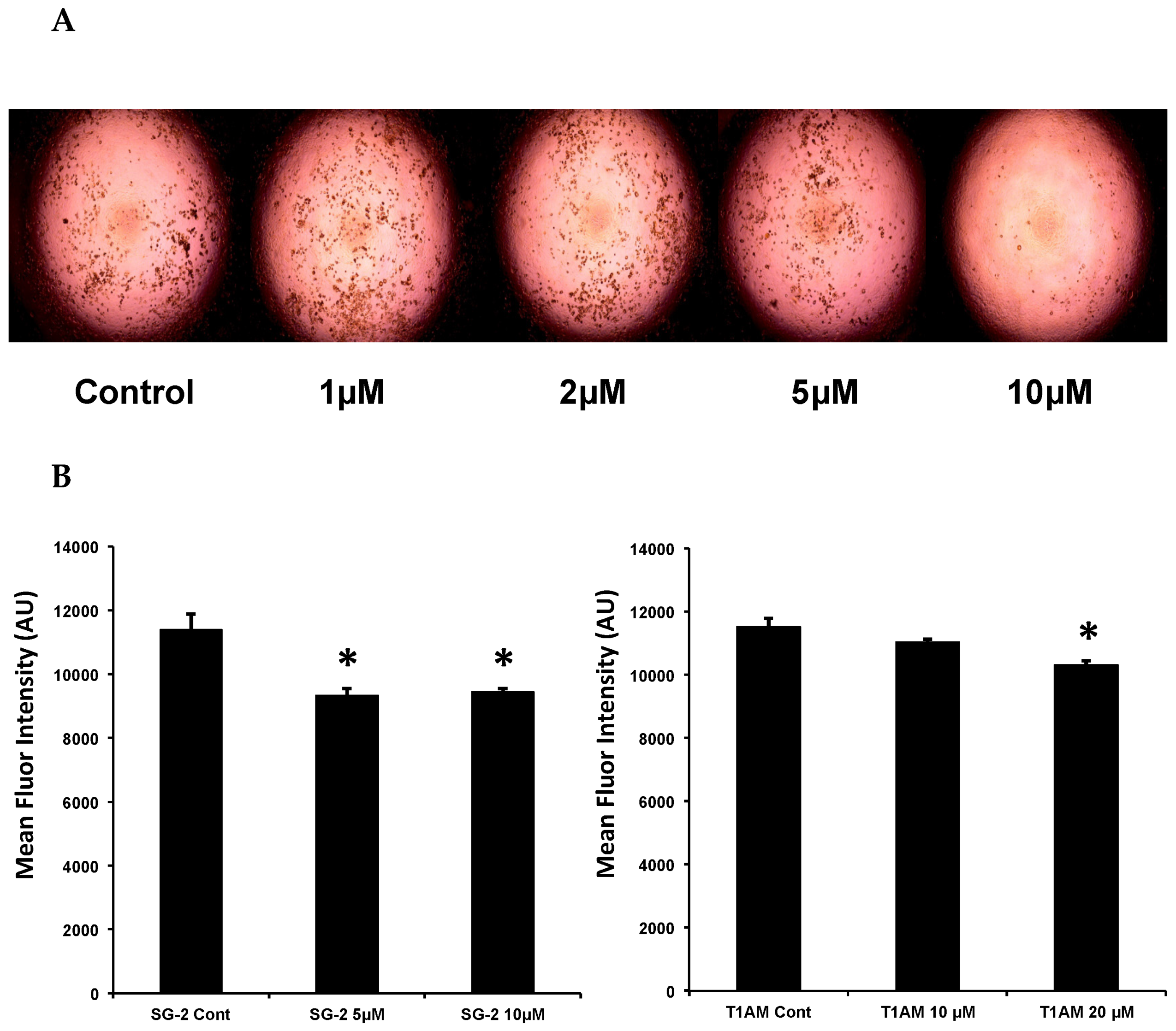
2.4. SG-2 and T1AM Show a Different Efficacy on Lipid Accumulation in Mature Adipocytes
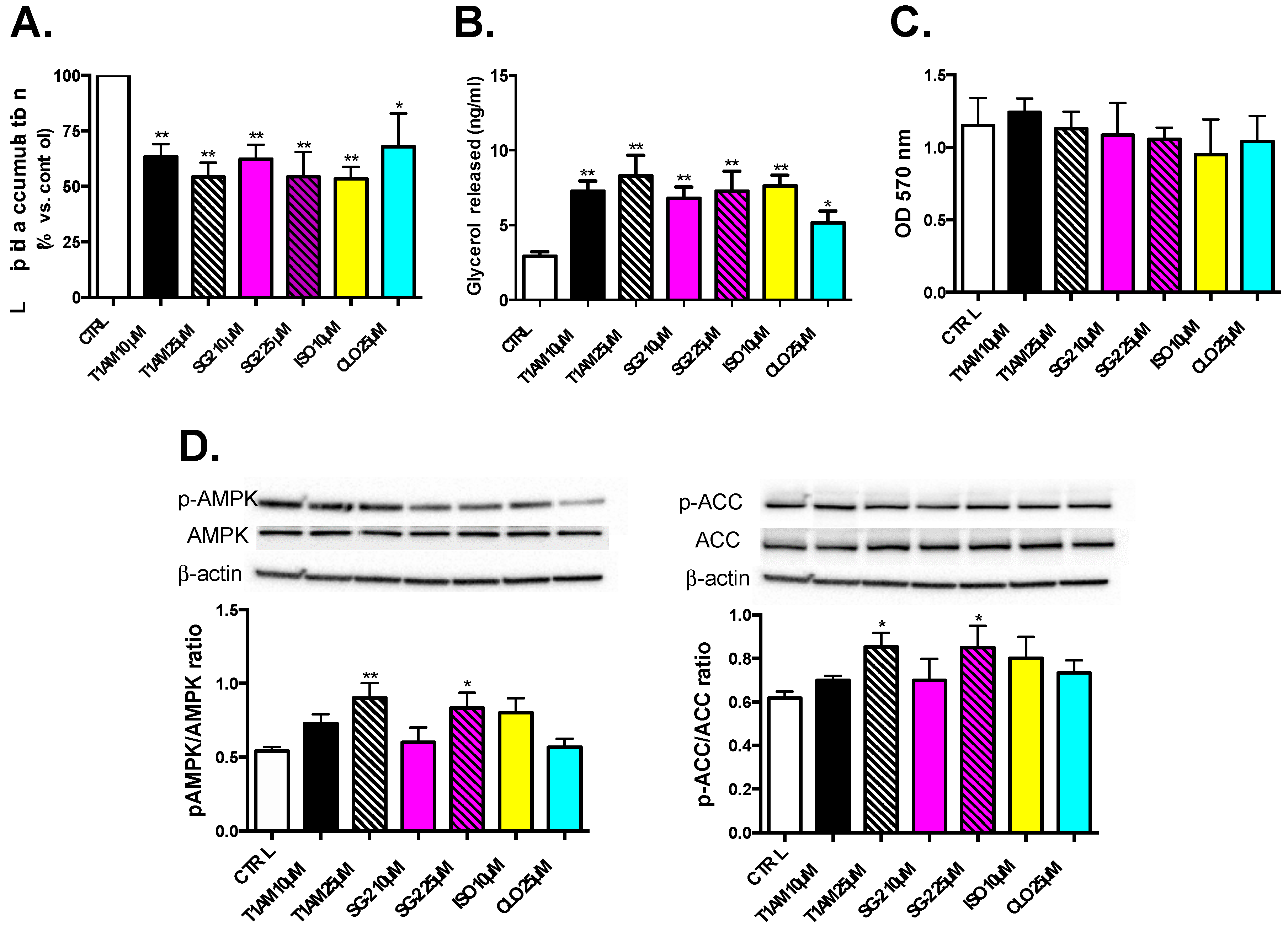
2.5. T1AM and SG-2 Induce Lipolysis in HepG2 Cells
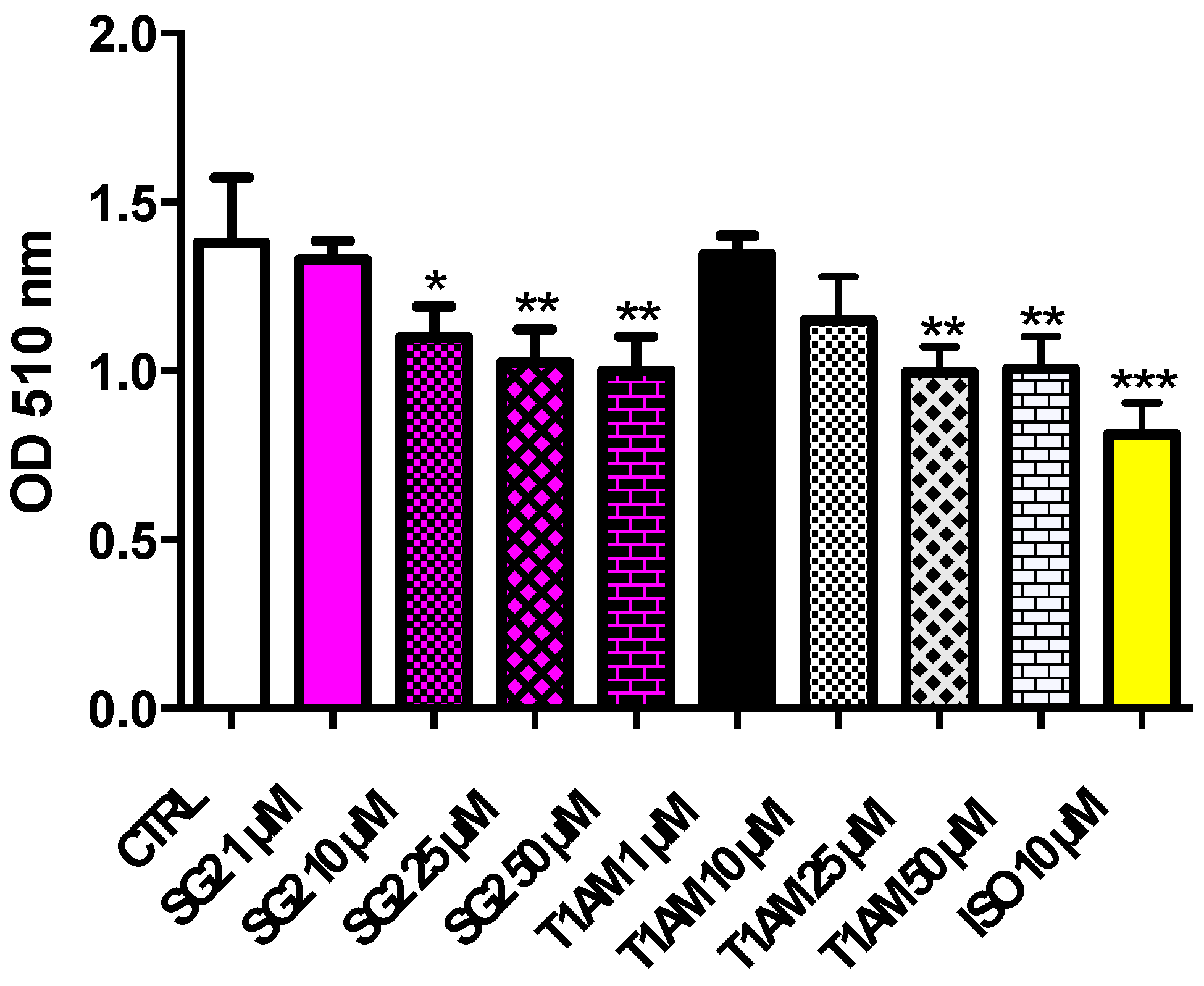
2.6. Effects of T1AM and SG-2 on HepG2 Cell Viability
2.7. Effects of T1AM and SG-2 on AMPK Activation
3. Discussion
4. Materials and Methods
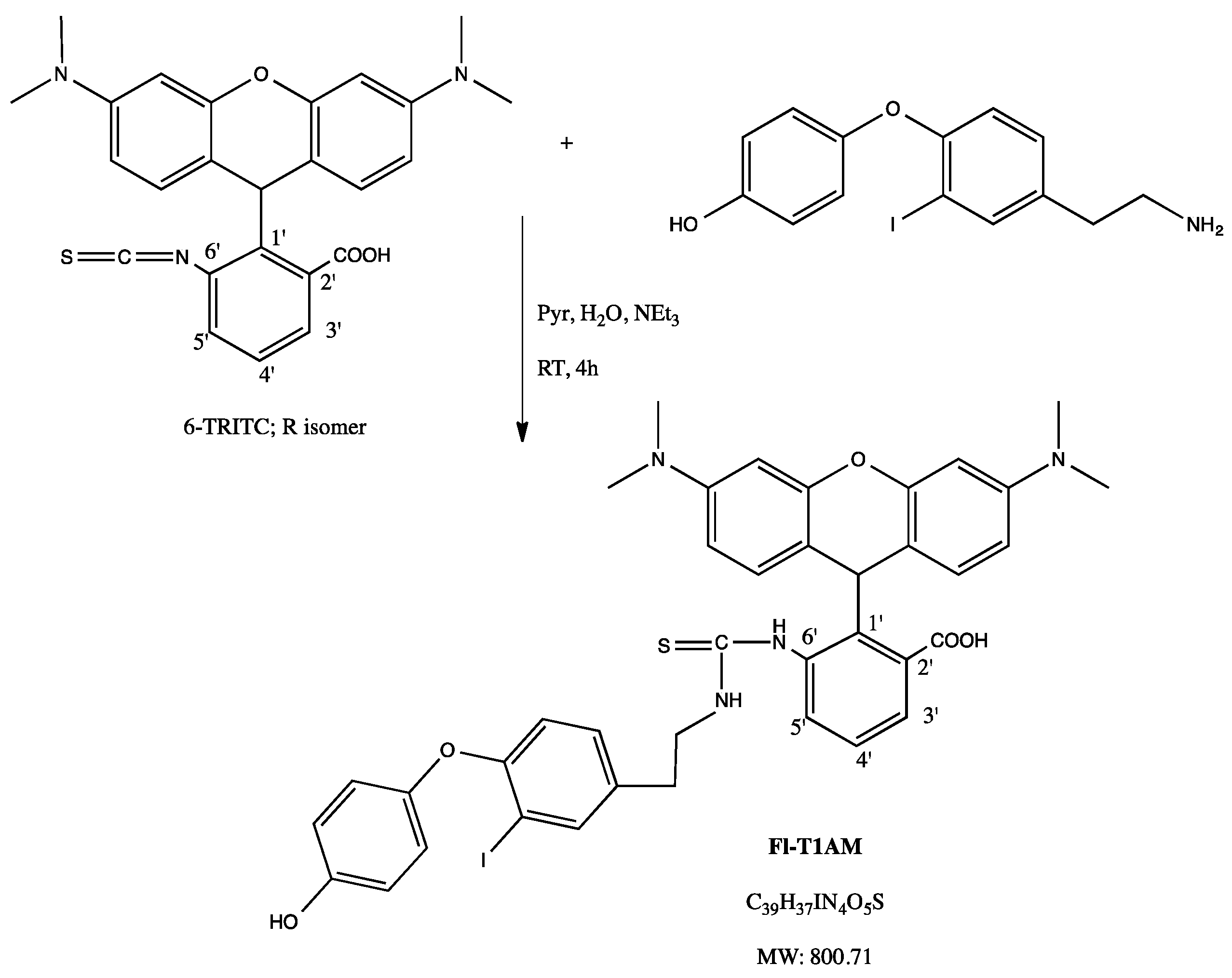
4.1. Development of Fluorescently Labeled T1AM (FL-T1AM)
4.2. Cell’s Culture and Treatment
4.2.2. Intracellular Localization of FL-T1AM
4.2.3. FL-T1AM Cellular Uptake by Flow Cytometry
4.2.4. Oil Red O (ORO) Staining of Lipid Accumulation in 3T3-L1 Adipocytes after Treatment with Test Compounds.
4.2.5. Human Hepatocellular Carcinoma (HepG2) Cell Culture and Treatment with T1AM and SG2
4.2.6. Western Blotting Analysis
4.2.7. Oil Red O (ORO) Staining of Lipid Accumulation in HepG2 Cells
4.2.8. Determination of Glycerol Release from HepG2 Cells
4.2.9. Cell Viability by MTT Assay
4.2.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scanlan, T.S.; Suchland, K.L.; Hart, M.E.; Chiellini, G.; Huang, Y.; Kruzich, P.J.; Frascarelli, S.; Crossley, D.A.; Bunzow, J.R.; Ronca-Testoni, S.; et al. 3-iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat. Med. 2004, 10, 638–642. [Google Scholar] [CrossRef]
- Hoefig, C.S.; Kohrle, J.; Brabant, G.; Dixit, K.; Yap, B.; Strasburger, C.J.; Wu, Z. Evidence for extrathyroidal formation of 3-iodothyronamine in humans as provided by a novel monoclonal antibody-based chemiluminescent serum immunoassay. J. Clin. Endocrinol. Metab. 2011, 96, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Haviland, J.A.; Reiland, H.; Butz, D.E.; Tonelli, M.; Porter, W.P.; Zucchi, R.; Scanlan, T.S.; Chiellini, G.; Assadi-Porter, F.M. Nmr-based metabolomics and breath studies show lipid and protein catabolism during low dose chronic t1 am treatment. Obesity 2013, 21, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Braulke, L.J.; Klingenspor, M.; DeBarber, A.; Tobias, S.C.; Grandy, D.K.; Scanlan, T.S.; Heldmaier, G. 3-iodothyronamine: A novel hormone controlling the balance between glucose and lipid utilisation. J. Comp. Physiol. B. 2008, 178, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Panas, H.N.; Lynch, L.J.; Vallender, E.J.; Xie, Z.; Chen, G.L.; Lynn, S.K.; Scanlan, T.S.; Miller, G.M. Normal thermoregulatory responses to 3-iodothyronamine, trace amines and amphetamine-like psychostimulants in trace amine associated receptor 1 knockout mice. J. Neurosci. Res. 2010, 88, 1962–1969. [Google Scholar] [CrossRef]
- Assadi-Porter, F.M.; Reiland, H.; Sabatini, M.; Lorenzini, L.; Carnicelli, V.; Rogowski, M.; Selen Alpergin, E.S.; Tonelli, M.; Ghelardoni, S.; Saba, A.; et al. Metabolic reprogramming by 3-iodothyronamine (t1am): A new perspective to reverse obesity through co-regulation of sirtuin 4 and 6 expression. Int. J. Mol. Sci. 2018, 19, 1535. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, G.; Erba, P.; Carnicelli, V.; Manfredi, C.; Frascarelli, S.; Ghelardoni, S.; Mariani, G.; Zucchi, R. Distribution of exogenous [125I]-3-iodothyronamine in mouse in vivo: Relationship with trace amine-associated receptors. J. Endocrinol. 2012, 213, 223–230. [Google Scholar] [CrossRef]
- Mariotti, V.; Melissari, E.; Iofrida, C.; Righi, M.; Di Russo, M.; Donzelli, R.; Saba, A.; Frascarelli, S.; Chiellini, G.; Zucchi, R.; et al. Modulation of gene expression by 3-iodothyronamine: Genetic evidence for a lipolytic pattern. PLoS ONE 2014, 9, e106923. [Google Scholar] [CrossRef]
- Frascarelli, S.; Ghelardoni, S.; Chiellini, G.; Galli, E.; Ronca, F.; Scanlan, T.S.; Zucchi, R. Cardioprotective effect of 3-iodothyronamine in perfused rat heart subjected to ischemia and reperfusion. Cardiovasc. Drugs Ther. 2011, 25, 307–313. [Google Scholar] [CrossRef]
- Roy, G.; Placzek, E.; Scanlan, T.S. Apob-100-containing lipoproteins are major carriers of 3-iodothyronamine in circulation. J. Biol. Chem. 2012, 287, 1790–1800. [Google Scholar] [CrossRef]
- Chiellini, G.; Nesi, G.; Digiacomo, M.; Malvasi, R.; Espinoza, S.; Sabatini, M.; Frascarelli, S.; Laurino, A.; Cichero, E.; Macchia, M.; et al. Design, synthesis, and evaluation of thyronamine analogues as novel potent mouse trace amine associated receptor 1 (mtaar1) agonists. J. Med. Chem. 2015, 58, 5096–5107. [Google Scholar] [CrossRef] [PubMed]
- Bellusci, L.; Laurino, A.; Sabatini, M.; Sestito, S.; Lenzi, P.; Raimondi, L.; Rapposelli, S.; Biagioni, F.; Fornai, F.; Salvetti, A.; et al. New insights into the potential roles of 3-iodothyronamine (t1am) and newly developed thyronamine-like taar1 agonists in neuroprotection. Front. Pharmacol. 2017, 8, 905. [Google Scholar] [CrossRef] [PubMed]
- Ghelardoni, S.; Chiellini, G.; Frascarelli, S.; Saba, A.; Zucchi, R. Uptake and metabolic effects of 3-iodothyronamine in hepatocytes. J. Endocrinol. 2014, 221, 101–110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rogowski, M.; Gollahon, L.; Chellini, G.; Assadi-Porter, F.M. Uptake of 3-iodothyronamine hormone analogs inhibits the growth and viability of cancer cells. FEBS Open Bio 2017, 7, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, K.Y.; Reasor, M.; Yazaki, P.J. Chloroquine-induced phospholipid fatty liver. Measurement of drug and lipid concentrations in rat liver lysosomes. J. Biol. Chem. 1985, 260, 215–219. [Google Scholar] [PubMed]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of amp-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef]
- Kemp, B.E.; Stapleton, D.; Campbell, D.J.; Chen, Z.P.; Murthy, S.; Walter, M.; Gupta, A.; Adams, J.J.; Katsis, F.; van Denderen, B.; et al. Amp-activated protein kinase, super metabolic regulator. Biochem. Soc. Trans. 2003, 31, 162–168. [Google Scholar] [CrossRef]
- Visser, T.J. Cellular uptake of thyroid hormones. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., Eds.; MDText.com, Inc.: Dartmouth, MA, USA,, 2000. [Google Scholar]
- Kruth, H. Localization of unesterified cholesterol in human atherosclerotic lesions. Demonstration of filipin-positive, oil-red-o-negative particles. Am. J. Pathol. 1984, 114, 201. [Google Scholar]
- Mitrofanova, A.; Molina, J.; Santos, J.V.; Guzman, J.; Morales, X.A.; Ducasa, G.M.; Bryn, J.; Sloan, A.; Volosenco, I.; Kim, J.J. Hydroxypropyl-β-cyclodextrin protects from kidney disease in experimental alport syndrome and focal segmental glomerulosclerosis. Kidney Int. 2018, 94, 1151–1159. [Google Scholar] [CrossRef]
- Cumero, S.; Fogolari, F.; Domenis, R.; Zucchi, R.; Mavelli, I.; Contessi, S. Mitochondrial f(0) f(1) -atp synthase is a molecular target of 3-iodothyronamine, an endogenous metabolite of thyroid hormone. Br. J. Pharm. 2012, 166, 2331–2347. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Eberhardt, N.L.; Robbins, J.; Baxter, J.D.; Pastan, I. Fluorescent rhodamine-labeled thyroid hormone derivatives: Synthesis and binding to the thyroid hormone nuclear receptor. FEBS Lett. 1979, 100, 113–116. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Maxfield, F.R.; Robbins, J.; Willingham, M.C.; Pastan, I.H. Receptor-mediated uptake of 3, 3’, 5-triiodo-l-thyronine by cultured fibroblasts. Proc. Natl. Acad. Sci. USA 1980, 77, 3425–3429. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. Nad+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogowski, M.; Bellusci, L.; Sabatini, M.; Rapposelli, S.; Rahman, S.M.; Chiellini, G.; Assadi-Porter, F.M. Lipolytic Effects of 3-Iodothyronamine (T1AM) and a Novel Thyronamine-Like Analog SG-2 through the AMPK Pathway. Int. J. Mol. Sci. 2019, 20, 4054. https://doi.org/10.3390/ijms20164054
Rogowski M, Bellusci L, Sabatini M, Rapposelli S, Rahman SM, Chiellini G, Assadi-Porter FM. Lipolytic Effects of 3-Iodothyronamine (T1AM) and a Novel Thyronamine-Like Analog SG-2 through the AMPK Pathway. International Journal of Molecular Sciences. 2019; 20(16):4054. https://doi.org/10.3390/ijms20164054
Chicago/Turabian StyleRogowski, Michael, Lorenza Bellusci, Martina Sabatini, Simona Rapposelli, Shaikh M. Rahman, Grazia Chiellini, and Fariba M. Assadi-Porter. 2019. "Lipolytic Effects of 3-Iodothyronamine (T1AM) and a Novel Thyronamine-Like Analog SG-2 through the AMPK Pathway" International Journal of Molecular Sciences 20, no. 16: 4054. https://doi.org/10.3390/ijms20164054
APA StyleRogowski, M., Bellusci, L., Sabatini, M., Rapposelli, S., Rahman, S. M., Chiellini, G., & Assadi-Porter, F. M. (2019). Lipolytic Effects of 3-Iodothyronamine (T1AM) and a Novel Thyronamine-Like Analog SG-2 through the AMPK Pathway. International Journal of Molecular Sciences, 20(16), 4054. https://doi.org/10.3390/ijms20164054