Stereoselective Synthesis and Investigation of Isopulegol-Based Chiral Ligands

 , ,
, ,  , and
, and 
Abstract
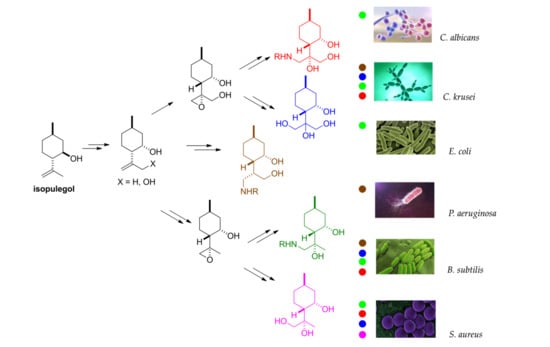
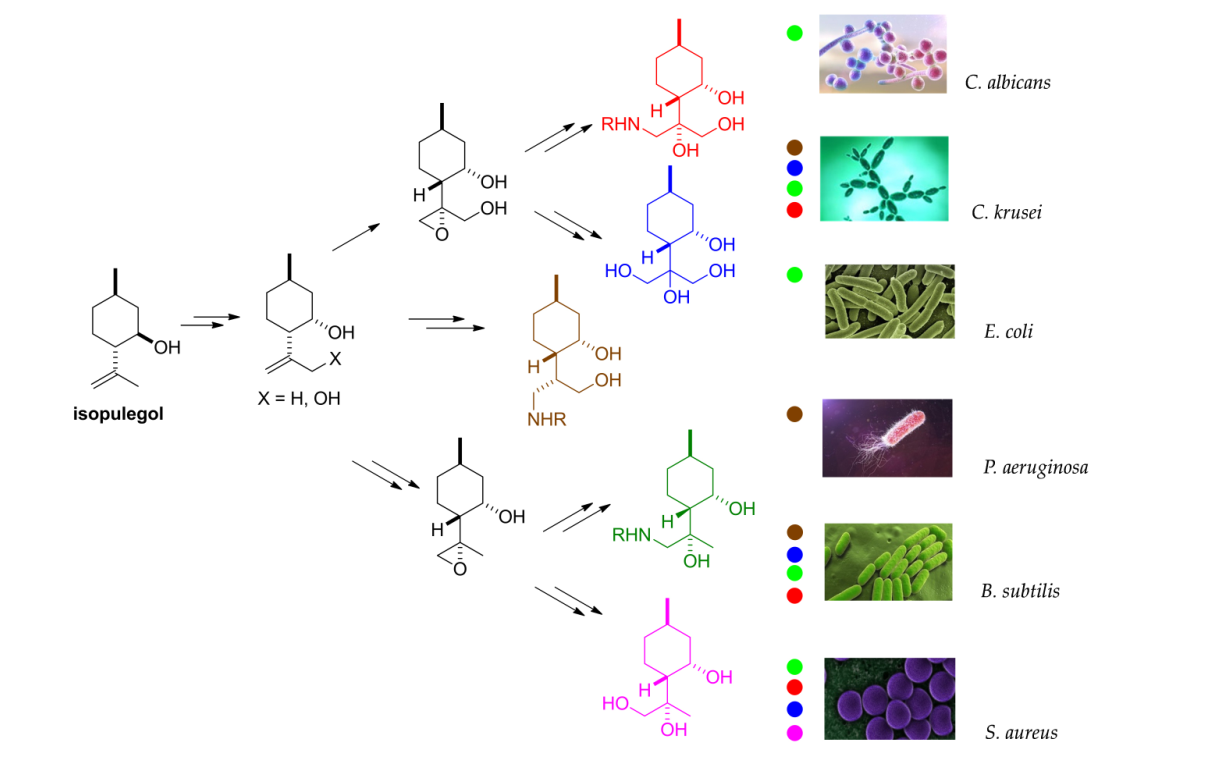
1. Introduction
2. Results
2.1. Synthesis of (−)-α-methylene-γ-butyrolactone 4
2.2. Synthesis of Isopulegol-based Aminodiols
2.3. Synthesis of Isopulegol-based Aminotriols
2.4. Application of Aminodiol Derivatives and Aminotriols as Chiral Ligands for Catalytic Addition of Diethylzinc to Benzaldehyde
2.5. Antimicrobial Effects
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Starting Materials
(3S,3aS,6R,7aS)-3-((Isopropylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (8)
4.3. General Procedure for Reduction with LiAlH4
4.3.1. (1S,2S,5R)-2-((S)-1-(Benzylamino)-3-hydroxypropan-2-yl)-5-methylcyclohexanol (9)
4.3.2. (1S,2S,5R)-2-((S)-1-Hydroxy-3-(((R)-1-phenylethyl)amino)propan-2-yl)-5-methylcyclohexanol (10)
4.3.3. (1S,2S,5R)-2-((S)-1-Hydroxy-3-(((S)-1-phenylethyl)amino)propan-2-yl)-5-methylcyclohexanol (11)
4.3.4. (1S,2S,5R)-2-((S)-1-Hydroxy-3-(isopropylamino)propan-2-yl)-5-methylcyclohexanol (12)
4.4. General Procedure of Epoxidation
4.4.1. (1S,2R,5R)-5-Methyl-2-((S)-2-methyloxiran-2-yl)cyclohexanol (19)
4.4.2. (1S,2R,5R)-2-((R)-2-(Hydroxymethyl)oxiran-2-yl)-5-methylcyclohexanol (31)
4.5. General Procedure for Ring-Opening of Epoxide with Primary Amines
4.5.1. (1S,2R,5R)-2-((S)-1-(Benzylamino)-2-hydroxypropan-2-yl)-5-methylcyclohexanol (20)
4.5.2. (1S,2R,5R)-2-((S)-2-Hydroxy-1-(((R)-1-phenylethyl)amino)propan-2-yl)-5-methylcyclohexanol (21)
4.5.3. (1S,2R,5R)-2-((S)-2-hydroxy-1-(((S)-1-phenylethyl)amino)propan-2-yl)-5-methylcyclohexanol (22)
4.5.4. (1S,2R,5R)-2-((S)-2-Hydroxy-1-(isopropylamino)propan-2-yl)-5-methylcyclohexanol (23)
4.5.5. (S)-3-(Benzylamino)-2-((1R,2S,4R)-2-hydroxy-4-methylcyclohexyl)propane-1,2-diol (32)
4.5.6. (S)-2-((1R,2S,4R)-2-Hydroxy-4-methylcyclohexyl)-3-(((R)-1-phenylethyl)amino)propane-1,2-diol (33)
4.5.7. (S)-2-((1R,2S,4R)-2-Hydroxy-4-methylcyclohexyl)-3-(((S)-1-phenylethyl)amino)propane-1,2-diol (34)
4.5.8. (S)-2-((1R,2S,4R)-2-Hydroxy-4-methylcyclohexyl)-3-(isopropylamino)propane-1,2-diol (35)
4.6. General Procedure for Ring Closure with Formaldehyde
4.6.1. (1S,2S,5R)-2-((S)-3-Benzyl-1,3-oxazinan-5-yl)-5-methylcyclohexanol (14)
4.6.2. (1S,2S,5R)-5-Methyl-2-((S)-3-((R)-1-phenylethyl)-1,3-oxazinan-5-yl)cyclohexanol (15)
4.6.3. (1S,2S,5R)-5-Methyl-2-((S)-3-((S)-1-phenylethyl)-1,3-oxazinan-5-yl)cyclohexanol (16)
4.6.4. (1S,2S,5R)-2-((S)-3-Isopropyl-1,3-oxazinan-5-yl)-5-methylcyclohexanol (17)
4.6.5. (1S,2R,5R)-2-((S)-3-Benzyl-5-methyloxazolidin-5-yl)-5-methylcyclohexanol (25)
4.6.6. (1S,2R,5R)-5-Methyl-2-((S)-5-methyl-3-((R)-1-phenylethyl)oxazolidin-5-yl)cyclohexanol (26)
4.6.7. (1S,2R,5R)-5-Methyl-2-((S)-5-methyl-3-((S)-1-phenylethyl)oxazolidin-5-yl)cyclohexanol (27)
4.6.8. (1S,2R,5R)-2-((S)-3-Isopropyl-5-methyloxazolidin-5-yl)-5-methylcyclohexanol (28)
4.7. General Procedure for Debenzylation
4.7.1. (1S,2S,5R)-2-((S)-1-Amino-3-hydroxypropan-2-yl)-5-methylcyclohexanol (13)
4.7.2. (1S,2R,5R)-2-((S)-1-Amino-2-hydroxypropan-2-yl)-5-methylcyclohexanol (24)
4.7.3. (S)-3-Amino-2-((1R,2S,4R)-2-hydroxy-4-methylcyclohexyl)propane-1,2-diol (36)
4.8. General Procedure for Dihydroxylation
4.8.1. (3R,3aR,6R,7aS)-3-Hydroxy-3-(hydroxymethyl)-6-methylhexahydrobenzofuran-2(3H)-one (18)
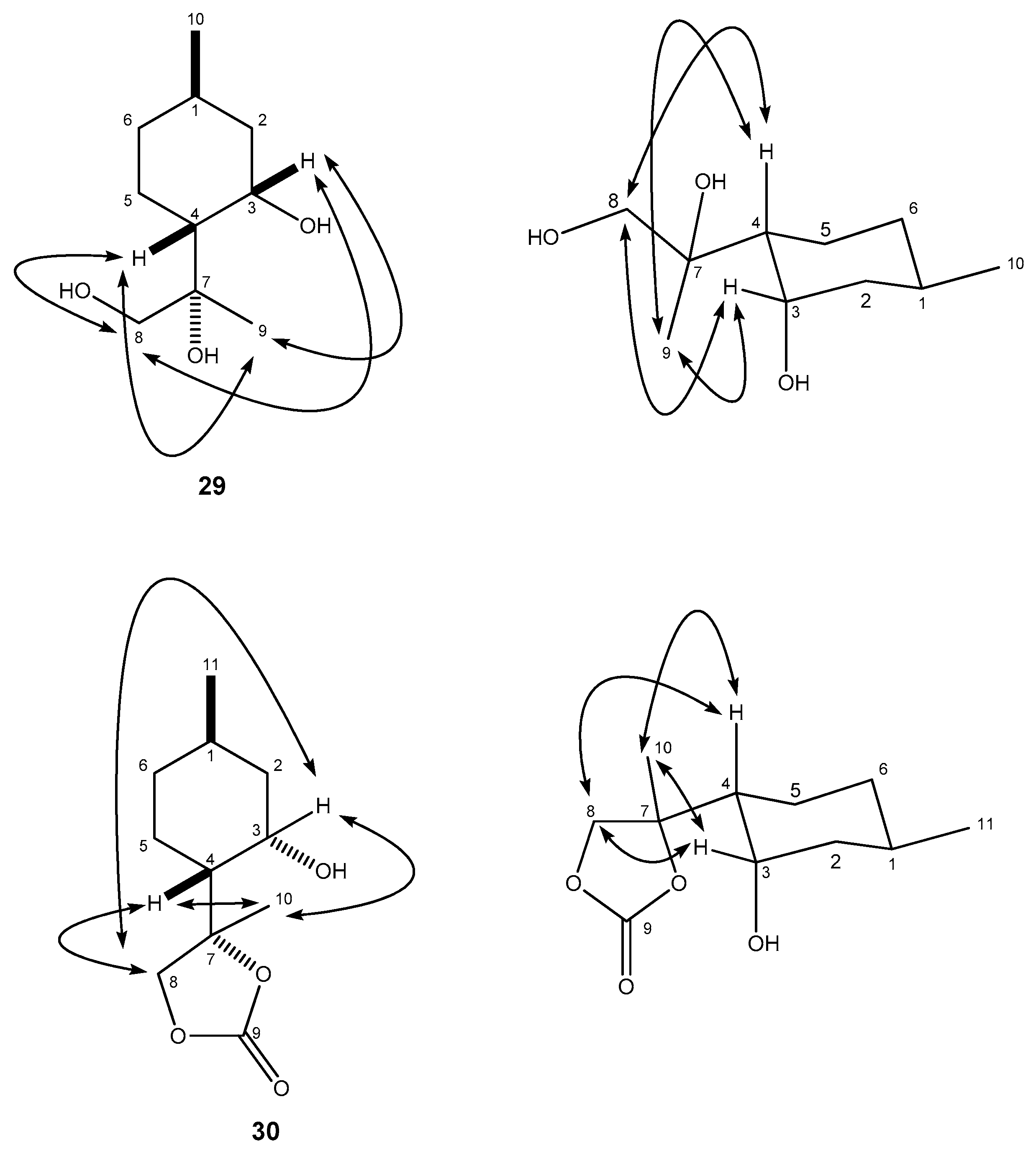
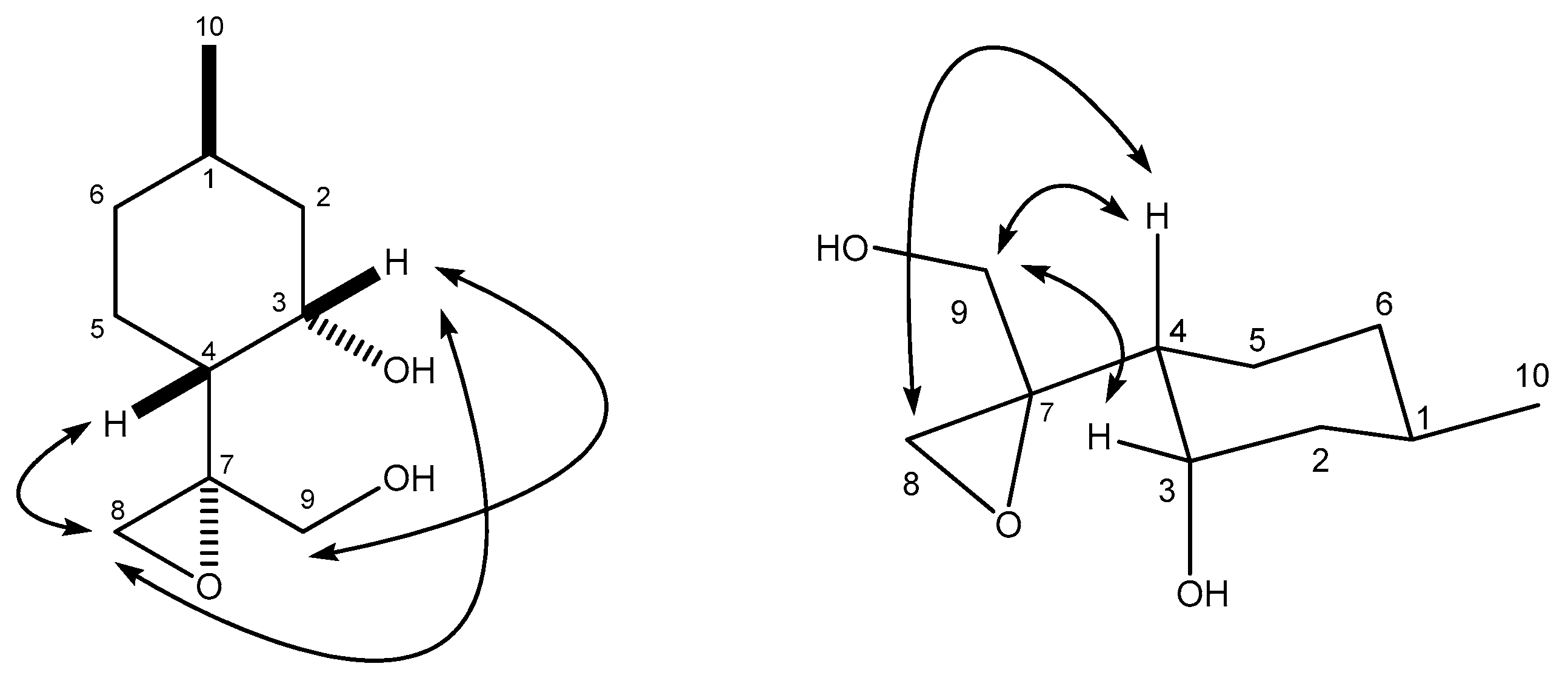
4.8.2. (R)-2-((1R,2S,4R)-2-Hydroxy-4-methylcyclohexyl)propane-1,2-diol (29)
4.8.3. 2-((1R,2S,4R)-2-Hydroxy-4-methylcyclohexyl)propane-1,2,3-triol (37)
4.9. (S)-4-((1R,2S,4R)-2-Hydroxy-4-methylcyclohexyl)-4-methyl-1,3-dioxolan-2-one (30)
(R)-2-((1R,2S,4R)-2-Hydroxy-4-methylcyclohexyl)propane-1,2-diol (29)
4.10. General Procedure for the Reaction of Benzaldehyde with Diethylzinc in the Presence of Chiral Catalysts
4.11. Antimicrobial Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Et2O | Diethyl ether |
| EtOH | Ethanol |
| HCHO | Formaldehyde |
| EtOAc | Ethyl acetate |
| t-BuOOH | tert-Butyl hydroperoxide |
| VO(acac)2 | Vanadyl aceatylacetonate |
| NMO | 4-Methylmorpholine N-oxide |
| MeCN | Acetonitrile |
| THF | Tetrahydrofuran |
| LiAlH4 | Lithium aluminum hydride |
References
- Largy, E.; Liu, W.; Hasan, A.; Perrin, D.M. A pyrimidopyrimidine Janus-AT nucleoside with improved base-pairing properties to both A and T within a DNA duplex: The stabilizing effect of a second endocyclic ring nitrogen. Chem. Eur. J. 2014, 20, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Grajewska, A.; Rozwadowska, M.D. Stereoselective synthesis of cytoxazone and its analogues. Tetrahedron Asymmetry 2007, 18, 803–813. [Google Scholar] [CrossRef]
- Mishra, R.K.; Coates, C.M.; Revell, K.D.; Turos, E. Synthesis of 2-Oxazolidinones from β-lactams: Stereospecific total synthesis of (−)-cytoxazone and all of its stereoisomers. Org. Lett. 2007, 9, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Allepuz, A.C.; Badorrey, R.; Díaz-de-Villegas, M.D.; Gálvez, J.A. Diastereoselective reduction of Ketimines derived from (R)-3,4-dihydroxybutan-2-one: An alternative route to key intermediates for the synthesis of anticancer agent ES-285. Tetrahedron Asymmetry 2010, 21, 503–506. [Google Scholar] [CrossRef]
- Richelle-Maurer, E.; Braekman, J.C.; Kluijver, M.; Gomez, R.; de Vyver, G.; Soest, R.; Devijver, C. Cellular location of (2R,3R,7Z)-2-aminotetradec-7-ene-1, 3-diol, a potent antimicrobial metabolite produced by the Caribbean sponge Haliclona vansoesti. Cell Tissue Res. 2001, 306, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, H.D.; Rosenberg, S.H.; Baker, W.R.; Stein, H.H.; Klinghofer, V.; Barlow, J.; Spina, K.; Polakowski, J.; Kovar, P.; Cohen, J. Discovery of a peptide-based renin inhibitor with oral bioavailability and efficacy. Science 1992, 257, 1940–1943. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, S.; Mohapatra, S.; Yadav, J.S. Practical synthesis of Abbott amino-diol: A core unit of the potent renin inhibitor Zankiren. Tetrahedron 1999, 55, 4763–4768. [Google Scholar] [CrossRef]
- Toribatake, K.; Miyata, S.; Naganawa, Y.; Nishiyama, H. Asymmetric synthesis of optically active 3-amino-1,2-diols from N-acyl-protected allylamines via catalytic diboration with Rh[bis(oxazolinyl)phenyl] catalysts. Tetrahedron 2015, 71, 3203–3208. [Google Scholar] [CrossRef]
- Jaime-Figueroa, S.; Greenhouse, R.; Padilla, F.; Dillon, M.P.; Gever, J.R.; Ford, A.P.D.W. Discovery and synthesis of a novel and selective drug-like P2X1 antagonist. Bioorg. Med. Chem. Lett. 2005, 15, 3292–3295. [Google Scholar] [CrossRef]
- Paraskar, A.S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone, novel cytokine modulators via Sharpless asymmetric epoxidation and L-proline catalyzed Mannich reaction. Tetrahedron 2006, 62, 5756–5762. [Google Scholar] [CrossRef]
- Kakeya, H.; Morishita, M.; Koshino, H.; Morita Ti, T.; Kobayashi, K.; Osada, H. Cytoxazone: A novel cytokine modulator containing a 2-Oxazolidinone ring produced by Streptomyces sp. J. Org. Chem. 1999, 64, 1052–1053. [Google Scholar] [CrossRef]
- Zhu, W.; Burnette, A.; Dorjsuren, D.; Roberts, P.E.; Huleihel, M.; Shoemaker, R.H.; Marquez, V.E.; Agbaria, R.; Sei, S. Potent antiviral activity of north-methanocarbathymidine against Kaposi’s sarcoma-associated herpesvirus. Antimicrob. Agents Chemother. 2005, 49, 4965–4973. [Google Scholar] [CrossRef]
- Lin, G.Q.; You, Q.D.; Cheng, J.F. Chiral Drugs: Chemistry and Biological Action; Wiley: Hoboken, NJ, USA, 2011; ISBN 978-0-470-58720-1. [Google Scholar]
- Tanaka, T.; Yasuda, Y.; Hayashi, M. New chiral Schiff base as a tridentate ligand for catalytic enantioselective addition of diethylzinc to aldehydes. J. Org. Chem. 2006, 71, 7091–7093. [Google Scholar] [CrossRef] [PubMed]
- Koneva, E.A.; Korchagina, D.V.; Gatilov, Y.V.; Genaev, A.M.; Krysin, A.P.; Volcho, K.P.; Tolstikov, A.G.; Salakhutdinov, N.F. New chiral ligands based on (+)-α-pinene. Russ. J. Org. Chem. 2010, 46, 1109–1115. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Csőr, Á.; Csámpai, A.; Fülöp, F. Stereoselective synthesis and modelling-driven optimisation of carane-based aminodiols and 1,3-oxazines as catalysts for the enantioselective addition of diethylzinc to benzaldehyde. Chem. Eur. J. 2016, 22, 7163–7173. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, Z.; Csillag, K.; Fülöp, F. Stereoselective synthesis of carane-based aminodiols as chiral ligands for the catalytic addition of diethylzinc to aldehydes. Tetrahedron Asymmetry 2011, 22, 1021–1027. [Google Scholar] [CrossRef]
- Panev, S.; Linden, A.; Dimitrov, V. Chiral aminoalcohols with a menthane skeleton as catalysts for the enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 2001, 12, 1313–1321. [Google Scholar] [CrossRef]
- Goldfuss, B.; Steigelmann, M.; Khan, S.I.; Houk, K.N. Rationalization of enantioselectivities in dialkylzinc additions to benzaldehyde catalyzed by fenchone derivatives. J. Org. Chem. 2000, 65, 77–82. [Google Scholar] [CrossRef]
- Tashenov, Y.; Daniels, M.; Robeyns, K.; Van Meervelt, L.; Dehaen, W.; Suleimen, Y.; Szakonyi, Z. Stereoselective syntheses and application of chiral bi- and tridentate ligands derived from (+)-sabinol. Molecules 2018, 23, 771. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Gonda, T.; Ötvös, S.B.; Fülöp, F. Stereoselective syntheses and transformations of chiral 1,3-aminoalcohols and 1,3-diols derived from nopinone. Tetrahedron Asymmetry 2014, 25, 1138–1145. [Google Scholar] [CrossRef]
- Gonda, T.; Szakonyi, Z.; Csámpai, A.; Haukka, M.; Fülöp, F. Stereoselective synthesis and application of tridentate aminodiols derived from (+)-pulegone. Tetrahedron Asymmetry 2016, 27, 480–486. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Duque-Soladana, J.P.; Rosón, C.D. Regio- and stereoselective 6- exo - trig radical cyclisations onto chiral perhydro-1,3-benzoxazines: Synthesis of enantiopure 3-alkylpiperidines. Tetrahedron Asymmetry 2000, 11, 2809–2821. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Nieto, J.; del Pozo, S. Synthesis of enantiopure 3-azabicyclo[3.2.0]heptanes by diastereoselective intramolecular [2+2] photocycloaddition reactions on chiral perhydro-1,3-benzoxazines. J. Org. Chem. 2003, 68, 4923–4931. [Google Scholar] [CrossRef]
- Alberola, A.; Andrés, C.; Pedrosa, R. Diastereoselective ring opening of 2-substituted N-benzyl-4,4, 7α-trimethyl-trans-octahydro-1,3-benzoxazines by Grignard reagents. Highly enantioselective synthesis of primary amines. Synlett 1990, 1990, 763–765. [Google Scholar] [CrossRef]
- Andrés, C.; Nieto, J.; Pedrosa, R.; Villamañán, N. Synthesis of enantiopure primary amines by stereoselective ring opening of chiral octahydro-1,3-benzoxazines by Grignard and organoaluminum reagents. J. Org. Chem. 1996, 61, 4130–4135. [Google Scholar] [CrossRef]
- Cherng, Y.J.; Fang, J.M.; Lu, T.J. Pinane-type tridentate reagents for enantioselective reactions: Reduction of ketones and addition of diethylzinc to aldehydes. J. Org. Chem. 1999, 64, 3207–3212. [Google Scholar] [CrossRef]
- Roy, C.D.; Brown, H.C. A study of transesterification of chiral (−)-pinanediol methylboronic ester with various structurally modified diols. Monatshefte Für Chem. Chem. Mon. 2007, 138, 747–753. [Google Scholar] [CrossRef]
- Ardashov, O.V.; Pavlova, A.V.; Il’ina, I.V.; Morozova, E.A.; Korchagina, D.V.; Karpova, E.V.; Volcho, K.P.; Tolstikova, T.G.; Salakhutdinov, N.F. Highly potent activity of (1R,2R,6S)-3-methyl-6-(prop-1-en-2-yl)cyclohex-3-ene-1,2-diol in animal models of Parkinson’s Disease. J. Med. Chem. 2011, 54, 3866–3874. [Google Scholar] [CrossRef]
- Radulović, N.S.; Mladenović, M.Z.; Randjelovic, P.J.; Stojanović, N.M.; Dekić, M.S.; Blagojević, P.D. Toxic essential oils. Part IV: The essential oil of Achillea falcata L. as a source of biologically/pharmacologically active trans-sabinyl esters. Food Chem. Toxicol. 2015, 80, 114–129. [Google Scholar] [CrossRef]
- Tuberoso, C.I.G.; Kowalczyk, A.; Coroneo, V.; Russo, M.T.; Dessì, S.; Cabras, P. Chemical composition and antioxidant, antimicrobial, and antifungal activities of the essential oil of Achillea ligustica All. J. Agric. Food Chem. 2005, 53, 10148–10153. [Google Scholar] [CrossRef]
- Brocksom, T.J.; dos Santos, R.B.; Varanda, N.A.; Brocksom, U. An Efficient synthesis of monoterpene α-methylene-γ-butyrolactones. Synth. Commun. 1988, 18, 1403–1410. [Google Scholar] [CrossRef]
- Brocksom, T.J.; Tercio, J.; Ferreira, B. A biomimetic synthesis of α-methylene-γ-butyrolactones. Synth. Commun. 1981, 11, 105–119. [Google Scholar] [CrossRef]
- Carda, M.; Marco, J.A. Total synthesis of the monoterpenes (−)-mintlactone and (+)-isomintlactone. Tetrahedron 1992, 48, 9789–9800. [Google Scholar] [CrossRef]
- Friedrich, D.; Bohlmann, F. Total synthesis of various elemanolides. Tetrahedron 1988, 44, 1369–1392. [Google Scholar] [CrossRef]
- Schlosser, M.; Kotthaus, M. Isopulegol as a model compound: Metalation and substitution of an allylic position in the presence of an unprotected hydroxy function. Eur. J. Org. Chem. 1999, 1999, 459–462. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C. Enzyme-mediated preparation of enantiomerically pure p-menthan- 3,9-diols and their use for the synthesis of natural p-menthane lactones and ethers. Helv. Chim. Acta 2002, 85, 2489–2502. [Google Scholar] [CrossRef]
- Le, T.; Bérdi, P.; Zupkó, I.; Fülöp, F.; Szakonyi, Z. Synthesis and transformation of (-)-isopulegol-based chiral β-aminolactones and β-aminoamides. Int. J. Mol. Sci. 2018, 19, 3522. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Fessler, D.C.; Eakin, M.A.; Giacobbe, T.J. Reactions of alpha methylene lactone tumor inhibitors with model biological nucleophiles. Science 1970, 168, 376–378. [Google Scholar] [CrossRef]
- Lawrence, N.J.; McGown, A.T.; Nduka, J.; Hadfield, J.A.; Pritchard, R.G. Cytotoxic michael-type amine adducts of α-methylene lactones alantolactone and isoalantolactone. Bioorg. Med. Chem. Lett. 2001, 11, 429–431. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Hetényi, A.; Fülöp, F. Synthesis and application of monoterpene-based chiral aminodiols. Tetrahedron 2008, 64, 1034–1039. [Google Scholar] [CrossRef]
- Kocienski, P.J.; Pontiroli, A.; Qun, L. Enantiospecific syntheses of pseudopterosin aglycones. Part 2. Synthesis of pseudopterosin K–L aglycone and pseudopterosin A–F aglycone via a B→BA→BAC annulation strategy. J. Chem. Soc. Perkin Trans. 1 2001, 2356–2366. [Google Scholar] [CrossRef]
- Rodríguez-Berríos, R.R.; Torres, G.; Prieto, J.A. Stereoselective VO(acac)(2) catalyzed epoxidation of acyclic homoallylic diols. Complementary preparation of C2-syn-3,4-epoxy alcohols. Tetrahedron 2011, 67, 830–836. [Google Scholar] [CrossRef]
- Carman, R.M.; Handley, P.N. 9,10-Dihydroxy-1,8-cineole (1,3,3-Trimethyl-2-oxabicyclo[2.2.2]octane-10,11-diol). Aust. J. Chem. 2001, 54, 769. [Google Scholar] [CrossRef]
- Shivani; Pujala, B.; Chakraborti, A.K. Zinc(II) perchlorate hexahydrate catalyzed opening of epoxide ring by amines: Applications to synthesis of (RS)/(R)-propranolols and (RS)/(R)/(S)-naftopidils. J. Org. Chem. 2007, 72, 3713–3722. [Google Scholar] [CrossRef]
- Bergmeier, S.C. The Synthesis of vicinal amino alcohols. Tetrahedron 2000, 56, 2561–2576. [Google Scholar] [CrossRef]
- Chen, K.; Baran, P.S. Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature 2009, 459, 824–828. [Google Scholar] [CrossRef]
- Morikawa, H.; Yamaguchi, J.; Sugimura, S.; Minamoto, M.; Gorou, Y.; Morinaga, H.; Motokucho, S. Systematic synthetic study of four diastereomerically distinct limonene-1,2-diols and their corresponding cyclic carbonates. Beilstein J. Org. Chem. 2019, 15, 130–136. [Google Scholar] [CrossRef]
- Paterson, I.; Anderson, E.A.; Dalby, S.M.; Lim, J.H.; Maltas, P. The stereocontrolled total synthesis of spirastrellolide A methyl ester. Fragment coupling studies and completion of the synthesis. Org. Biomol. Chem. 2012, 10, 5873. [Google Scholar] [CrossRef]
- Superchi, S.; Donnoli, M.I.; Proni, G.; Spada, G.P.; Rosini, C. Induction of cholesteric mesophases by simple cyclic derivatives of p,p’-disubstituted 1,2-diphenylethane-1,2-diols: Importance of shape and polarizability effects. J. Org. Chem. 1999, 64, 4762–4767. [Google Scholar] [CrossRef]
- Morikawa, H.; Minamoto, M.; Gorou, Y.; Yamaguchi, J.; Morinaga, H.; Motokucho, S. Two diastereomers of d-limonene-derived cyclic carbonates from d-limonene oxide and carbon dioxide with a tetrabutylammonium chloride catalyst. Bull. Chem. Soc. Jpn. 2018, 91, 92–94. [Google Scholar] [CrossRef]
- Wender, P.A.; Buschmann, N.; Cardin, N.B.; Jones, L.R.; Kan, C.; Kee, J.M.; Kowalski, J.A.; Longcore, K.E. Gateway synthesis of daphnane congeners and their protein kinase C affinities and cell-growth activities. Nat. Chem. 2011, 3, 615–619. [Google Scholar] [CrossRef][Green Version]
- Marcos, I.S.; Castañeda, L.; Basabe, P.; Díez, D.; Urones, J.G. Synthesis of sibiricinone A, sibiricinone B and leoheterin. Tetrahedron 2008, 64, 10860–10866. [Google Scholar] [CrossRef]
- Jimeno, C.; Pastó, M.; Riera, A.; Pericàs, M.A. Modular amino alcohol ligands containing bulky alkyl groups as chiral controllers for Et2Zn addition to aldehydes: Illustration of a design principle. J. Org. Chem. 2003, 68, 3130–3138. [Google Scholar] [CrossRef]
- Weinstein, M.P. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; Clinical and Laboratory: Wayne, PA, USA, 2018; ISBN 978-1-56238-836-3. [Google Scholar]
- Béni, Z.; Dékány, M.; Kovács, B.; Csupor-Löffler, B.; Zomborszki, Z.; Kerekes, E.; Szekeres, A.; Urbán, E.; Hohmann, J.; Ványolós, A. Bioactivity-guided isolation of antimicrobial and antioxidant metabolites from the mushroom. Tapinella Atrotomentosa Mol. 2018, 23, 1082. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ligand | Yield a (%) | eeb (%) | Configuration c |
|---|---|---|---|---|
| 1 | 9 | 90 | 18 | (R) |
| 2 | 10 | 92 | 60 | (R) |
| 3 | 11 | 95 | 43 | (R) |
| 4 | 12 | 95 | 17 | (R) |
| 5 | 22 | 97 | 18 | (R) |
| 6 | 25 | 93 | 37 | (R) |
| 7 | 32 | 86 | 19 | (S) |
| 8 | 34 | 87 | 28 | (S) |
| 9 | 35 | 80 | 16 | (S) |
| Inhibitory Effect (%) ± RSD (%) | |||||||
|---|---|---|---|---|---|---|---|
| Yeast | Gram-negative | Gram-positive | |||||
| Analogue | Conc. (µg/mL) | C. albicans | C. krusei | E. coli | P. aeruginosa | B. subtilis | S. aureus |
| 9 | 10 | − | 38.2 ± 4.2 | − | − | − | − |
| 100 | − | 41.8 ± 1.2 | 24.8 ± 1.3 | − | − | − | |
| 10 | 10 | − | − | 21.1 ± 8.0 | − | − | − |
| 100 | − | − | 25.8 ± 10.1 | 19.5 ± 0.5 | − | − | |
| 11 | 10 | 0.2 ± 0.9 | 0.9 ± 0.5 | − | 22.6 ± 2.1 | 18.4 ± 0.7 | 8.5 ± 0.6 |
| 100 | 2.2 ± 2.3 | 3.3 ± 4.7 | 0.2 ± 1.8 | 41.6 ± 12.2 | 21.0 ± 7.5 | 9.8 ± 4.0 | |
| 12 | 10 | − | − | − | 5.4 ± 0.3 | − | 8.5 ± 6.3 |
| 100 | − | − | − | 14.3 ± 4.5 | − | 9.4 ± 5.4 | |
| 13 | 10 | − | − | − | − | 26.1 ± 8.3 | 24.5 ± 15.6 |
| 100 | − | 40.4 ± 2.4 | − | − | 42.9 ± 20.0 | 25.2 ± 1.1 | |
| 14 | 10 | − | 8.4 ± 4.1 | − | 3.2 ± 7.1 | − | − |
| 100 | − | 6.7 ± 2.0 | 25.6 ± 2.1 | 4.4 ± 5.8 | − | 12.6 ± 4.5 | |
| 15 | 10 | − | 1.9 ± 0.7 | − | 8.6 ± 2.5 | − | 3.6 ± 1.4 |
| 100 | − | 2.8 ± 4.2 | 18.0 ± 1.8 | 20.1 ± 0.2 | − | 10.4 ± 1.5 | |
| 16 | 10 | − | − | − | 4.4 ± 5.8 | − | − |
| 100 | − | 48.9 ± 0.1 | 15.7 ± 1.7 | 8.4 ± 5.1 | 4.6 ± 12.5 | − | |
| 17 | 10 | − | − | − | 15.2 ± 10.4 | − | − |
| 100 | − | 33.1 ± 0.4 | 8.5 ± 2.06 | 16.7 ± 7.2 | − | − | |
| 18 | 10 | − | − | − | − | 16.9 ± 17.7 | 34.4 ± 11.7 |
| 100 | − | − | − | − | 27.1 ± 16.0 | 34.2 ± 2.6 | |
| 20 | 10 | − | − | − | 2.0 ± 0.9 | 10.6 ± 6.4 | − |
| 100 | 4.1 ± 1.6 | − | 35.4 ± 0.8 | 19.9 ± 4.8 | 11.5 ± 1.3 | 19.7 ± 7.2 | |
| 24 | 10 | − | 34.6 ± 3.3 | − | − | 47.6 ± 10.6 | 30.0 ± 2.0 |
| 100 | 23.6 ± 1.2 | 37.8 ± 3.6 | − | − | 55.1 ± 19.9 | 33.9 ± 4.0 | |
| 29 | 10 | − | − | − | − | − | 32.4 ± 4.1 |
| 100 | − | − | − | − | 21.2 ± 5.2 | 34.7 ± 6.6 | |
| 32 | 10 | − | − | − | − | − | − |
| 100 | − | − | − | − | − | 24.6 ± 11.9 | |
| 10 | − | − | − | − | 14.9 ± 13.8 | 31.2 ± 7.9 | |
| 36 | 100 | 39.9 ± 4.1 | − | − | 43.1 ± 2.6 | 33.3 ± 2.1 | |
| 37 | 10 | − | − | − | − | − | 31.6 ± 15.1 |
| 100 | − | 26.8 ± 7.9 | − | − | 40.9 ± 16.6 | 32.8 ± 8.2 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.M.; Szilasi, T.; Volford, B.; Szekeres, A.; Fülöp, F.; Szakonyi, Z. Stereoselective Synthesis and Investigation of Isopulegol-Based Chiral Ligands. Int. J. Mol. Sci. 2019, 20, 4050. https://doi.org/10.3390/ijms20164050
Le TM, Szilasi T, Volford B, Szekeres A, Fülöp F, Szakonyi Z. Stereoselective Synthesis and Investigation of Isopulegol-Based Chiral Ligands. International Journal of Molecular Sciences. 2019; 20(16):4050. https://doi.org/10.3390/ijms20164050
Chicago/Turabian StyleLe, Tam Minh, Tamás Szilasi, Bettina Volford, András Szekeres, Ferenc Fülöp, and Zsolt Szakonyi. 2019. "Stereoselective Synthesis and Investigation of Isopulegol-Based Chiral Ligands" International Journal of Molecular Sciences 20, no. 16: 4050. https://doi.org/10.3390/ijms20164050
APA StyleLe, T. M., Szilasi, T., Volford, B., Szekeres, A., Fülöp, F., & Szakonyi, Z. (2019). Stereoselective Synthesis and Investigation of Isopulegol-Based Chiral Ligands. International Journal of Molecular Sciences, 20(16), 4050. https://doi.org/10.3390/ijms20164050