Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders



Abstract
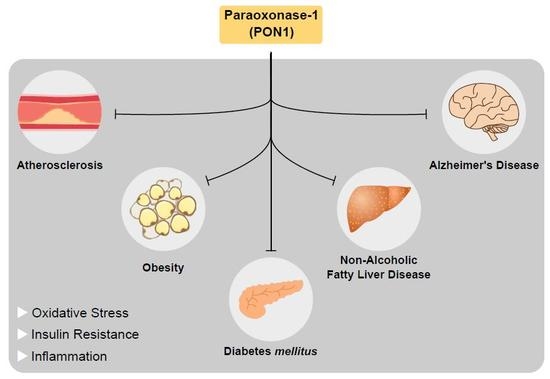
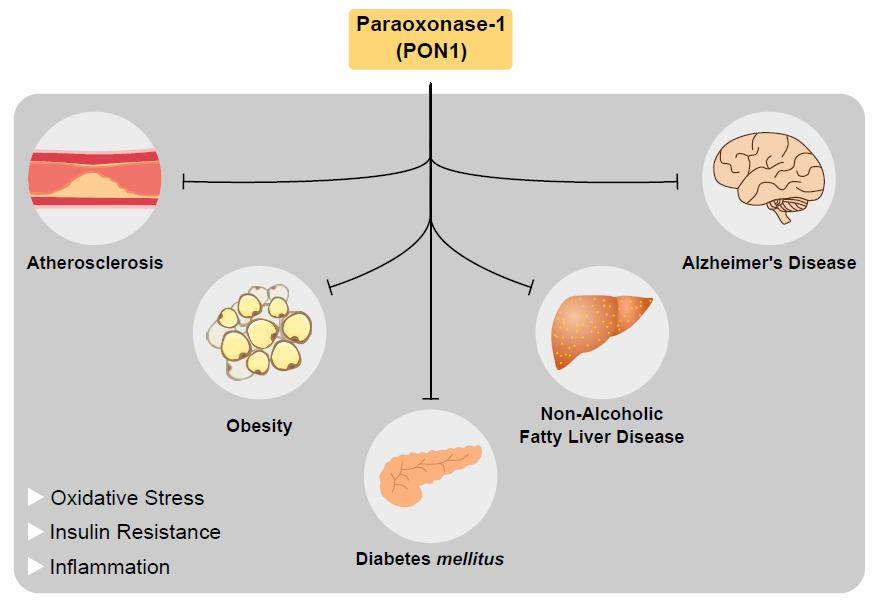{kind=link}
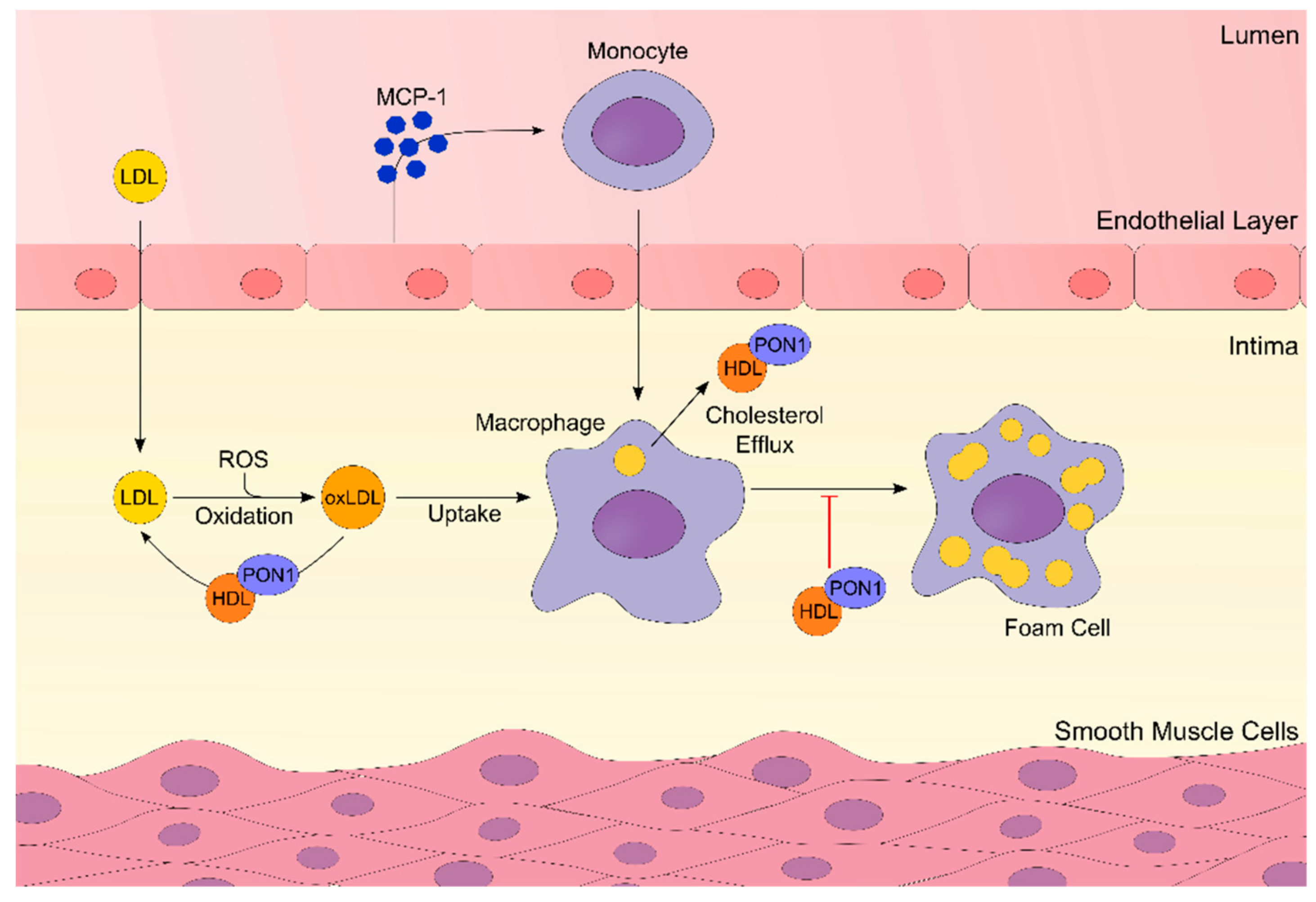{kind=link}
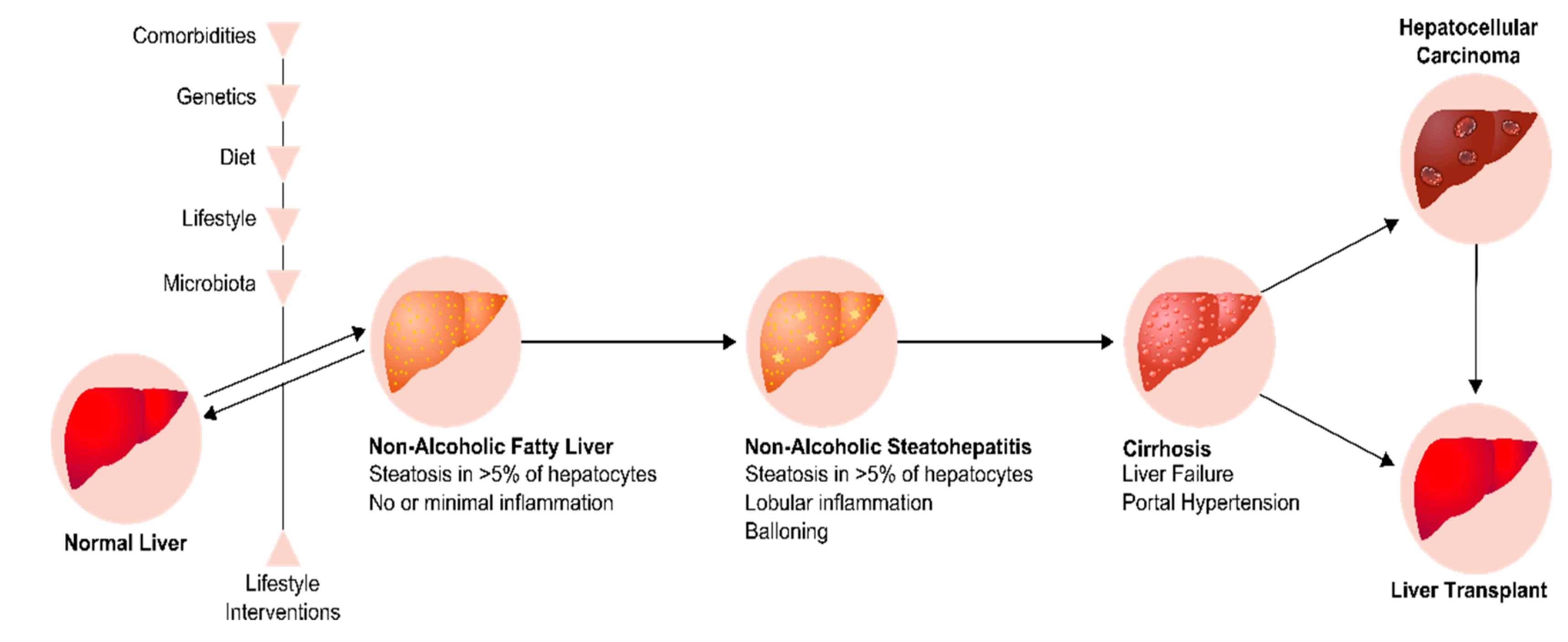{kind=link}
1. Introduction
2. Hallmarks of Metabolic Disorders
3. Paraoxonase Family
3.1. Paraoxonase-1 (PON1): An Antioxidant Enzyme
3.2. PON1 and Lipid Metabolism
3.3. PON1 and Glucose Homeostasis
3.4. PON1 and Pancreatic β Cells
3.5. PON1 and Diet
3.6. PON1 and Disease
3.6.1. PON1 and Atherosclerosis
3.6.2. PON1 and Diabetes
3.6.3. PON1 and Obesity
3.6.4. PON1 and Non-Alcoholic Fatty Liver Disease
3.6.5. PON1 and Cancer
3.6.6. PON1 and Alzheimer’s Disease
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| ApoA-I | Apolipoprotein A - I |
| ApoJ | Apolipoprotein |
| BMI | Body Mass Index |
| CVD | Cardiovascular Disease |
| DM | Diabetes mellitus |
| DK | Double Knockout |
| FLI | Fatty Liver Index |
| FFA | Free Fatty Acids |
| GLUTs | Glucose Transporters |
| HDL | High Density Lipoproteins |
| IR | Insulin Resistance |
| LDL | Low Density Lipoproteins |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MUFA | Monounsaturated Fatty Acids |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| NASH | Non-Alcoholic Steatohepatitis |
| PON1 | Paraoxonase 1 |
| PPAR | Peroxisome Proliferator Activated Receptors |
| PUFA | Polyunsaturated Fatty Acids |
| ROS | Reactive Oxygen Species |
| SNP | Single Nucleotide Polymorphisms |
| Sp1 | Specificity protein 1 |
| SREBP2 | Sterol Regulatory Element-binding Protein 2 |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TNFα | Tumor Necrosis Factor α |
| VLDL | Very Low Density Lipoprotein |
| WHO | World Health Organization |
References
- World Health Organization. Obesity and Overweight. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 12 December 2017).
- World Health Organization. Cardiovascular Diseases (CVDs). Available online: http://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 23 November 2018).
- World Health Organization. Diabetes. Available online: http://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 23 November 2018).
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Furlong, C.E.; Marsillach, J.; Jarvik, G.P.; Costa, L.G. Paraoxonases-1, -2 and -3: What are their functions? Chem. Biol. Interact. 2016, 259, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Rosenblat, M.; Bisgaier, C.L.; Newton, R.S.; Primo-Parmo, S.L.; La Du, B.N. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J. Clin. Investig. 1998, 101, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Bisbal, C.; Lambert, K.; Avignon, A. Antioxidants and glucose metabolism disorders. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Kota, S.K.; Meher, L.K.; Kota, S.K.; Jammula, S.; Krishna, S.V.; Modi, K.D. Implications of serum paraoxonase activity in obesity, diabetes mellitus, and dyslipidemia. Indian J. Endocrinol. Metab. 2013, 17, 402–412. [Google Scholar] [CrossRef]
- McEvoy, B.; Sumayao, R.; Slattery, C.; McMorrow, T.; Newsholme, P. Cystine accumulation attenuates insulin release from the pancreatic beta-cell due to elevated oxidative stress and decreased ATP levels. J. Physiol. 2015, 593, 5167–5182. [Google Scholar] [CrossRef]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef]
- Weiss, M.; Steiner, D.F.; Philipson, L.H. Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Robinson, S.D.; Safavi-Hemami, H. Insulin as a weapon. Toxicon 2016, 123, 56–61. [Google Scholar] [CrossRef]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Macedo, M.P.; Lima, I.S.; Gaspar, J.M.; Afonso, R.A.; Patarrao, R.S.; Kim, Y.B.; Ribeiro, R.T. Risk of postprandial insulin resistance: The liver/vagus rapport. Rev. Endocr. Metab. Disord. 2014, 15, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.V.B.; Kolka, C.M.; Kim, S.P.; Bergman, R.N. Obesity, insulin resistance and comorbidities—Mechanisms of association. Arq. Bras. Endocrinol. Metab. 2014, 58, 600–609. [Google Scholar] [CrossRef]
- Kawai, M.; de Paula, F.J.; Rosen, C.J. New insights into osteoporosis: The bone-fat connection. J. Intern. Med. 2012, 272, 317–329. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Leroith, D.; Karnieli, E. Insulin resistance in obesity as the underlying cause for the metabolic syndrome. Mt. Sinai J. Med. 2010, 77, 511–523. [Google Scholar] [CrossRef]
- Sesti, G. Pathophysiology of insulin resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 665–679. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D. Beta-cell dysfunction and insulin resistance in type 2 diabetes: Role of metabolic and genetic abnormalities. Am. J. Med. 2002, 113 (Suppl. 6A), 3S–11S. [Google Scholar] [CrossRef]
- Haldar, S.R.; Chakrabarty, A.; Chowdhury, S.; Haldar, A.; Sengupta, S.; Bhattacharyya, M. Oxidative stress-related genes in type 2 diabetes: Association analysis and their clinical impact. Biochem. Genet. 2015, 53, 93–119. [Google Scholar] [CrossRef]
- Okin, D.; Medzhitov, R. Evolution of Inflammatory Diseases. Curr. Biol. 2012, 22, R733–R740. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Yki-Jarvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Hwang, J.H.; Stein, D.T.; Barzilai, N.; Cui, M.H.; Tonelli, J.; Kishore, P.; Hawkins, M. Increased intrahepatic triglyceride is associated with peripheral insulin resistance: In vivo MR imaging and spectroscopy studies. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1663–E1669. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646; viii–ix. [Google Scholar] [CrossRef] [PubMed]
- Primo-Parmo, S.L.; Sorenson, R.C.; Teiber, J.; Du, B.N.L. The Human Serum Paraoxonase/Arylesterase Gene (PON1) Is One Member of a Multigene Family. Genomics 1996, 33, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Rajkovic, M.G.; Rumora, L.; Barisic, K. The paraoxonase 1, 2 and 3 in humans. Biochem. Med. 2011, 21, 122–130. [Google Scholar] [CrossRef]
- Dias, C.G.; Batuca, J.R.; Marinho, A.T.; Caixas, U.; Monteiro, E.C.; Antunes, A.M.; Pereira, S.A. Quantification of the arylesterase activity of paraoxonase-1 in human blood. Anal. Methods 2014, 6, 289–294. [Google Scholar] [CrossRef]
- Reddy, S.T.; Wadleigh, D.J.; Grijalva, V.; Ng, C.; Hama, S.; Gangopadhyay, A.; Shih, D.M.; Lusis, A.J.; Navab, M.; Fogelman, A.M. Human paraoxonase-3 is an HDL-associated enzyme with biological activity similar to paraoxonase-1 protein but is not regulated by oxidized lipids. Arter. Thromb. Vasc. Biol. 2001, 21, 542–547. [Google Scholar] [CrossRef]
- Davies, H.G.; Richter, R.J.; Keifer, M.; Broomfield, C.A.; Sowalla, J.; Furlong, C.E. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat. Genet. 1996, 14, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Mackness, M.; Mackness, B. Human paraoxonase-1 (PON1): Gene structure and expression, promiscuous activities and multiple physiological roles. Gene 2015, 567, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.; Bacchetti, T. Effect of dietary lipids on paraoxonase-1 activity and gene expression. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.J.; Wadleigh, D.J.; Gangopadhyay, A.; Hama, S.; Grijalva, V.R.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Paraoxonase-2 is a ubiquitously expressed protein with antioxidant properties and is capable of preventing cell-mediated oxidative modification of low density lipoprotein. J. Biol. Chem. 2001, 276, 44444–44449. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox. Signal. 2011, 14, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Mazur, A. An enzyme in animal tissues capable of hydrolysing the phosphorus-fluorine bond of alkyl fluorophosphates. J. Biol. Chem. 1946, 164, 271–289. [Google Scholar] [PubMed]
- Sorenson, R.C.; Primo-Parmo, S.L.; Camper, S.A.; La Du, B.N. The genetic mapping and gene structure of mouse paraoxonase/arylesterase. Genomics 1995, 30, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Humbert, R.; Adler, D.A.; Disteche, C.M.; Hassett, C.; Omiecinski, C.J.; Furlong, C.E. The molecular basis of the human serum paraoxonase activity polymorphism. Nat. Genet. 1993, 3, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Arii, K.; Suehiro, T.; Ikeda, Y.; Kumon, Y.; Inoue, M.; Inada, S.; Takata, H.; Ishibashi, A.; Hashimoto, K.; Terada, Y. Role of protein kinase C in pitavastatin-induced human paraoxonase I expression in Huh7 cells. Metabolism 2010, 59, 1287–1293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tan, N.Y.; Khachigian, L.M. Sp1 phosphorylation and its regulation of gene transcription. Mol. Cell. Biol. 2009, 29, 2483–2488. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Brophy, V.H.; Jampsa, R.L.; Clendenning, J.B.; McKinstry, L.A.; Jarvik, G.P.; Furlong, C.E. Effects of 5′ regulatory-region polymorphisms on paraoxonase-gene (PON1) expression. Am. J. Hum. Genet. 2001, 68, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Osaki, F.; Ikeda, Y.; Suehiro, T.; Ota, K.; Tsuzura, S.; Arii, K.; Kumon, Y.; Hashimoto, K. Roles of Sp1 and protein kinase C in regulation of human serum paraoxonase 1 (PON1) gene transcription in HepG2 cells. Atherosclerosis 2004, 176, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Richter, R.J.; Jarvik, G.P.; Furlong, C.E. Paraoxonase 1 status as a risk factor for disease or exposure. Adv. Exp. Med. Biol. 2010, 660, 29–35. [Google Scholar] [PubMed]
- Leviev, I.; Negro, F.; James, R.W. Two alleles of the human paraoxonase gene produce different amounts of mRNA. An explanation for differences in serum concentrations of paraoxonase associated with the (Leu-Met54) polymorphism. Arter. Thromb. Vasc. Biol. 1997, 17, 2935–2939. [Google Scholar] [CrossRef]
- Garin, M.C.; James, R.W.; Dussoix, P.; Blanche, H.; Passa, P.; Froguel, P.; Ruiz, J. Paraoxonase polymorphism Met-Leu54 is associated with modified serum concentrations of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease in diabetes. J. Clin. Investig. 1997, 99, 62–66. [Google Scholar] [CrossRef]
- Adkins, S.; Gan, K.N.; Mody, M.; la Du, B.N. Molecular basis for the polymorphic forms of human serum paraoxonase/arylesterase: Glutamine or arginine at position 191, for the respective A or B allozymes. Am. J. Hum. Genet. 1993, 52, 598–608. [Google Scholar] [PubMed]
- Rainwater, D.L.; Rutherford, S.; Dyer, T.D.; Rainwater, E.D.; Cole, S.A.; Vandeberg, J.L.; Almasy, L.; Blangero, J.; Maccluer, J.W.; Mahaney, M.C. Determinants of variation in human serum paraoxonase activity. Heredity 2009, 102, 147–154. [Google Scholar] [CrossRef]
- James, R.W.; Leviev, I.; Ruiz, J.; Passa, P.; Froguel, P.; Garin, M.C. Promoter polymorphism T(-107)C of the paraoxonase PON1 gene is a risk factor for coronary heart disease in type 2 diabetic patients. Diabetes 2000, 49, 1390–1393. [Google Scholar] [CrossRef][Green Version]
- Schrader, C.; Rimbach, G. Determinants of paraoxonase 1 status: Genes, drugs and nutrition. Curr. Med. Chem. 2011, 18, 5624–5643. [Google Scholar] [CrossRef]
- Costa, L.G.; Vitalone, A.; Cole, T.B.; Furlong, C.E. Modulation of paraoxonase (PON1) activity. Biochem. Pharm. 2005, 69, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.I.; La Du, B.N. Pharmacogenetics of paraoxonases: A brief review. Naunyn. Schmiedebergs Arch. Pharm. 2004, 369, 78–88. [Google Scholar] [CrossRef] [PubMed]
- She, Z.-G.; Chen, H.-Z.; Yan, Y.; Li, H.; Liu, D.-P. The Human Paraoxonase Gene Cluster as a Target in the Treatment of Atherosclerosis. Antioxid. Redox Signal. 2012, 16, 597–632. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Brumshtein, B.; Meged, R.; Dvir, H.; Ravelli, R.B.; McCarthy, A.; Toker, L.; Silman, I.; Sussman, J.L. 3-D structure of serum paraoxonase 1 sheds light on its activity, stability, solubility and crystallizability. Arh. Hig. Rada Toksikol. 2007, 58, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Aharoni, A.; Gaidukov, L.; Brumshtein, B.; Khersonsky, O.; Meged, R.; Dvir, H.; Ravelli, R.B.; McCarthy, A.; Toker, L.; et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004, 11, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; La Du, B.N. Calcium binding by human and rabbit serum paraoxonases. Structural stability and enzymatic activity. Drug Metab. Dispos. 1998, 26, 653–660. [Google Scholar] [PubMed]
- Moya, C.; Manez, S. Paraoxonases: Metabolic role and pharmacological projection. Naunyn. Schmiedebergs Arch. Pharm. 2018, 391, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.C.; Bisgaier, C.L.; Aviram, M.; Hsu, C.; Billecke, S.; la Du, B.N. Human serum Paraoxonase/Arylesterase’s retained hydrophobic N-terminal leader sequence associates with HDLs by binding phospholipids: Apolipoprotein A-I stabilizes activity. Arter. Thromb. Vasc. Biol. 1999, 19, 2214–2225. [Google Scholar] [CrossRef]
- Deakin, S.; Leviev, I.; Gomaraschi, M.; Calabresi, L.; Franceschini, G.; James, R.W. Enzymatically active paraoxonase-1 is located at the external membrane of producing cells and released by a high affinity, saturable, desorption mechanism. J. Biol. Chem. 2002, 277, 4301–4308. [Google Scholar] [CrossRef]
- Fuhrman, B.; Volkova, N.; Aviram, M. Paraoxonase 1 (PON1) is present in postprandial chylomicrons. Atherosclerosis 2005, 180, 55–61. [Google Scholar] [CrossRef]
- James, R.W.; Deakin, S.P. The importance of high-density lipoproteins for paraoxonase-1 secretion, stability, and activity. Free Radic. Biol. Med. 2004, 37, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Mathis, K.W.; Lee, I.K. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev. Endocr. Metab. Disord. 2014, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.A.; Kang, M.C.; Ciaraldi, T.P.; Kim, S.S.; Park, K.S.; Choe, C.; Hwang, W.M.; Lim, D.M.; Farr, O.; Mantzoros, C.; et al. Circulating ApoJ is closely associated with insulin resistance in human subjects. Metabolism 2018, 78, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Daimon, M.; Oizumi, T.; Karasawa, S.; Kaino, W.; Takase, K.; Tada, K.; Jimbu, Y.; Wada, K.; Kameda, W.; Susa, S.; et al. Association of the clusterin gene polymorphisms with type 2 diabetes mellitus. Metabolism 2011, 60, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Won, J.C.; Park, C.Y.; Oh, S.W.; Lee, E.S.; Youn, B.S.; Kim, M.S. Plasma clusterin (ApoJ) levels are associated with adiposity and systemic inflammation. PLoS ONE 2014, 9, e103351. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.I.; Teiber, J.F.; Speelman, A.; Osawa, Y.; Sunahara, R.; La Du, B.N. Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J. Lipid Res. 2005, 46, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry 2005, 44, 6371–6382. [Google Scholar] [CrossRef]
- Pahan, K. Lipid-lowering drugs. Cell. Mol. Life Sci. 2006, 63, 1165–1178. [Google Scholar] [CrossRef]
- Aslan, M.; Horoz, M.; Sabuncu, T.; Celik, H.; Selek, S. Serum paraoxonase enzyme activity and oxidative stress in obese subjects. Pol. Arch. Med. Wewn. 2011, 121, 181–186. [Google Scholar]
- Fedelesova, M.; Kupcova, V.; Luha, J.; Turecky, L. Paraoxonase activity in sera of patients with non-alcoholic fatty liver disease. Bratisl. Lek. Listy 2017, 118, 719–720. [Google Scholar] [CrossRef]
- Garcia-Heredia, A.; Kensicki, E.; Mohney, R.P.; Rull, A.; Triguero, I.; Marsillach, J.; Tormos, C.; Mackness, B.; Mackness, M.; Shih, D.M.; et al. Paraoxonase-1 deficiency is associated with severe liver steatosis in mice fed a high-fat high-cholesterol diet: A metabolomic approach. J. Proteome Res. 2013, 12, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Rosenblat, M.; Bisgaier, C.L.; Newton, R.S. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis 1998, 138, 271–280. [Google Scholar] [CrossRef]
- Deakin, S.; Leviev, I.; Guernier, S.; James, R.W. Simvastatin modulates expression of the PON1 gene and increases serum paraoxonase: A role for sterol regulatory element-binding protein-2. Arter. Thromb. Vasc. Biol. 2003, 23, 2083–2089. [Google Scholar] [CrossRef] [PubMed]
- Rozenberg, O.; Shih, D.M.; Aviram, M. Human serum paraoxonase 1 decreases macrophage cholesterol biosynthesis: Possible role for its phospholipase-A2-like activity and lysophosphatidylcholine formation. Arter. Thromb. Vasc. Biol. 2003, 23, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Tavori, H.; Aviram, M.; Khatib, S.; Musa, R.; Nitecki, S.; Hoffman, A.; Vaya, J. Human carotid atherosclerotic plaque increases oxidative state of macrophages and low-density lipoproteins, whereas paraoxonase 1 (PON1) decreases such atherogenic effects. Free Radic. Biol. Med. 2009, 46, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Tavori, H.; Aviram, M.; Khatib, S.; Musa, R.; Mannheim, D.; Karmeli, R.; Vaya, J. Human carotid lesion linoleic acid hydroperoxide inhibits paraoxonase 1 (PON1) activity via reaction with PON1 free sulfhydryl cysteine 284. Free Radic. Biol. Med. 2011, 50, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Koren-Gluzer, M.; Aviram, M.; Hayek, T. Paraoxonase1 (PON1) reduces insulin resistance in mice fed a high-fat diet, and promotes GLUT4 overexpression in myocytes, via the IRS-1/Akt pathway. Atherosclerosis 2013, 229, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. The histidine 115-histidine 134 dyad mediates the lactonase activity of mammalian serum paraoxonases. J. Biol. Chem. 2006, 281, 7649–7656. [Google Scholar] [CrossRef]
- Kulkarni, R.N. The islet beta-cell. Int. J. Biochem. Cell Biol. 2004, 36, 365–371. [Google Scholar] [CrossRef]
- Anuradha, R.; Saraswati, M.; Kumar, K.G.; Rani, S.H. Apoptosis of beta cells in diabetes mellitus. DNA Cell Biol. 2014, 33, 743–748. [Google Scholar] [CrossRef]
- Laybutt, R.; Hasenkamp, W.; Groff, A.; Grey, S.; Jonas, J.C.; Kaneto, H.; Sharma, A.; Bonner-Weir, S.; Weir, G. beta-cell adaptation to hyperglycemia. Diabetes 2001, 50 (Suppl. 1), S180–S181. [Google Scholar] [CrossRef]
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 2001, 50 (Suppl. 1), S154–S159. [Google Scholar] [CrossRef]
- Koren-Gluzer, M.; Aviram, M.; Meilin, E.; Hayek, T. The antioxidant HDL-associated paraoxonase-1 (PON1) attenuates diabetes development and stimulates beta-cell insulin release. Atherosclerosis 2011, 219, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Billecke, S.; Sorenson, R.; Bisgaier, C.; Newton, R.; Rosenblat, M.; Erogul, J.; Hsu, C.; Dunlop, C.; la Du, B. Paraoxonase active site required for protection against LDL oxidation involves its free sulfhydryl group and is different from that required for its arylesterase/paraoxonase activities: Selective action of human paraoxonase allozymes Q and R. Arter. Thromb. Vasc. Biol. 1998, 18, 1617–1624. [Google Scholar] [CrossRef]
- Gaidukov, L.; Tawfik, D.S. High affinity, stability, and lactonase activity of serum paraoxonase PON1 anchored on HDL with ApoA-I. Biochemistry 2005, 44, 11843–11854. [Google Scholar] [CrossRef] [PubMed]
- De Keyzer, D.; Karabina, S.A.; Wei, W.; Geeraert, B.; Stengel, D.; Marsillach, J.; Camps, J.; Holvoet, P.; Ninio, E. Increased PAFAH and oxidized lipids are associated with inflammation and atherosclerosis in hypercholesterolemic pigs. Arter. Thromb. Vasc. Biol. 2009, 29, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Kris-Etherton, P.M.; Yu, S.; Etherton, T.D.; Morgan, R.; Moriarty, K.; Shaffer, D. Fatty acids and progression of coronary artery disease. Am. J. Clin. Nutr. 1997, 65, 1088–1090. [Google Scholar] [CrossRef]
- Marsillach, J.; Camps, J.; Ferre, N.; Beltran, R.; Rull, A.; Mackness, B.; Mackness, M.; Joven, J. Paraoxonase-1 is related to inflammation, fibrosis and PPAR delta in experimental liver disease. BMC Gastroenterol. 2009, 9, 3. [Google Scholar] [CrossRef]
- Bonen, A.; Dohm, G.L.; van Loon, L.J. Lipid metabolism, exercise and insulin action. Essays Biochem. 2006, 42, 47–59. [Google Scholar] [CrossRef]
- Diniz, Y.S.; Rocha, K.K.; Souza, G.A.; Galhardi, C.M.; Ebaid, G.M.; Rodrigues, H.G.; Novelli Filho, J.L.; Cicogna, A.C.; Novelli, E.L. Effects of N-acetylcysteine on sucrose-rich diet-induced hyperglycaemia, dyslipidemia and oxidative stress in rats. Eur. J. Pharm. 2006, 543, 151–157. [Google Scholar] [CrossRef]
- Flatt, J.P. Conversion of carbohydrate to fat in adipose tissue: An energy-yielding and, therefore, self-limiting process. J. Lipid Res. 1970, 11, 131–143. [Google Scholar] [PubMed]
- Kannan, R.; Baker, N.; Bruckdorfer, K.R. Secretion and turnover of very low density lipoprotein triacylglycerols in rats fed chronically diets rich in glucose and fructose. J. Nutr. 1981, 111, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Deakin, S.; Moren, X.; James, R.W. Very low density lipoproteins provide a vector for secretion of paraoxonase-1 from cells. Atherosclerosis 2005, 179, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Zimowska, W.; Rock, E.; Rayssiguier, Y.; Mazur, A. Rats fed a high sucrose diet have altered heart antioxidant enzyme activity and gene expression. Life Sci. 2002, 71, 1303–1312. [Google Scholar] [CrossRef]
- Macan, M.; Vrkic, N.; Vrdoljak, A.L.; Radic, B.; Bradamante, V. Effects of high sucrose diet, gemfibrozil, and their combination on plasma paraoxonase 1 activity and lipid levels in rats. Acta Biochim. Pol. 2010, 57, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Blum, S.; Aviram, M.; Ben-Amotz, A.; Levy, Y. Effect of a Mediterranean meal on postprandial carotenoids, paraoxonase activity and C-reactive protein levels. Ann. Nutr. Metab. 2006, 50, 20–24. [Google Scholar] [CrossRef]
- Calabresi, L.; Villa, B.; Canavesi, M.; Sirtori, C.R.; James, R.W.; Bernini, F.; Franceschini, G. An omega-3 polyunsaturated fatty acid concentrate increases plasma high-density lipoprotein 2 cholesterol and paraoxonase levels in patients with familial combined hyperlipidemia. Metabolism 2004, 53, 153–158. [Google Scholar] [CrossRef]
- Loued, S.; Berrougui, H.; Componova, P.; Ikhlef, S.; Helal, O.; Khalil, A. Extra-virgin olive oil consumption reduces the age-related decrease in HDL and paraoxonase 1 anti-inflammatory activities. Br. J. Nutr. 2013, 110, 1272–1284. [Google Scholar] [CrossRef]
- Lou-Bonafonte, J.M.; Gabás-Rivera, C.; Navarro, M.A.; Osada, J. PON1 and Mediterranean Diet. Nutrients 2015, 7, 4068–4092. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- La Du, B.N.; Aviram, M.; Billecke, S.; Navab, M.; Primo-Parmo, S.; Sorenson, R.C.; Standiford, T.J. On the physiological role(s) of the paraoxonases. Chem. Biol. Interact. 1999, 119–120, 379–388. [Google Scholar] [CrossRef]
- Tward, A.; Xia, Y.-R.; Wang, X.-P.; Shi, Y.-S.; Park, C.; Castellani, L.W.; Lusis, A.J.; Shih, D.M. Decreased Atherosclerotic Lesion Formation in Human Serum Paraoxonase Transgenic Mice. Circulation 2002, 106, 484–490. [Google Scholar] [CrossRef]
- Shih, D.M.; Xia, Y.-R.; Wang, X.-P.; Miller, E.; Castellani, L.W.; Subbanagounder, G.; Cheroutre, H.; Faull, K.F.; Berliner, J.A.; Witztum, J.L.; et al. Combined Serum Paraoxonase Knockout/Apolipoprotein E Knockout Mice Exhibit Increased Lipoprotein Oxidation and Atherosclerosis. J. Biol. Chem. 2000, 275, 17527–17535. [Google Scholar] [CrossRef]
- Rozenberg, O.; Rosenblat, M.; Coleman, R.; Shih, D.M.; Aviram, M. Paraoxonase (PON1) deficiency is associated with increased macrophage oxidative stress: Studies in PON1-knockout mice. Free Radic. Biol. Med. 2003, 34, 774–784. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M. Paraoxonases 1, 2, and 3, oxidative stress, and macrophage foam cell formation during atherosclerosis development. Free Radic. Biol. Med. 2004, 37, 1304–1316. [Google Scholar] [CrossRef]
- Mackness, B.; Hine, D.; Liu, Y.; Mastorikou, M.; Mackness, M. Paraoxonase-1 inhibits oxidised LDL-induced MCP-1 production by endothelial cells. Biochem. Biophys. Res. Commun. 2004, 318, 680–683. [Google Scholar] [CrossRef]
- Rosenblat, M.; Vaya, J.; Shih, D.; Aviram, M. Paraoxonase 1 (PON1) enhances HDL-mediated macrophage cholesterol efflux via the ABCA1 transporter in association with increased HDL binding to the cells: A possible role for lysophosphatidylcholine. Atherosclerosis 2005, 179, 69–77. [Google Scholar] [CrossRef]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Ansell, B.J.; Fogelman, A.M. Mechanisms of Disease: Proatherogenic HDL—An evolving field. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 504. [Google Scholar] [CrossRef]
- Watson, A.D.; Berliner, J.A.; Hama, S.Y.; La Du, B.N.; Faull, K.F.; Fogelman, A.M.; Navab, M. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J. Clin. Investig. 1995, 96, 2882–2891. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M.; Billecke, S.; Erogul, J.; Sorenson, R.; Bisgaier, C.L.; Newton, R.S.; La Du, B. Human serum paraoxonase (PON 1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic. Biol. Med. 1999, 26, 892–904. [Google Scholar] [CrossRef]
- Mackness, M.I.; Mackness, B.; Arrol, S.; Wood, G.; Bhatnagar, D.; Durrington, P.N. Presence of paraoxonase in human interstitial fluid. FEBS Lett. 1997, 416, 377–380. [Google Scholar] [CrossRef]
- Garcia-Heredia, A.; Marsillach, J.; Rull, A.; Triguero, I.; Fort, I.; Mackness, B.; Mackness, M.; Shih, D.M.; Joven, J.; Camps, J. Paraoxonase-1 inhibits oxidized low-density lipoprotein-induced metabolic alterations and apoptosis in endothelial cells: A nondirected metabolomic study. Mediat. Inflamm. 2013, 2013, 156053. [Google Scholar] [CrossRef]
- Mackness, B.; Mackness, M. Anti-inflammatory properties of paraoxonase-1 in atherosclerosis. Adv. Exp. Med. Biol. 2010, 660, 143–151. [Google Scholar]
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev. Cardiovasc. 2012, 10, 353–361. [Google Scholar] [CrossRef]
- Clay, M.A.; Pyle, D.H.; Rye, K.A.; Vadas, M.A.; Gamble, J.R.; Barter, P.J. Time sequence of the inhibition of endothelial adhesion molecule expression by reconstituted high density lipoproteins. Atherosclerosis 2001, 157, 23–29. [Google Scholar] [CrossRef]
- Barter, P.J.; Rye, K.A. High density lipoproteins and coronary heart disease. Atherosclerosis 1996, 121, 1–12. [Google Scholar] [CrossRef]
- Taskinen, M.R. Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia 2003, 46, 733–749. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef]
- Bardini, G.; Rotella, C.M.; Giannini, S. Dyslipidemia and diabetes: Reciprocal impact of impaired lipid metabolism and Beta-cell dysfunction on micro- and macrovascular complications. Rev. Diabet. Stud. 2012, 9, 82–93. [Google Scholar] [CrossRef]
- Alam, U.; Asghar, O.; Azmi, S.; Malik, R.A. General aspects of diabetes mellitus. Handb. Clin. Neurol. 2014, 126, 211–222. [Google Scholar]
- Xia, Y.; Xie, Z.; Huang, G.; Zhou, Z. Incidence and trend of type 1 diabetes and the underlying environmental determinants. Diabetes Metab. Res. Rev. 2018, e3075. [Google Scholar] [CrossRef]
- Merino, J.; Udler, M.S.; Leong, A.; Meigs, J.B. A Decade of Genetic and Metabolomic Contributions to Type 2 Diabetes Risk Prediction. Curr. Diab. Rep. 2017, 17, 135. [Google Scholar] [CrossRef]
- De Vegt, F.; Dekker, J.M.; Jager, A.; Hienkens, E.; Kostense, P.J.; Stehouwer, C.D.; Nijpels, G.; Bouter, L.M.; Heine, R.J. Relation of impaired fasting and postload glucose with incident type 2 diabetes in a Dutch population: The Hoorn Study. JAMA 2001, 285, 2109–2113. [Google Scholar] [CrossRef]
- Shaw, J.E.; Zimmet, P.Z.; de Courten, M.; Dowse, G.K.; Chitson, P.; Gareeboo, H.; Hemraj, F.; Fareed, D.; Tuomilehto, J.; Alberti, K.G. Impaired fasting glucose or impaired glucose tolerance. What best predicts future diabetes in Mauritius? Diabetes Care 1999, 22, 399–402. [Google Scholar] [CrossRef]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef]
- Craciun, E.C.; Leucuta, D.C.; Rusu, R.L.; David, B.A.; Cret, V.; Dronca, E. Paraoxonase-1 activities in children and adolescents with type 1 diabetes mellitus. Acta Biochim. Pol. 2016, 63, 511–515. [Google Scholar] [CrossRef]
- Boemi, M.; Leviev, I.; Sirolla, C.; Pieri, C.; Marra, M.; James, R.W. Serum paraoxonase is reduced in type 1 diabetic patients compared to non-diabetic, first degree relatives; influence on the ability of HDL to protect LDL from oxidation. Atherosclerosis 2001, 155, 229–235. [Google Scholar] [CrossRef]
- Letellier, C.; Durou, M.R.; Jouanolle, A.M.; le Gall, J.Y.; Poirier, J.Y.; Ruelland, A. Serum paraoxonase activity and paraoxonase gene polymorphism in type 2 diabetic patients with or without vascular complications. Diabetes Metab. 2002, 28, 297–304. [Google Scholar]
- Mackness, B.; Durrington, P.N.; Abuashia, B.; Boulton, A.J.; Mackness, M.I. Low paraoxonase activity in type II diabetes mellitus complicated by retinopathy. Clin. Sci. 2000, 98, 355–363. [Google Scholar] [CrossRef]
- Deakin, S.; Leviev, I.; Nicaud, V.; Brulhart Meynet, M.C.; Tiret, L.; James, R.W. Paraoxonase-1 L55M polymorphism is associated with an abnormal oral glucose tolerance test and differentiates high risk coronary disease families. J. Clin. Endocrinol. Metab. 2002, 87, 1268–1273. [Google Scholar] [CrossRef][Green Version]
- Chiu, K.C.; Chuang, L.M.; Chu, A.; Lu, J.; Hu, J.; Fernando, S. Association of paraoxonase 1 polymorphism with beta-cell function: A case of molecular heterosis. Pancreas 2004, 28, e96–e103. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamauchi, K.; Shigematsu, S.; Ikeo, S.; Komatsu, M.; Aizawa, T.; Hashizume, K. Selective attenuation of metabolic branch of insulin receptor down-signaling by high glucose in a hepatoma cell line, HepG2 cells. J. Biol. Chem. 2000, 275, 20880–20886. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Schmitz-Peiffer, C.; Saha, A.K.; Ruderman, N.B.; Biden, T.J.; Kraegen, E.W. Muscle lipid accumulation and protein kinase C activation in the insulin-resistant chronically glucose-infused rat. Am. J. Physiol. 1999, 277, E1070–E1076. [Google Scholar] [CrossRef]
- Mastorikou, M.; Mackness, B.; Liu, Y.; Mackness, M. Glycation of paraoxonase-1 inhibits its activity and impairs the ability of high-density lipoprotein to metabolize membrane lipid hydroperoxides. Diabet. Med. 2008, 25, 1049–1055. [Google Scholar] [CrossRef]
- Ikeda, Y.; Suehiro, T.; Arii, K.; Kumon, Y.; Hashimoto, K. High glucose induces transactivation of the human paraoxonase 1 gene in hepatocytes. Metabolism 2008, 57, 1725–1732. [Google Scholar] [CrossRef]
- Gateva, A.; Assyov, Y.; Tsakova, A.; Kamenov, Z. Serum Paraoxonase-1 Levels are Significantly Decreased in the Presence of Insulin Resistance. Exp. Clin. Endocrinol. Diabetes 2016, 124, 444–447. [Google Scholar] [CrossRef]
- Deakin, S.P.; James, R.W. Genetic and environmental factors modulating serum concentrations and activities of the antioxidant enzyme paraoxonase-1. Clin. Sci. 2004, 107, 435–447. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.O.; Taskinen, M.R.; Boren, J. Diabetic dyslipidaemia. Curr. Opin. Lipidol. 2006, 17, 238–246. [Google Scholar] [CrossRef]
- Fonseca, M.I.H.; da Silva, I.T.; Ferreira, S.R.G. Impact of menopause and diabetes on atherogenic lipid profile: Is it worth to analyse lipoprotein subfractions to assess cardiovascular risk in women? Diabetol. Metab. Syndr. 2017, 9, 22. [Google Scholar] [CrossRef]
- Verges, B. New insight into the pathophysiology of lipid abnormalities in type 2 diabetes. Diabetes Metab. 2005, 31, 429–439. [Google Scholar] [CrossRef]
- Mastorikou, M.; Mackness, M.; Mackness, B. Defective metabolism of oxidized phospholipid by HDL from people with type 2 diabetes. Diabetes 2006, 55, 3099–3103. [Google Scholar] [CrossRef][Green Version]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef]
- Patel, S.; Drew, B.G.; Nakhla, S.; Duffy, S.J.; Murphy, A.J.; Barter, P.J.; Rye, K.A.; Chin-Dusting, J.; Hoang, A.; Sviridov, D.; et al. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J. Am. Coll. Cardiol. 2009, 53, 962–971. [Google Scholar] [CrossRef]
- Yu, W.; Liu, X.; Feng, L.; Yang, H.; Yu, W.; Feng, T.; Wang, S.; Wang, J.; Liu, N. Glycation of paraoxonase 1 by high glucose instigates endoplasmic reticulum stress to induce endothelial dysfunction in vivo. Sci. Rep. 2017, 7, 45827. [Google Scholar] [CrossRef]
- Witztum, J.L.; Fisher, M.; Pietro, T.; Steinbrecher, U.P.; Elam, R.L. Nonenzymatic glucosylation of high-density lipoprotein accelerates its catabolism in guinea pigs. Diabetes 1982, 31, 1029–1032. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T.; Marchionni, C.; Caldarelli, L.; Curatola, G. Effect of glycation of high density lipoproteins on their physicochemical properties and on paraoxonase activity. Acta Diabetol. 2001, 38, 163–169. [Google Scholar] [CrossRef]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef]
- Rashid, M.N.; Fuentes, F.; Touchon, R.C.; Wehner, P.S. Obesity and the risk for cardiovascular disease. Prev. Cardiol. 2003, 6, 42–47. [Google Scholar] [CrossRef]
- NHLBI Obesity Education Initiative Expert Panel on the Identification, Evaluation, and Treatment of Obesity in Adults (US). Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults: The Evidence Report. Bethesda (Md): National Heart, Lung, and Blood Institute. 1998. Available online: https://www.ncbi.nlm.nih.gov/books/NBK2003HLBI (accessed on 25 June 2019).
- Klein, S.; Allison, D.B.; Heymsfield, S.B.; Kelley, D.E.; Leibel, R.L.; Nonas, C.; Kahn, R. Waist Circumference and Cardiometabolic Risk: A Consensus Statement from Shaping America’s Health: Association for Weight Management and Obesity Prevention; NAASO, The Obesity Society; the American Society for Nutrition; and the American Diabetes Association. Obesity 2007, 15, 1061–1067. [Google Scholar]
- Grundy, S.M. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 2004, 89, 2595–2600. [Google Scholar] [CrossRef]
- Mutlu-Turkoglu, U.; Oztezcan, S.; Telci, A.; Orhan, Y.; Aykac-Toker, G.; Sivas, A.; Uysal, M. An increase in lipoprotein oxidation and endogenous lipid peroxides in serum of obese women. Clin. Exp. Med. 2003, 2, 171–174. [Google Scholar] [CrossRef]
- Bergman, R.N.; Kim, S.P.; Catalano, K.J.; Hsu, I.R.; Chiu, J.D.; Kabir, M.; Hucking, K.; Ader, M. Why visceral fat is bad: Mechanisms of the metabolic syndrome. Obesity 2006, 14, 16S–19S. [Google Scholar] [CrossRef]
- Kim, F.; Pham, M.; Maloney, E.; Rizzo, N.O.; Morton, G.J.; Wisse, B.E.; Kirk, E.A.; Chait, A.; Schwartz, M.W. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1982–1988. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; Kahn, C.R. Tissue–specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arter. Thromb. Vasc. Biol. 2012, 32, 2052–2059. [Google Scholar] [CrossRef]
- Semenkovich, C.F. Insulin resistance and atherosclerosis. J. Clin. Investig. 2006, 116, 1813. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T.; Moroni, C.; Savino, S.; Liuzzi, A.; Balzola, F.; Bicchiega, V. Paraoxonase activity in high-density lipoproteins: A comparison between healthy and obese females. J. Clin. Endocrinol. Metab. 2005, 90, 1728–1733. [Google Scholar] [CrossRef]
- Cervellati, C.; Bonaccorsi, G.; Trentini, A.; Valacchi, G.; Sanz, J.M.; Squerzanti, M.; Spagnolo, M.; Massari, L.; Crivellari, I.; Greco, P.; et al. Paraoxonase, arylesterase and lactonase activities of paraoxonase-1 (PON1) in obese and severely obese women. Scand. J. Clin. Lab. Investig. 2018, 78, 18–24. [Google Scholar] [CrossRef]
- Bajnok, L.; Seres, I.; Varga, Z.; Jeges, S.; Peti, A.; Karanyi, Z.; Juhasz, A.; Csongradi, E.; Mezosi, E.; Nagy, E.V.; et al. Relationship of serum resistin level to traits of metabolic syndrome and serum paraoxonase 1 activity in a population with a broad range of body mass index. Exp. Clin. Endocrinol. Diabetes 2008, 116, 592–599. [Google Scholar] [CrossRef]
- Koncsos, P.; Seres, I.; Harangi, M.; Illyes, I.; Jozsa, L.; Gonczi, F.; Bajnok, L.; Paragh, G. Human paraoxonase-1 activity in childhood obesity and its relation to leptin and adiponectin levels. Pediatr. Res. 2010, 67, 309–313. [Google Scholar] [CrossRef]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Scheen, A.J. Pathophysiology of type 2 diabetes. Acta Clin. Belg. 2003, 58, 335–341. [Google Scholar] [CrossRef]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab. 2002, 87, 3023–3028. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Duarte, N.; Coelho, I.C.; Patarrão, R.S.; Almeida, J.I.; Penha-Gonçalves, C.; Macedo, M.P. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. Biomed. Res. Int. 2015, 2015, 11. [Google Scholar] [CrossRef]
- Duarte, N.; Coelho, I.; Holovanchuk, D.; Ines Almeida, J.; Penha-Goncalves, C.; Paula Macedo, M. Dipeptidyl Peptidase-4 Is a Pro-Recovery Mediator During Acute Hepatotoxic Damage and Mirrors Severe Shifts in Kupffer Cells. Hepatol. Commun. 2018, 2, 1080–1094. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Yu, J.; Shen, J.; Sun, T.T.; Zhang, X.; Wong, N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin. Cancer Biol. 2013, 23, 483–491. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef]
- van den Berg, E.H.; Gruppen, E.G.; James, R.W.; Bakker, S.J.L.; Dullaart, R.P.F. Serum paraoxonase 1 activity is paradoxically maintained in nonalcoholic fatty liver disease despite low HDL cholesterol. J. Lipid Res. 2019, 60, 168–175. [Google Scholar] [CrossRef]
- Atamer, A.; Bilici, A.; Yenice, N.; Selek, S.; Ilhan, N.; Atamer, Y. The importance of paraoxonase 1 activity, nitric oxide and lipid peroxidation in hepatosteatosis. J. Int. Med. Res. 2008, 36, 771–776. [Google Scholar] [CrossRef]
- Long, J.; Zhang, C.-J.; Zhu, N.; Du, K.; Yin, Y.-F.; Tan, X.; Liao, D.-F.; Qin, L. Lipid metabolism and carcinogenesis, cancer development. Am. J. Cancer Res. 2018, 8, 778–791. [Google Scholar]
- Elkiran, E.T.; Mar, N.; Aygen, B.; Gursu, F.; Karaoglu, A.; Koca, S. Serum paraoxonase and arylesterase activities in patients with lung cancer in a Turkish population. BMC Cancer 2007, 7, 48. [Google Scholar] [CrossRef]
- Akcay, M.N.; Polat, M.F.; Yilmaz, I.; Akcay, G. Serum paraoxonase levels in pancreatic cancer. Hepatogastroenterology 2003, 50 (Suppl. 2), ccxxv–ccxxvii. [Google Scholar]
- Akcay, M.N.; Yilmaz, I.; Polat, M.F.; Akcay, G. Serum paraoxonase levels in gastric cancer. Hepatogastroenterology 2003, 50 (Suppl. 2), cclxxiii–cclxxv. [Google Scholar]
- Bulbuller, N.; Eren, E.; Ellidag, H.Y.; Oner, O.Z.; Sezer, C.; Aydin, O.; Yilmaz, N. Diagnostic value of thiols, paraoxonase 1, arylesterase and oxidative balance in colorectal cancer in human. Neoplasma 2013, 60, 419–424. [Google Scholar] [CrossRef]
- Camuzcuoglu, H.; Arioz, D.T.; Toy, H.; Kurt, S.; Celik, H.; Erel, O. Serum paraoxonase and arylesterase activities in patients with epithelial ovarian cancer. Gynecol. Oncol. 2009, 112, 481–485. [Google Scholar] [CrossRef]
- Saadat, M. Paraoxonase 1 genetic polymorphisms and susceptibility to breast cancer: A meta-analysis. Cancer Epidemiol. 2012, 36, e101–e103. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Caselli, R.J.; Chen, K.; Lee, W.; Alexander, G.E.; Reiman, E.M. Correlating cerebral hypometabolism with future memory decline in subsequent converters to amnestic pre-mild cognitive impairment. Arch. Neurol. 2008, 65, 1231–1236. [Google Scholar] [CrossRef]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef]
- Aluganti Narasimhulu, C.; Mitra, C.; Bhardwaj, D.; Burge, K.Y.; Parthasarathy, S. Alzheimer’s Disease Markers in Aged ApoE-PON1 Deficient Mice. J. Alzheimers Dis. 2019, 67, 1353–1365. [Google Scholar] [CrossRef]
- Erlich, P.M.; Lunetta, K.L.; Cupples, L.A.; Huyck, M.; Green, R.C.; Baldwin, C.T.; Farrer, L.A. Polymorphisms in the PON gene cluster are associated with Alzheimer disease. Hum. Mol. Genet. 2006, 15, 77–85. [Google Scholar] [CrossRef]
- Cellini, E.; Tedde, A.; Bagnoli, S.; Nacmias, B.; Piacentini, S.; Bessi, V.; Bracco, L.; Sorbi, S. Association analysis of the paraoxonase-1 gene with Alzheimer’s disease. Neurosci. Lett. 2006, 408, 199–202. [Google Scholar] [CrossRef]
- Leduc, V.; Theroux, L.; Dea, D.; Robitaille, Y.; Poirier, J. Involvement of paraoxonase 1 genetic variants in Alzheimer’s disease neuropathology. Eur. J. Neurosci. 2009, 30, 1823–1830. [Google Scholar] [CrossRef]
- Rabi, D.M.; Edwards, A.L.; Southern, D.A.; Svenson, L.W.; Sargious, P.M.; Norton, P.; Larsen, E.T.; Ghali, W.A. Association of socio-economic status with diabetes prevalence and utilization of diabetes care services. BMC Health Serv. Res. 2006, 6, 124. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meneses, M.J.; Silvestre, R.; Sousa-Lima, I.; Macedo, M.P. Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders. Int. J. Mol. Sci. 2019, 20, 4049. https://doi.org/10.3390/ijms20164049
Meneses MJ, Silvestre R, Sousa-Lima I, Macedo MP. Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders. International Journal of Molecular Sciences. 2019; 20(16):4049. https://doi.org/10.3390/ijms20164049
Chicago/Turabian StyleMeneses, Maria João, Regina Silvestre, Inês Sousa-Lima, and Maria Paula Macedo. 2019. "Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders" International Journal of Molecular Sciences 20, no. 16: 4049. https://doi.org/10.3390/ijms20164049
APA StyleMeneses, M. J., Silvestre, R., Sousa-Lima, I., & Macedo, M. P. (2019). Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders. International Journal of Molecular Sciences, 20(16), 4049. https://doi.org/10.3390/ijms20164049