Mitigating RNA Toxicity in Myotonic Dystrophy using Small Molecules


Abstract

{kind=link}
{kind=link}
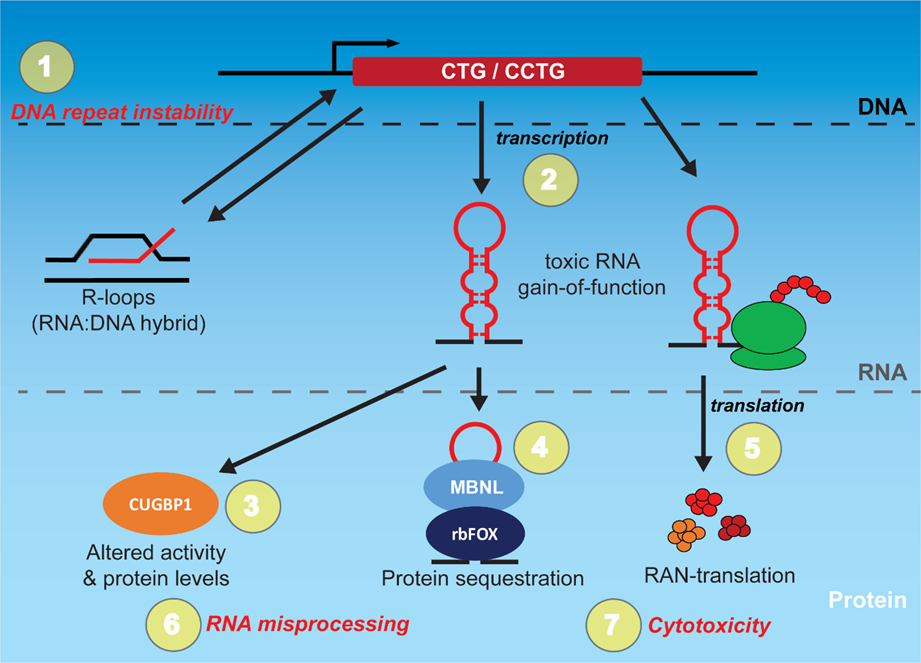
1. Introduction
2. Targeting the Toxic RNA in DM through Rational Design
2.1. Triaminotriazine-Based Designs
2.2. 2,9-Diaminoalkyl-1,10-phenanthroline (DAP)-Based Designs—An Independent Rationale Design Approach
2.3. Kanamycin and other Derivatives to Target the DM2 CCUG RNA
2.4. The Development of Cugamycin
3. Screening Small Molecule Libraries to Target Toxic CUG RNA
3.1. Combinatorial Chemistry Screen
3.2. The Identification of Diamidines from Screens of Nucleic Acid Binding Molecules
3.3. Repurposing Drug Screens
3.4. Screening for Disruption of the RNA:RBP Interaction
3.5. Ribonuclear Foci as a Screening Measure
4. Upregulating MBNL Protein Levels as a Therapeutic
5. Leveraging Mis-Splicing as a Read-Out in High-Throughput Screens
6. Restoring CUGBP1 for Therapeutic Benefit in DM1
7. Blocking Transcription of the CTG/CCTG Expansions
8. Targeting Repeat-Associated Non-ATG (RAN) Translation
9. Modulating DNA Repeat Instability for Therapeutic Benefit in DM
10. The Future Direction of DM Therapeutics
Supplementary Materials
Funding
Conflicts of Interest
References
- Lin, Y.; Dent, S.Y.; Wilson, J.H.; Wells, R.D.; Napierala, M. R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl. Acad. Sci. USA 2010, 107, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Tam, M.; Bowater, R.P.; Barber, M.; Tomlinson, M.; Nichol Edamura, K.; Wang, Y.H.; Pearson, C.E. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 2011, 39, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Schmidt, M.H.; Geist, J.M.; Thakkar, N.P.; Panigrahi, G.B.; Wang, Y.H.; Pearson, C.E. Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC).(GGCCCC) repeats causes instability. Nucleic Acids Res. 2014, 42, 10473–10487. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Cerro-Herreros, E.; Blatter, M.; Freyermuth, F.; Gaucherot, A.; Ruffenach, F.; Sarkar, P.; Puymirat, J.; Udd, B.; Day, J.W.; et al. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun. 2018, 9, 2009. [Google Scholar] [CrossRef] [PubMed]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Konieczny, P.; Selma-Soriano, E.; Rapisarda, A.S.; Fernandez-Costa, J.M.; Perez-Alonso, M.; Artero, R. Myotonic dystrophy: Candidate small molecule therapeutics. Drug Discov. Today 2017, 22, 1740–1748. [Google Scholar] [CrossRef]
- Lopez-Morato, M.; Brook, J.D.; Wojciechowska, M. Small Molecules Which Improve Pathogenesis of Myotonic Dystrophy Type 1. Front. Neurol. 2018, 9, 349. [Google Scholar] [CrossRef]
- Napierala, M.; Krzyzosiak, W.J. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J. Biol. Chem. 1997, 272, 31079–31085. [Google Scholar] [CrossRef]
- Tian, B.; White, R.J.; Xia, T.; Welle, S.; Turner, D.H.; Mathews, M.B.; Thornton, C.A. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA 2000, 6, 79–87. [Google Scholar] [CrossRef]
- Mooers, B.H.; Logue, J.S.; Berglund, J.A. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. USA 2005, 102, 16626–16631. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Yildirim, I.; Park, H.; Lohman, J.R.; Guan, L.; Tran, T.; Sarkar, P.; Schatz, G.C.; Disney, M.D. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem. Biol. 2014, 9, 538–550. [Google Scholar] [CrossRef]
- Blaszczyk, L.; Rypniewski, W.; Kiliszek, A. Structures of RNA repeats associated with neurological diseases. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef]
- Arambula, J.F.; Ramisetty, S.R.; Baranger, A.M.; Zimmerman, S.C. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc. Natl. Acad. Sci. USA 2009, 106, 16068–16073. [Google Scholar] [CrossRef]
- Wong, C.H.; Nguyen, L.; Peh, J.; Luu, L.M.; Sanchez, J.S.; Richardson, S.L.; Tuccinardi, T.; Tsoi, H.; Chan, W.Y.; Chan, H.Y.; et al. Targeting toxic RNAs that cause myotonic dystrophy type 1 (DM1) with a bisamidinium inhibitor. J. Am. Chem. Soc. 2014, 136, 6355–6361. [Google Scholar] [CrossRef]
- Nguyen, L.; Luu, L.M.; Peng, S.; Serrano, J.F.; Chan, H.Y.; Zimmerman, S.C. Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1. J. Am. Chem. Soc. 2015, 137, 14180–14189. [Google Scholar] [CrossRef]
- Lee, J.; Bai, Y.; Chembazhi, U.V.; Peng, S.; Yum, K.; Luu, L.M.; Hagler, L.D.; Serrano, J.F.; Chan, H.Y.E.; Kalsotra, A.; et al. Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Li, J.; Matsumoto, J.; Bai, L.P.; Murata, A.; Dohno, C.; Nakatani, K. A Ligand That Targets CUG Trinucleotide Repeats. Chemistry 2016, 22, 14881–14889. [Google Scholar] [CrossRef]
- Li, J.; Nakamori, M.; Matsumoto, J.; Murata, A.; Dohno, C.; Kiliszek, A.; Taylor, K.; Sobczak, K.; Nakatani, K. A Dimeric 2,9-Diamino-1,10-phenanthroline Derivative Improves Alternative Splicing in Myotonic Dystrophy Type 1 Cell and Mouse Models. Chemistry 2018, 24, 18115–18122. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Wu, M.; Pushechnikov, A.; Aminova, O.; Disney, M.D. A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem. Biol. 2007, 2, 745–754. [Google Scholar] [CrossRef]
- Disney, M.D.; Childs-Disney, J.L. Using selection to identify and chemical microarray to study the RNA internal loops recognized by 6’-N-acylated kanamycin A. Chembiochem 2007, 8, 649–656. [Google Scholar] [CrossRef]
- Lee, M.M.; Pushechnikov, A.; Disney, M.D. Rational and modular design of potent ligands targeting the RNA that causes myotonic dystrophy 2. ACS Chem. Biol. 2009, 4, 345–355. [Google Scholar] [CrossRef]
- Lee, M.M.; Childs-Disney, J.L.; Pushechnikov, A.; French, J.M.; Sobczak, K.; Thornton, C.A.; Disney, M.D. Controlling the specificity of modularly assembled small molecules for RNA via ligand module spacing: Targeting the RNAs that cause myotonic muscular dystrophy. J. Am. Chem. Soc. 2009, 131, 17464–17472. [Google Scholar] [CrossRef]
- Wong, C.H.; Fu, Y.; Ramisetty, S.R.; Baranger, A.M.; Zimmerman, S.C. Selective inhibition of MBNL1-CCUG interaction by small molecules toward potential therapeutic agents for myotonic dystrophy type 2 (DM2). Nucleic Acids Res. 2011, 39, 8881–8890. [Google Scholar] [CrossRef][Green Version]
- Nguyen, L.; Lee, J.; Wong, C.H.; Zimmerman, S.C. Small molecules that target the toxic RNA in myotonic dystrophy type 2. ChemMedChem 2014, 9, 2455–2462. [Google Scholar] [CrossRef]
- Disney, M.D.; Labuda, L.P.; Paul, D.J.; Poplawski, S.G.; Pushechnikov, A.; Tran, T.; Velagapudi, S.P.; Wu, M.; Childs-Disney, J.L. Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J. Am. Chem. Soc. 2008, 130, 11185–11194. [Google Scholar] [CrossRef]
- Pushechnikov, A.; Lee, M.M.; Childs-Disney, J.L.; Sobczak, K.; French, J.M.; Thornton, C.A.; Disney, M.D. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: Application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J. Am. Chem. Soc. 2009, 131, 9767–9779. [Google Scholar] [CrossRef]
- Velagapudi, S.P.; Seedhouse, S.J.; French, J.; Disney, M.D. Defining the RNA internal loops preferred by benzimidazole derivatives via 2D combinatorial screening and computational analysis. J. Am. Chem. Soc. 2011, 133, 10111–10118. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Hoskins, J.; Rzuczek, S.G.; Thornton, C.A.; Disney, M.D. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem. Biol. 2012, 7, 856–862. [Google Scholar] [CrossRef]
- Rzuczek, S.G.; Gao, Y.; Tang, Z.Z.; Thornton, C.A.; Kodadek, T.; Disney, M.D. Features of modularly assembled compounds that impart bioactivity against an RNA target. ACS Chem. Biol. 2013, 8, 2312–2321. [Google Scholar] [CrossRef]
- Rzuczek, S.G.; Colgan, L.A.; Nakai, Y.; Cameron, M.D.; Furling, D.; Yasuda, R.; Disney, M.D. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 2017, 13, 188–193. [Google Scholar] [CrossRef]
- Angelbello, A.J.; Rzuczek, S.G.; McKee, K.K.; Chen, J.L.; Olafson, H.; Cameron, M.D.; Moss, W.N.; Wang, E.T.; Disney, M.D. Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 7799–7804. [Google Scholar] [CrossRef]
- McNaughton, B.R.; Gareiss, P.C.; Miller, B.L. Identification of a selective small-molecule ligand for HIV-1 frameshift-inducing stem-loop RNA from an 11,325 member resin bound dynamic combinatorial library. J. Am. Chem. Soc. 2007, 129, 11306–11307. [Google Scholar] [CrossRef]
- Gareiss, P.C.; Sobczak, K.; McNaughton, B.R.; Palde, P.B.; Thornton, C.A.; Miller, B.L. Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNA-MBNL1 interaction in vitro: Discovery of lead compounds targeting myotonic dystrophy (DM1). J. Am. Chem. Soc. 2008, 130, 16254–16261. [Google Scholar] [CrossRef]
- Ofori, L.O.; Hoskins, J.; Nakamori, M.; Thornton, C.A.; Miller, B.L. From dynamic combinatorial ‘hit’ to lead: In vitro and in vivo activity of compounds targeting the pathogenic RNAs that cause myotonic dystrophy. Nucleic Acids Res. 2012, 40, 6380–6390. [Google Scholar] [CrossRef]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18551–18556. [Google Scholar] [CrossRef]
- Frayha, G.J.; Smyth, J.D.; Gobert, J.G.; Savel, J. The mechanisms of action of antiprotozoal and anthelmintic drugs in man. Gen. Pharm. 1997, 28, 273–299. [Google Scholar] [CrossRef]
- Miletti, K.E.; Leibowitz, M.J. Pentamidine inhibition of group I intron splicing in Candida albicans correlates with growth inhibition. Antimicrob Agents Chemother 2000, 44, 958–966. [Google Scholar] [CrossRef]
- Zhang, Y.; Bell, A.; Perlman, P.S.; Leibowitz, M.J. Pentamidine inhibits mitochondrial intron splicing and translation in Saccharomyces cerevisiae. RNA 2000, 6, 937–951. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Pilch, D.S.; Leibowitz, M.J. Pentamidine inhibits catalytic activity of group I intron Ca.LSU by altering RNA folding. Nucleic Acids Res. 2002, 30, 2961–2971. [Google Scholar] [CrossRef][Green Version]
- Edwards, K.J.; Jenkins, T.C.; Neidle, S. Crystal structure of a pentamidine-oligonucleotide complex: Implications for DNA-binding properties. Biochemistry 1992, 31, 7104–7109. [Google Scholar] [CrossRef]
- Girard, R.M.; Crispim, M.; Stolic, I.; Damasceno, F.S.; Santos da Silva, M.; Pral, E.M.; Elias, M.C.; Bajic, M.; Silber, A.M. An Aromatic Diamidine That Targets Kinetoplast DNA, Impairs the Cell Cycle in Trypanosoma cruzi, and Diminishes Trypomastigote Release from Infected Mammalian Host Cells. Antimicrob Agents Chemother 2016, 60, 5867–5877. [Google Scholar] [CrossRef]
- Coonrod, L.A.; Nakamori, M.; Wang, W.; Carrell, S.; Hilton, C.L.; Bodner, M.J.; Siboni, R.B.; Docter, A.G.; Haley, M.M.; Thornton, C.A.; et al. Reducing levels of toxic RNA with small molecules. ACS Chem. Biol. 2013, 8, 2528–2537. [Google Scholar] [CrossRef]
- Siboni, R.B.; Bodner, M.J.; Khalifa, M.M.; Docter, A.G.; Choi, J.Y.; Nakamori, M.; Haley, M.M.; Berglund, J.A. Biological Efficacy and Toxicity of Diamidines in Myotonic Dystrophy Type 1 Models. J. Med. Chem. 2015, 58, 5770–5780. [Google Scholar] [CrossRef]
- Jenquin, J.R.; Coonrod, L.A.; Silverglate, Q.A.; Pellitier, N.A.; Hale, M.A.; Xia, G.; Nakamori, M.; Berglund, J.A. Furamidine Rescues Myotonic Dystrophy Type I Associated Mis-Splicing through Multiple Mechanisms. ACS Chem. Biol. 2018, 13, 2708–2718. [Google Scholar] [CrossRef]
- Pohlig, G.; Bernhard, S.C.; Blum, J.; Burri, C.; Mpanya, A.; Lubaki, J.P.; Mpoto, A.M.; Munungu, B.F.; N’Tombe P, M.; Deo, G.K.; et al. Efficacy and Safety of Pafuramidine versus Pentamidine Maleate for Treatment of First Stage Sleeping Sickness in a Randomized, Comparator-Controlled, International Phase 3 Clinical Trial. PLoS Negl. Trop. Dis. 2016, 10, e0004363. [Google Scholar] [CrossRef]
- Nakamori, M.; Taylor, K.; Mochizuki, H.; Sobczak, K.; Takahashi, M.P. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 2016, 3, 42–54. [Google Scholar] [CrossRef]
- Chen, C.Z.; Sobczak, K.; Hoskins, J.; Southall, N.; Marugan, J.J.; Zheng, W.; Thornton, C.A.; Austin, C.P. Two high-throughput screening assays for aberrant RNA-protein interactions in myotonic dystrophy type 1. Anal. Bioanal. Chem. 2012, 402, 1889–1898. [Google Scholar] [CrossRef]
- Hoskins, J.W.; Ofori, L.O.; Chen, C.Z.; Kumar, A.; Sobczak, K.; Nakamori, M.; Southall, N.; Patnaik, S.; Marugan, J.J.; Zheng, W.; et al. Lomofungin and dilomofungin: Inhibitors of MBNL1-CUG RNA binding with distinct cellular effects. Nucleic Acids Res. 2014, 42, 6591–6602. [Google Scholar] [CrossRef]
- Garcia-Lopez, A.; Llamusi, B.; Orzaez, M.; Perez-Paya, E.; Artero, R.D. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl. Acad. Sci. USA 2011, 108, 11866–11871. [Google Scholar] [CrossRef]
- Garcia-Lopez, A.; Monferrer, L.; Garcia-Alcover, I.; Vicente-Crespo, M.; Alvarez-Abril, M.C.; Artero, R.D. Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS ONE 2008, 3, e1595. [Google Scholar] [CrossRef]
- Chakraborty, M.; Sellier, C.; Ney, M.; Pascal, V.; Charlet-Berguerand, N.; Artero, R.; Llamusi, B. Daunorubicin reduces MBNL1 sequestration caused by CUG-repeat expansion and rescues cardiac dysfunctions in a Drosophila model of myotonic dystrophy. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef]
- Ho, T.H.; Charlet, B.N.; Poulos, M.G.; Singh, G.; Swanson, M.S.; Cooper, T.A. Muscleblind proteins regulate alternative splicing. EMBO J. 2004, 23, 3103–3112. [Google Scholar] [CrossRef]
- Mankodi, A.; Urbinati, C.R.; Yuan, Q.P.; Moxley, R.T.; Sansone, V.; Krym, M.; Henderson, D.; Schalling, M.; Swanson, M.S.; Thornton, C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001, 10, 2165–2170. [Google Scholar] [CrossRef]
- Mankodi, A.; Teng-Umnuay, P.; Krym, M.; Henderson, D.; Swanson, M.; Thornton, C.A. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann. Neurol. 2003, 54, 760–768. [Google Scholar] [CrossRef]
- Fardaei, M.; Rogers, M.T.; Thorpe, H.M.; Larkin, K.; Hamshere, M.G.; Harper, P.S.; Brook, J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002, 11, 805–814. [Google Scholar] [CrossRef]
- Ketley, A.; Chen, C.Z.; Li, X.; Arya, S.; Robinson, T.E.; Granados-Riveron, J.; Udosen, I.; Morris, G.E.; Holt, I.; Furling, D.; et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum. Mol. Genet. 2014, 23, 1551–1562. [Google Scholar] [CrossRef]
- Wang, G.S.; Kuyumcu-Martinez, M.N.; Sarma, S.; Mathur, N.; Wehrens, X.H.; Cooper, T.A. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Invest. 2009, 119, 3797–3806. [Google Scholar] [CrossRef]
- Berman, E.; Brown, S.C.; James, T.L.; Shafer, R.H. NMR studies of chromomycin A3 interaction with DNA. Biochemistry 1985, 24, 6887–6893. [Google Scholar] [CrossRef]
- Kaziro, Y.; Kamiyama, M. Inhibition of Rna Polymerase Reaction by Chromomycin A3. Biochem. Biophys. Res. Commun. 1965, 19, 433–437. [Google Scholar] [CrossRef]
- Charizanis, K.; Lee, K.Y.; Batra, R.; Goodwin, M.; Zhang, C.; Yuan, Y.; Shiue, L.; Cline, M.; Scotti, M.M.; Xia, G.; et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012, 75, 437–450. [Google Scholar] [CrossRef]
- Lee, K.Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef]
- Thomas, J.D.; Sznajder, L.J.; Bardhi, O.; Aslam, F.N.; Anastasiadis, Z.P.; Scotti, M.M.; Nishino, I.; Nakamori, M.; Wang, E.T.; Swanson, M.S. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017, 31, 1122–1133. [Google Scholar] [CrossRef]
- Yadava, R.S.; Kim, Y.K.; Mandal, M.; Mahadevan, K.; Gladman, J.T.; Yu, Q.; Mahadevan, M.S. MBNL1 overexpression is not sufficient to rescue the phenotypes in a mouse model of RNA toxicity. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Shin, J.; Yuan, Y.; Beattie, S.G.; Wheeler, T.M.; Thornton, C.A.; Swanson, M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11748–11753. [Google Scholar] [CrossRef]
- Chamberlain, C.M.; Ranum, L.P. Mouse model of muscleblind-like 1 overexpression: Skeletal muscle effects and therapeutic promise. Hum. Mol. Genet. 2012, 21, 4645–4654. [Google Scholar] [CrossRef]
- Zhang, F.; Bodycombe, N.E.; Haskell, K.M.; Sun, Y.L.; Wang, E.T.; Morris, C.A.; Jones, L.H.; Wood, L.D.; Pletcher, M.T. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum. Mol. Genet. 2017, 26, 3056–3068. [Google Scholar] [CrossRef]
- Chen, G.; Masuda, A.; Konishi, H.; Ohkawara, B.; Ito, M.; Kinoshita, M.; Kiyama, H.; Matsuura, T.; Ohno, K. Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci. Rep. 2016, 6, 25317. [Google Scholar] [CrossRef]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Timchenko, L.T.; Miller, J.W.; Timchenko, N.A.; DeVore, D.R.; Datar, K.V.; Lin, L.; Roberts, R.; Caskey, C.T.; Swanson, M.S. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996, 24, 4407–4414. [Google Scholar] [CrossRef]
- Nakamori, M.; Sobczak, K.; Puwanant, A.; Welle, S.; Eichinger, K.; Pandya, S.; Dekdebrun, J.; Heatwole, C.R.; McDermott, M.P.; Chen, T.; et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013, 74, 862–872. [Google Scholar] [CrossRef]
- Wagner, S.D.; Struck, A.J.; Gupta, R.; Farnsworth, D.R.; Mahady, A.E.; Eichinger, K.; Thornton, C.A.; Wang, E.T.; Berglund, J.A. Dose-Dependent Regulation of Alternative Splicing by MBNL Proteins Reveals Biomarkers for Myotonic Dystrophy. PLoS Genet. 2016, 12, e1006316. [Google Scholar] [CrossRef]
- O’Leary, D.A.; Vargas, L.; Sharif, O.; Garcia, M.E.; Sigal, Y.J.; Chow, S.K.; Schmedt, C.; Caldwell, J.S.; Brinker, A.; Engels, I.H. HTS-Compatible Patient-Derived Cell-Based Assay to Identify Small Molecule Modulators of Aberrant Splicing in Myotonic Dystrophy Type 1. Curr. Chem. Genom. 2010, 4, 9–18. [Google Scholar] [CrossRef]
- Oana, K.; Oma, Y.; Suo, S.; Takahashi, M.P.; Nishino, I.; Takeda, S.; Ishiura, S. Manumycin A corrects aberrant splicing of Clcn1 in myotonic dystrophy type 1 (DM1) mice. Sci. Rep. 2013, 3, 2142. [Google Scholar] [CrossRef]
- Garcia-Alcover, I.; Colonques-Bellmunt, J.; Garijo, R.; Tormo, J.R.; Artero, R.; Alvarez-Abril, M.C.; Lopez Castel, A.; Perez-Alonso, M. Development of a Drosophila melanogaster spliceosensor system for in vivo high-throughput screening in myotonic dystrophy type 1. Dis. Model. Mech. 2014, 7, 1297–1306. [Google Scholar] [CrossRef]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Charlet, B.N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Lueck, J.D.; Swanson, M.S.; Dirksen, R.T.; Thornton, C.A. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Invest. 2007, 117, 3952–3957. [Google Scholar] [CrossRef]
- Ho, T.H.; Bundman, D.; Armstrong, D.L.; Cooper, T.A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 2005, 14, 1539–1547. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Taylor, K.; Sobczak, K.; Napierala, M.; Krzyzosiak, W.J. Small molecule kinase inhibitors alleviate different molecular features of myotonic dystrophy type 1. RNA Biol. 2014, 11, 742–754. [Google Scholar] [CrossRef]
- Timchenko, L.T.; Salisbury, E.; Wang, G.L.; Nguyen, H.; Albrecht, J.H.; Hershey, J.W.; Timchenko, N.A. Age-specific CUGBP1-eIF2 complex increases translation of CCAAT/enhancer-binding protein beta in old liver. J. Biol. Chem. 2006, 281, 32806–32819. [Google Scholar] [CrossRef]
- Huichalaf, C.; Sakai, K.; Jin, B.; Jones, K.; Wang, G.L.; Schoser, B.; Schneider-Gold, C.; Sarkar, P.; Pereira-Smith, O.M.; Timchenko, N.; et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. 2010, 24, 3706–3719. [Google Scholar] [CrossRef]
- Jones, K.; Wei, C.; Iakova, P.; Bugiardini, E.; Schneider-Gold, C.; Meola, G.; Woodgett, J.; Killian, J.; Timchenko, N.A.; Timchenko, L.T. GSK3beta mediates muscle pathology in myotonic dystrophy. J. Clin. Invest. 2012, 122, 4461–4472. [Google Scholar] [CrossRef]
- Mei, W.; Wen-Chin, W.; Lauren, S.; Diana, L.; Ana, M.; Genevieve, G.; Nikolai, T.; Mike, S.; Lubov, T. Correction of GSK3beta in DM1 reduces the mutant RNA and improves postnatal survival of DMSXL mice. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef]
- Muller, W.; Crothers, D.M. Studies of the binding of actinomycin and related compounds to DNA. J. Mol. Biol. 1968, 35, 251–290. [Google Scholar] [CrossRef]
- Kamitori, S.; Takusagawa, F. Crystal structure of the 2:1 complex between d(GAAGCTTC) and the anticancer drug actinomycin D. J. Mol. Biol. 1992, 225, 445–456. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by actinomycin D: Characteristic dose-response of different RNA species. J. Cell Physiol. 1970, 76, 127–139. [Google Scholar] [CrossRef]
- Chen, F.M. Binding of actinomycin D to DNA oligomers of CXG trinucleotide repeats. Biochemistry 1998, 37, 3955–3964. [Google Scholar] [CrossRef]
- Hou, M.H.; Robinson, H.; Gao, Y.G.; Wang, A.H. Crystal structure of actinomycin D bound to the CTG triplet repeat sequences linked to neurological diseases. Nucleic Acids Res. 2002, 30, 4910–4917. [Google Scholar] [CrossRef]
- Siboni, R.B.; Nakamori, M.; Wagner, S.D.; Struck, A.J.; Coonrod, L.A.; Harriott, S.A.; Cass, D.M.; Tanner, M.K.; Berglund, J.A. Actinomycin D Specifically Reduces Expanded CUG Repeat RNA in Myotonic Dystrophy Models. Cell Rep. 2015, 13, 2386–2394. [Google Scholar] [CrossRef]
- Pinto, B.S.; Saxena, T.; Oliveira, R.; Mendez-Gomez, H.R.; Cleary, J.D.; Denes, L.T.; McConnell, O.; Arboleda, J.; Xia, G.; Swanson, M.S.; et al. Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol. Cell 2017, 68, 479–490.e475. [Google Scholar] [CrossRef] [PubMed]
- Banez-Coronel, M.; Ranum, L.P.W. Repeat-associated non-AUG (RAN) translation: Insights from pathology. Lab. Invest. 2019. [Google Scholar] [CrossRef]
- Nguyen, L.; Cleary, J.D.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Pattamatta, A.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation in Neurological Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Ranum, L.P. New developments in RAN translation: Insights from multiple diseases. Curr. Opin. Genet. Dev. 2017, 44, 125–134. [Google Scholar] [CrossRef]
- Zu, T.; Cleary, J.D.; Liu, Y.; Banez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 2017, 95, 1292–1305.e1295. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Zhang, Y.; Gendron, T.F.; Bauer, P.O.; Chew, J.; Yang, W.Y.; Fostvedt, E.; Jansen-West, K.; Belzil, V.V.; Desaro, P.; et al. Discovery of a Biomarker and Lead Small Molecules to Target r(GGGGCC)-Associated Defects in c9FTD/ALS. Neuron 2014, 84, 239. [Google Scholar] [CrossRef]
- Yang, W.Y.; Wilson, H.D.; Velagapudi, S.P.; Disney, M.D. Inhibition of Non-ATG Translational Events in Cells via Covalent Small Molecules Targeting RNA. J. Am. Chem. Soc. 2015, 137, 5336–5345. [Google Scholar] [CrossRef]
- Wang, Z.F.; Ursu, A.; Childs-Disney, J.L.; Guertler, R.; Yang, W.Y.; Bernat, V.; Rzuczek, S.G.; Fuerst, R.; Zhang, Y.J.; Gendron, T.F.; et al. The Hairpin Form of r(G4C2)(exp) in c9ALS/FTD Is Repeat-Associated Non-ATG Translated and a Target for Bioactive Small Molecules. Cell Chem. Biol. 2019, 26, 179–190.e112. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, S.; Mestre, A.A.; Fu, C.; Makarem, A.; Xian, F.; Hayes, L.R.; Lopez-Gonzalez, R.; Drenner, K.; Jiang, J.; et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat. Commun. 2018, 9, 51. [Google Scholar] [CrossRef]
- Cinesi, C.; Aeschbach, L.; Yang, B.; Dion, V. Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat. Commun. 2016, 7, 13272. [Google Scholar] [CrossRef]
- van Agtmaal, E.L.; Andre, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef]
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol. Nucleic Acids 2017, 9, 337–348. [Google Scholar] [CrossRef]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef]
- Lo Scrudato, M.; Poulard, K.; Sourd, C.; Tome, S.; Klein, A.F.; Corre, G.; Huguet, A.; Furling, D.; Gourdon, G.; Buj-Bello, A. Genome Editing of Expanded CTG Repeats within the Human DMPK Gene Reduces Nuclear RNA Foci in the Muscle of DM1 Mice. Mol. Ther. 2019. [Google Scholar] [CrossRef]
- Yang, Z.; Lau, R.; Marcadier, J.L.; Chitayat, D.; Pearson, C.E. Replication inhibitors modulate instability of an expanded trinucleotide repeat at the myotonic dystrophy type 1 disease locus in human cells. Am. J. Hum. Genet. 2003, 73, 1092–1105. [Google Scholar] [CrossRef]
- Schmidt, M.H.M.; Pearson, C.E. Disease-associated repeat instability and mismatch repair. DNA Repair (Amst) 2016, 38, 117–126. [Google Scholar] [CrossRef]
- Jenquin, J.R.Y.; Yang, H.; Huigens, R.W., III; Nakamori, M.; Berglund, J.A. Combination Treatment of Erythromycin and Furamidine Provides Additive and Synergistic Rescue of Mis-splicing in Myotonic Dystrophy Type 1 Models. ACS Pharmacol. Transl. Sci. 2019, 2, 247–263. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, K.; Jenquin, J.R.; Cleary, J.D.; Berglund, J.A. Mitigating RNA Toxicity in Myotonic Dystrophy using Small Molecules. Int. J. Mol. Sci. 2019, 20, 4017. https://doi.org/10.3390/ijms20164017
Reddy K, Jenquin JR, Cleary JD, Berglund JA. Mitigating RNA Toxicity in Myotonic Dystrophy using Small Molecules. International Journal of Molecular Sciences. 2019; 20(16):4017. https://doi.org/10.3390/ijms20164017
Chicago/Turabian StyleReddy, Kaalak, Jana R. Jenquin, John D. Cleary, and J. Andrew Berglund. 2019. "Mitigating RNA Toxicity in Myotonic Dystrophy using Small Molecules" International Journal of Molecular Sciences 20, no. 16: 4017. https://doi.org/10.3390/ijms20164017
APA StyleReddy, K., Jenquin, J. R., Cleary, J. D., & Berglund, J. A. (2019). Mitigating RNA Toxicity in Myotonic Dystrophy using Small Molecules. International Journal of Molecular Sciences, 20(16), 4017. https://doi.org/10.3390/ijms20164017