Regulation of Hedgehog signaling Offers A Novel Perspective for Bone Homeostasis Disorder Treatment

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
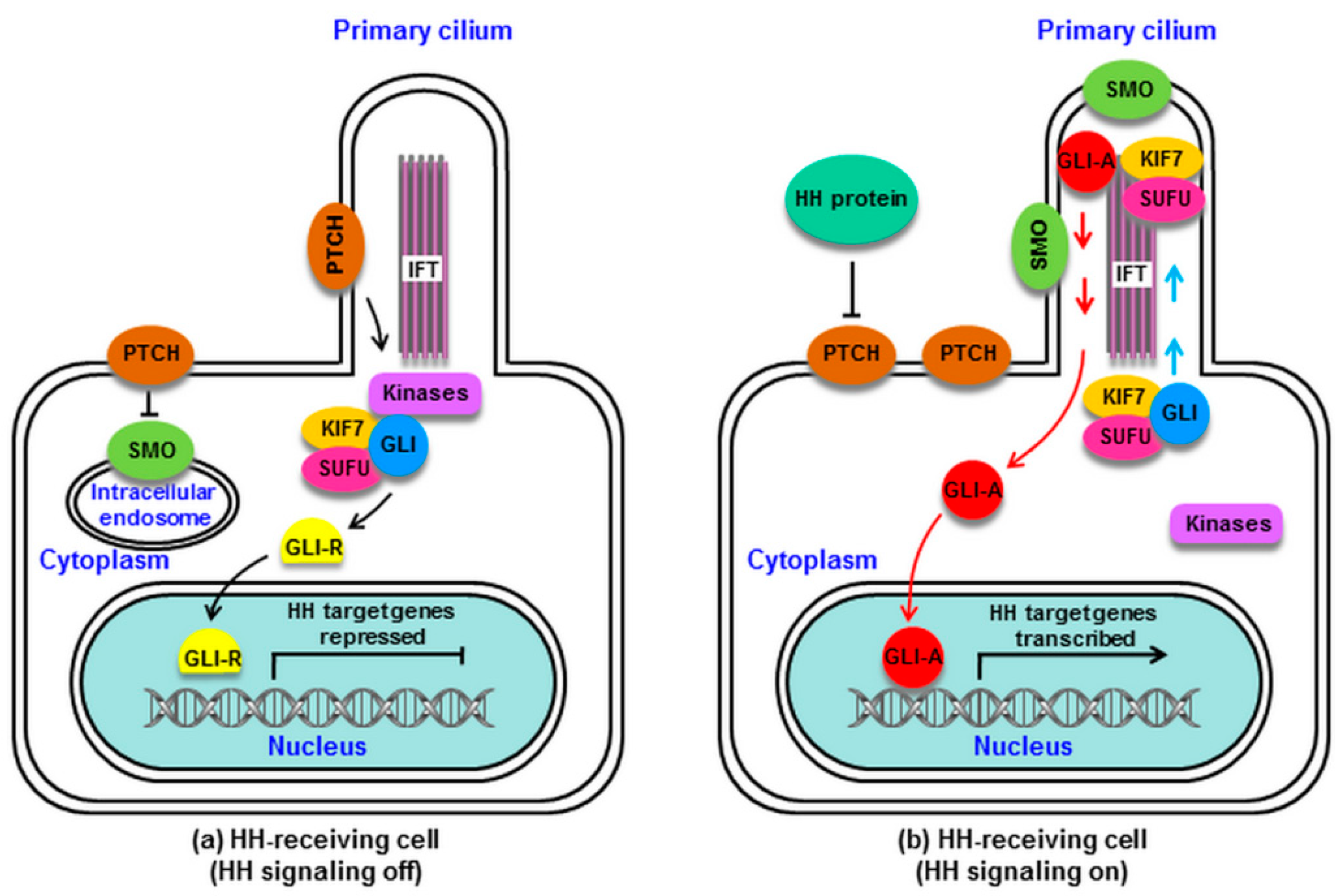
2. HH Signaling in Mammals
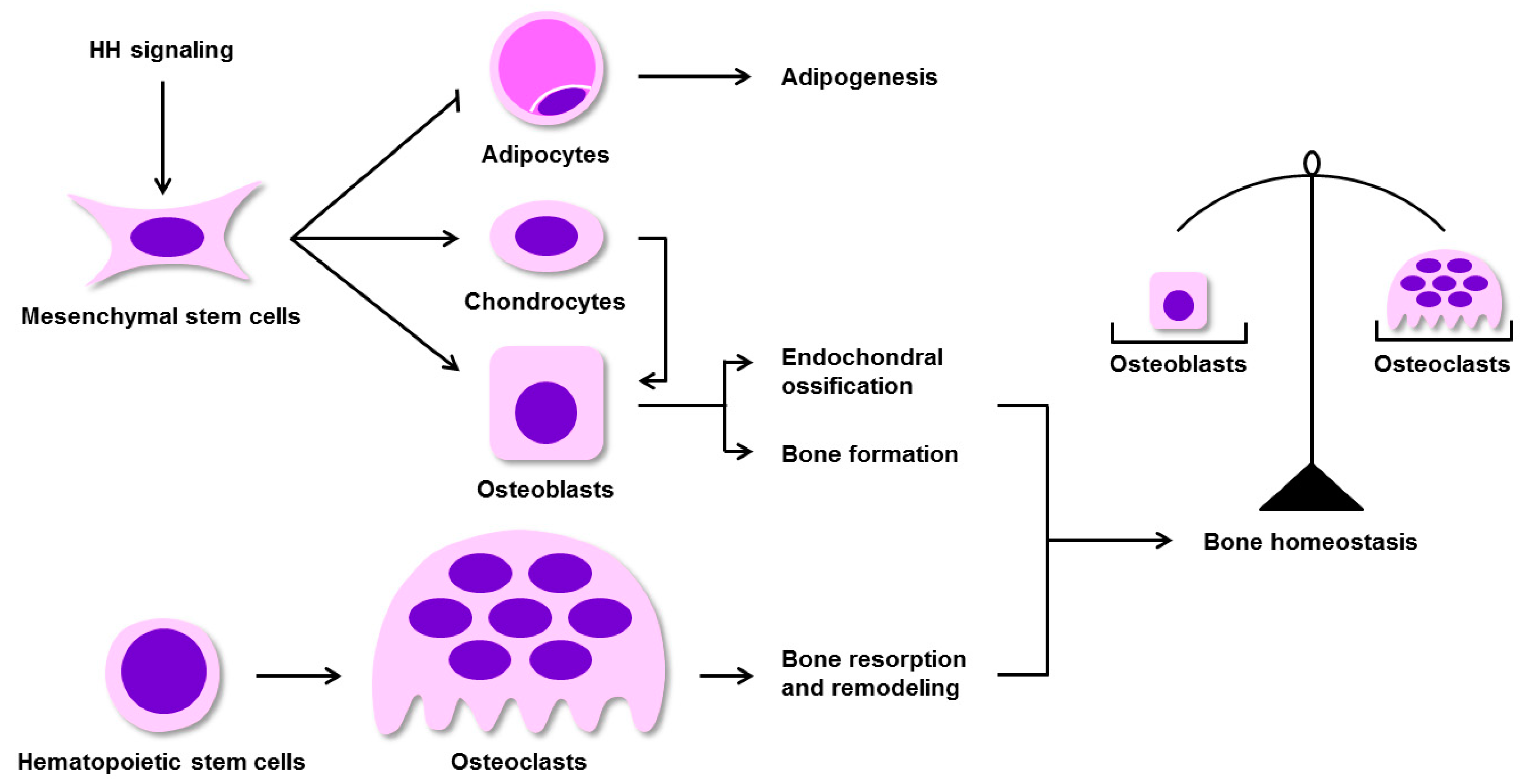
3. Regulation of BM-MSC Osteogenic Differentiation by HH Signaling in Mammals
4. Activation of HH Signaling Is Able to Increase Osteoblast Activity
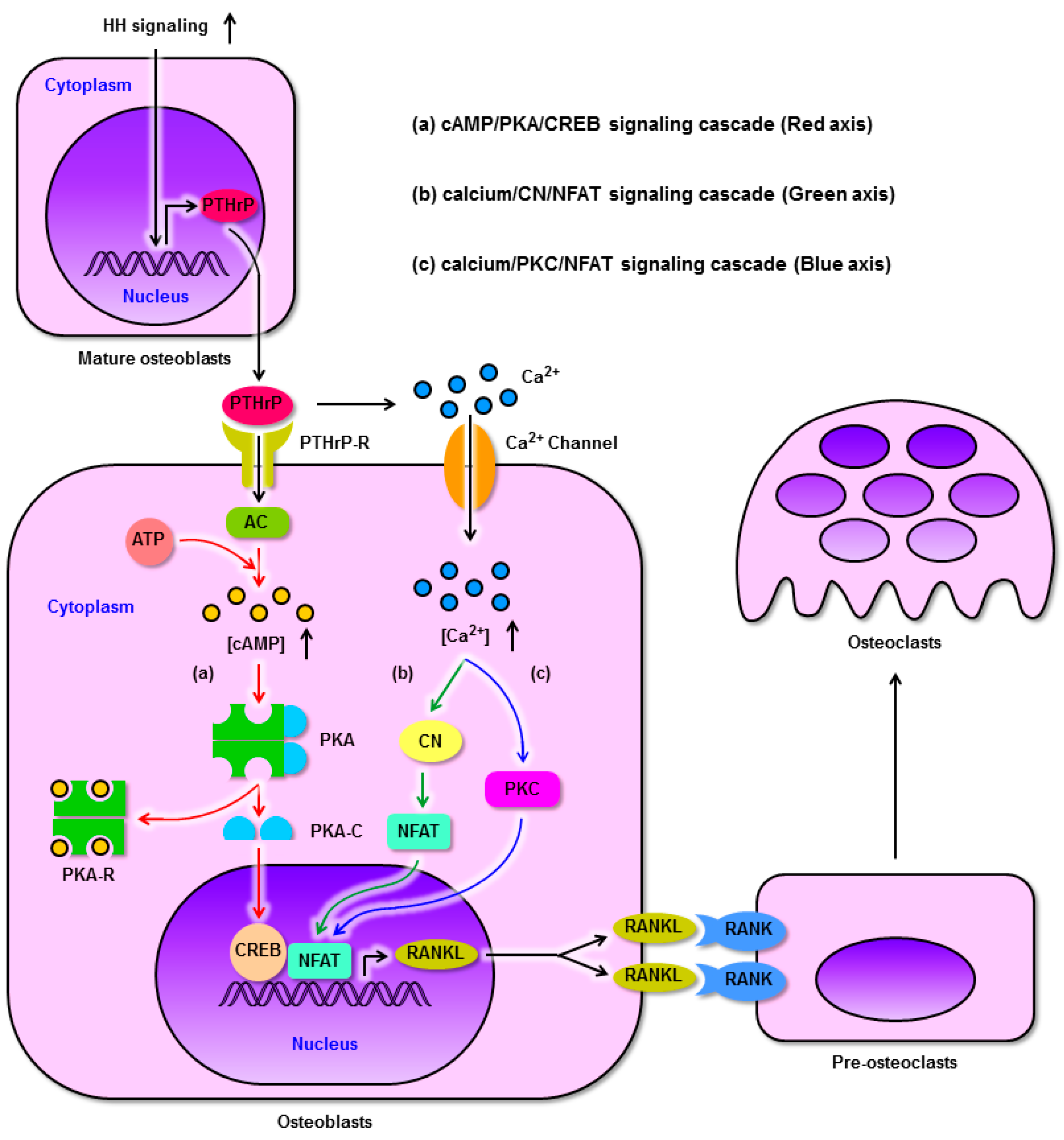
5. Activation of HH Signaling in Mature Osteoblasts Enhances Bone Resorption via Up-Regulation of RANKL
5.1. HH Signaling Promotes the Expression of RANKL through PTHrP
5.2. The Molecular Mechanisms by which PTHrP Regulates RANKL Expression
6. Pharmacologic Management of the OPG/RANK/RANKL Axis
7. The Effects of HH Signaling on Skeletal Homeostasis Remodeling and Repair
8. Conclusions and Future Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Dashti, M.; Peppelenbosch, M.P.; Rezaee, F. Hedgehog signalling as an antagonist of ageing and its associated diseases. Bioessays News Rev. Mol. Cell. Dev. Biol. 2012, 34, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Huang, H.X.; Huang, H.; Li, Z.T.; Lai, Y.Y. Hedgehog signaling pathway and osteoporosis. Zhongguo Gu Shang China J. Orthop. Traumatol. 2014, 27, 169–172. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Gradilla, A.-C.; Sanchez-Hernandez, D.; Brunt, L.; Scholpp, S. From top to bottom: Cell polarity in Hedgehog and Wnt trafficking. BMC Biol. 2018, 16, 37. [Google Scholar] [CrossRef]
- Yang, J.; Andre, P.; Ye, L.; Yang, Y.Z. The Hedgehog signalling pathway in bone formation. Int. J. Oral Sci. 2015, 7, 73–79. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharm. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Wilkinson, S.E.; Furic, L.; Buchanan, G.; Larsson, O.; Pedersen, J.; Frydenberg, M.; Risbridger, G.P.; Taylor, R.A. Hedgehog signaling is active in human prostate cancer stroma and regulates proliferation and differentiation of adjacent epithelium. Prostate 2013, 73, 1810–1823. [Google Scholar] [CrossRef]
- He, Y.; Guo, Q.; Cheng, Y.; Qu, Y.; Sun, L.; Kong, C.; Lei, L.; Zhang, G. Abnormal activation of the sonic hedgehog signaling pathway in endometriosis and its diagnostic potency. Fertil. Steril. 2018, 110, 128–136.e2. [Google Scholar] [CrossRef]
- Cannonier, S.A.; Sterling, J.A. The Role of Hedgehog Signaling in Tumor Induced Bone Disease. Cancers (Basel) 2015, 7, 1658–1683. [Google Scholar] [CrossRef]
- Lee, B.; Iwaniec, U.T.; Turner, R.T.; Lin, Y.W.; Clarke, B.L.; Gingery, A.; Wei, L.N. RIP140 in monocytes/macrophages regulates osteoclast differentiation and bone homeostasis. JCI Insight 2017, 2, e90517. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B. TNF and Bone Remodeling. Curr. Osteoporos. Rep. 2017, 15, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Roshandel, D.; Holliday, K.L.; Pye, S.R.; Boonen, S.; Borghs, H.; Vanderschueren, D.; Huhtaniemi, I.T.; Adams, J.E.; Ward, K.A.; Bartfai, G.; et al. Genetic variation in the RANKL/RANK/OPG signaling pathway is associated with bone turnover and bone mineral density in men. J. Bone Min. Res. 2010, 25, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Ledesma-Colunga, M.G.; Adán, N.; Ortiz, G.; Solís-Gutiérrez, M.; López-Barrera, F.; Martínez de la Escalera, G.; Clapp, C. Prolactin blocks the expression of receptor activator of nuclear factor κB ligand and reduces osteoclastogenesis and bone loss in murine inflammatory arthritis. Arthritis Res. Ther. 2017, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, S.; Zhao, Y.; Liu, D.; Liu, Y.; Chen, C.; Karray, S.; Shi, S.; Jin, Y. Osteoblast-induced osteoclast apoptosis by fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ. 2015, 22, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Menale, C.; Villa, A. The RANKL-RANK Axis: A Bone to Thymus Round Trip. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Bi, Y.; Wan, C.; Chuang, P.T.; Clemens, T.; Young, M.; Yang, Y. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev. Cell 2008, 14, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dong, Q.; Wang, Y.; Feng, Q.; Zhou, P.; Ou, X.; Meng, Q.; He, T.; Luo, J. Hedgehog signaling is involved in the BMP9-induced osteogenic differentiation of mesenchymal stem cells. Int. J. Mol. Med. 2015, 35, 1641–1650. [Google Scholar] [CrossRef]
- Ye, L.; Lou, F.; Yu, F.; Zhang, D.; Wang, C.; Wu, F.; Li, X.; Ping, Y.; Yang, X.; Yang, J.; et al. NUMB maintains bone mass by promoting degradation of PTEN and GLI1 via ubiquitination in osteoblasts. Bone Res. 2018, 6, 32. [Google Scholar] [CrossRef]
- Oliveira, F.S.; Bellesini, L.S.; Defino, H.L.; da Silva Herrero, C.F.; Beloti, M.M.; Rosa, A.L. Hedgehog signaling and osteoblast gene expression are regulated by purmorphamine in human mesenchymal stem cells. J. Cell Biochem. 2012, 113, 204–208. [Google Scholar] [CrossRef]
- Zhang, C. Molecular mechanisms of osteoblast-specific transcription factor Osterix effect on bone formation. Beijing Da Xue Xue Bao Yi Xue Ban 2012, 44, 659–665. [Google Scholar] [CrossRef]
- Ohata, Y.; Ozono, K. Bone and Stem Cells. The mechanism of osteogenic differentiation from mesenchymal stem cell. Clin. Calcium 2014, 24, 501–508. [Google Scholar]
- Komori, T. Roles of Runx2 in Skeletal Development. Adv. Exp. Med. Biol. 2017, 962, 83–93. [Google Scholar] [CrossRef]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef]
- Fernandes-Silva, H.; Correia-Pinto, J.; Moura, R.S. Canonical Sonic Hedgehog Signaling in Early Lung Development. J. Dev. Biol. 2017, 5, 3. [Google Scholar] [CrossRef]
- Dimou, A.; Bamias, A.; Gogas, H.; Syrigos, K. Inhibition of the Hedgehog pathway in lung cancer. Lung Cancer 2019, 133, 56–61. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Borycki, A.G.; Mendham, L.; Emerson, C.P., Jr. Control of somite patterning by Sonic hedgehog and its downstream signal response genes. Development 1998, 125, 777–790. [Google Scholar]
- Ho, K.S.; Scott, M.P. Sonic hedgehog in the nervous system: Functions, modifications and mechanisms. Curr. Opin. Neurobiol. 2002, 12, 57–63. [Google Scholar] [CrossRef]
- van den Brink, G.R.; Hardwick, J.C.; Nielsen, C.; Xu, C.; ten Kate, F.J.; Glickman, J.; van Deventer, S.J.; Roberts, D.J.; Peppelenbosch, M.P. Sonic hedgehog expression correlates with fundic gland differentiation in the adult gastrointestinal tract. Gut 2002, 51, 628–633. [Google Scholar] [CrossRef]
- Ellis, T.; Smyth, I.; Riley, E.; Bowles, J.; Adolphe, C.; Rothnagel, J.A.; Wicking, C.; Wainwright, B.J. Overexpression of Sonic Hedgehog suppresses embryonic hair follicle morphogenesis. Dev. Biol. 2003, 263, 203–215. [Google Scholar] [CrossRef]
- James, A.W. Review of Signaling Pathways Governing MSC Osteogenic and Adipogenic Differentiation. Scientifica 2013, 2013, 684736. [Google Scholar] [CrossRef]
- O’Hara, W.A.; Azar, W.J.; Behringer, R.R.; Renfree, M.B.; Pask, A.J. Desert hedgehog is a mammal-specific gene expressed during testicular and ovarian development in a marsupial. BMC Dev. Biol. 2011, 11, 72. [Google Scholar] [CrossRef]
- Maeda, Y.; Nakamura, E.; Nguyen, M.T.; Suva, L.J.; Swain, F.L.; Razzaque, M.S.; Mackem, S.; Lanske, B. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. USA 2007, 104, 6382–6387. [Google Scholar] [CrossRef]
- Bai, Y.; Bai, Y.; Dong, J.; Li, Q.; Jin, Y.; Chen, B.; Zhou, M. Hedgehog Signaling in Pancreatic Fibrosis and Cancer. Medicine 2016, 95, e2996. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, L.; Chi, C.; Lan, W.; Su, Y. The emerging roles of phosphatases in Hedgehog pathway. Cell Commun. Signal 2017, 15, 35. [Google Scholar] [CrossRef]
- Murone, M.; Rosenthal, A.; de Sauvage, F.J. Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr. Biol. 1999, 9, 76–84. [Google Scholar] [CrossRef]
- Dey, J.; Ditzler, S.; Knoblaugh, S.E.; Hatton, B.A.; Schelter, J.M.; Cleary, M.A.; Mecham, B.; Rorke-Adams, L.B.; Olson, J.M. A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol. Cell Biol. 2012, 32, 4104–4115. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Wilson, C.W.; Chuang, P.T. Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 2010, 137, 2079–2094. [Google Scholar] [CrossRef]
- Han, Y.; Xiong, Y.; Shi, X.; Wu, J.; Zhao, Y.; Jiang, J. Regulation of Gli ciliary localization and Hedgehog signaling by the PY-NLS/karyopherin-beta2 nuclear import system. PLoS Biol. 2017, 15, e2002063. [Google Scholar] [CrossRef]
- Kasper, M.; Regl, G.; Frischauf, A.M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Cox, B.; Briscoe, J.; Ulloa, F. SUMOylation by Pias1 regulates the activity of the Hedgehog dependent Gli transcription factors. PLoS ONE 2010, 5, e11996. [Google Scholar] [CrossRef]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef]
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef]
- Caro, I.; Low, J.A. The role of the hedgehog signaling pathway in the development of basal cell carcinoma and opportunities for treatment. Clin. Cancer Res. 2010, 16, 3335–3339. [Google Scholar] [CrossRef]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef]
- Zhao, Y.; Tong, C.; Jiang, J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 2007, 450, 252–258. [Google Scholar] [CrossRef]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef]
- Wang, L.; Jie, Q.; Yang, L. Chondrocytes-osteoblast transition in endochondral ossification. Ann. Jt. 2017, 2. [Google Scholar] [CrossRef]
- Lin, H.; Sohn, J.; Shen, H.; Langhans, M.T.; Tuan, R.S. Bone marrow mesenchymal stem cells: Aging and tissue engineering applications to enhance bone healing. Biomaterials 2019, 203, 96–110. [Google Scholar] [CrossRef]
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 2005, 8, 739–750. [Google Scholar] [CrossRef]
- Miclea, R.L.; Karperien, M.; Bosch, C.A.; van der Horst, G.; van der Valk, M.A.; Kobayashi, T.; Kronenberg, H.M.; Rawadi, G.; Akcakaya, P.; Lowik, C.W.; et al. Adenomatous polyposis coli-mediated control of beta-catenin is essential for both chondrogenic and osteogenic differentiation of skeletal precursors. BMC Dev. Biol. 2009, 9, 26. [Google Scholar] [CrossRef]
- Zhang, C. Transcriptional regulation of bone formation by the osteoblast-specific transcription factor Osx. J. Orthop. Surg. Res. 2010, 5, 37. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, J.; Karner, C.M.; Long, F. Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, 4678–4683. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef]
- Amano, K.; Densmore, M.J.; Lanske, B. Conditional Deletion of Indian Hedgehog in Limb Mesenchyme Results in Complete Loss of Growth Plate Formation but Allows Mature Osteoblast Differentiation. J. Bone Min. Res. 2015, 30, 2262–2272. [Google Scholar] [CrossRef]
- Komori, T. Regulation of osteoblast differentiation by transcription factors. J. Cell Biochem. 2006, 99, 1233–1239. [Google Scholar] [CrossRef]
- Baek, W.Y.; Lee, M.A.; Jung, J.W.; Kim, S.Y.; Akiyama, H.; de Crombrugghe, B.; Kim, J.E. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. J. Bone Min. Res. 2009, 24, 1055–1065. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Z.; Duan, C.; Liu, W.; Sun, J.; Han, B. Role of TCF/LEF Transcription Factors in Bone Development and Osteogenesis. Int. J. Med. Sci. 2018, 15, 1415–1422. [Google Scholar] [CrossRef]
- Pang, X.G.; Cong, Y.; Bao, N.R.; Li, Y.G.; Zhao, J.N. Quercetin Stimulates Bone Marrow Mesenchymal Stem Cell Differentiation through an Estrogen Receptor-Mediated Pathway. BioMed Res. Int. 2018, 2018, 4178021. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, Y.; Fu, Q.; Dong, Y. Osterix is required for Sonic hedgehog-induced osteoblastic MC3T3-E1 cell differentiation. Cell Biochem. Biophys. 2012, 64, 169–176. [Google Scholar] [CrossRef]
- Nakamura, T.; Naruse, M.; Chiba, Y.; Komori, T.; Sasaki, K.; Iwamoto, M.; Fukumoto, S. Novel hedgehog agonists promote osteoblast differentiation in mesenchymal stem cells. J. Cell Physiol. 2015, 230, 922–929. [Google Scholar] [CrossRef]
- Shimoyama, A.; Wada, M.; Ikeda, F.; Hata, K.; Matsubara, T.; Nifuji, A.; Noda, M.; Amano, K.; Yamaguchi, A.; Nishimura, R.; et al. Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol. Biol. Cell 2007, 18, 2411–2418. [Google Scholar] [CrossRef]
- Tu, X.; Joeng, K.S.; Long, F. Indian hedgehog requires additional effectors besides Runx2 to induce osteoblast differentiation. Dev. Biol. 2012, 362, 76–82. [Google Scholar] [CrossRef]
- Nakamura, T.; Aikawa, T.; Iwamoto-Enomoto, M.; Iwamoto, M.; Higuchi, Y.; Pacifici, M.; Kinto, N.; Yamaguchi, A.; Noji, S.; Kurisu, K.; et al. Induction of osteogenic differentiation by hedgehog proteins. Biochem. Biophys. Res. Commun. 1997, 237, 465–469. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Komori, T.; Suda, T. Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehogs, and Cbfa1. Endocr. Rev. 2000, 21, 393–411. [Google Scholar] [CrossRef]
- Hojo, H.; Ohba, S.; Taniguchi, K.; Shirai, M.; Yano, F.; Saito, T.; Ikeda, T.; Nakajima, K.; Komiyama, Y.; Nakagata, N.; et al. Hedgehog-Gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium. J. Biol. Chem. 2013, 288, 9924–9932. [Google Scholar] [CrossRef]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar]
- Horikiri, Y.; Shimo, T.; Kurio, N.; Okui, T.; Matsumoto, K.; Iwamoto, M.; Sasaki, A. Sonic hedgehog regulates osteoblast function by focal adhesion kinase signaling in the process of fracture healing. PLoS ONE 2013, 8, e76785. [Google Scholar] [CrossRef]
- Matsumoto, K.; Shimo, T.; Kurio, N.; Okui, T.; Obata, K.; Masui, M.; Pang, P.; Horikiri, Y.; Sasaki, A. Expression and Role of Sonic Hedgehog in the Process of Fracture Healing with Aging. In Vivo 2016, 30, 99–105. [Google Scholar]
- Baht, G.S.; Silkstone, D.; Nadesan, P.; Whetstone, H.; Alman, B.A. Activation of hedgehog signaling during fracture repair enhances osteoblastic-dependent matrix formation. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2014, 32, 581–586. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Bi, H.; Chen, X.; Gao, S.; Yu, X.; Xiao, J.; Zhang, B.; Liu, X.; Dai, M. Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review. Front. Med. 2017, 4, 234. [Google Scholar] [CrossRef]
- Otsuka, A.; Dreier, J.; Cheng, P.F.; Nageli, M.; Lehmann, H.; Felderer, L.; Frew, I.J.; Matsushita, S.; Levesque, M.P.; Dummer, R. Hedgehog pathway inhibitors promote adaptive immune responses in basal cell carcinoma. Clin. Cancer Res. 2015, 21, 1289–1297. [Google Scholar] [CrossRef]
- Martin, T.J.; Moseley, J.M.; Williams, E.D. Parathyroid hormone-related protein: Hormone and cytokine. J. Endocrinol. 1997, 154, S23–S37. [Google Scholar]
- Zhang, W.; Chen, J.; Zhang, S.; Ouyang, H.W. Inhibitory function of parathyroid hormone-related protein on chondrocyte hypertrophy: The implication for articular cartilage repair. Arthritis Res. 2012, 14, 221. [Google Scholar] [CrossRef]
- Martin, T.J. Parathyroid Hormone-Related Protein, Its Regulation of Cartilage and Bone Development, and Role in Treating Bone Diseases. Physiol. Rev. 2016, 96, 831–871. [Google Scholar] [CrossRef]
- Karaplis, A.C. PTHrP: Novel roles in skeletal biology. Curr. Pharm. Des. 2001, 7, 655–670. [Google Scholar] [CrossRef]
- Datta, N.S.; Abou-Samra, A.B. PTH and PTHrP signaling in osteoblasts. Cell Signal 2009, 21, 1245–1254. [Google Scholar] [CrossRef]
- Leder, B.Z. Parathyroid Hormone and Parathyroid Hormone-Related Protein Analogs in Osteoporosis Therapy. Curr. Osteoporos. Rep. 2017, 15, 110–119. [Google Scholar] [CrossRef]
- Kronenberg, H.M. PTHrP and skeletal development. Ann. N. Y. Acad Sci. 2006, 1068, 1–13. [Google Scholar] [CrossRef]
- Ono, W.; Sakagami, N.; Nishimori, S.; Ono, N.; Kronenberg, H.M. Parathyroid hormone receptor signalling in osterix-expressing mesenchymal progenitors is essential for tooth root formation. Nat. Commun. 2016, 7, 11277. [Google Scholar] [CrossRef]
- Thomas, R.J.; Guise, T.A.; Yin, J.J.; Elliott, J.; Horwood, N.J.; Martin, T.J.; Gillespie, M.T. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology 1999, 140, 4451–4458. [Google Scholar] [CrossRef]
- Das, S.; Samant, R.S.; Shevde, L.A. Hedgehog signaling induced by breast cancer cells promotes osteoclastogenesis and osteolysis. J. Biol. Chem. 2011, 286, 9612–9622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, Q.; Wang, Y.; Leung, P.S.; Mak, K.K. Hedgehog signaling in bone regulates whole-body energy metabolism through a bone-adipose endocrine relay mediated by PTHrP and adiponectin. Cell Death Differ. 2017, 24, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Takahashi, N.; Udagawa, N. New roles of osteoblasts involved in osteoclast differentiation. World J. Orthop. 2012, 3, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Dong, Y.; Huang, X.; Chen, P.; Guo, F.; Chen, A.; Huang, S. Pioglitazone affects the OPG/RANKL/RANK system and increase osteoclastogenesis. Mol. Med. Rep. 2016, 14, 2289–2296. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Branco, J.C. Rank/Rankl/opg: Literature review. Acta Reum. Port. 2011, 36, 209–218. [Google Scholar]
- Vega, D.; Maalouf, N.M.; Sakhaee, K. CLINICAL Review #: The role of receptor activator of nuclear factor-kappaB (RANK)/RANK ligand/osteoprotegerin: Clinical implications. J. Clin. Endocrinol. Metab. 2007, 92, 4514–4521. [Google Scholar] [CrossRef]
- Tat, S.K.; Pelletier, J.P.; Velasco, C.R.; Padrines, M.; Martel-Pelletier, J. New perspective in osteoarthritis: The OPG and RANKL system as a potential therapeutic target? Keio J. Med. 2009, 58, 29–40. [Google Scholar] [CrossRef]
- Park, H.J.; Baek, K.; Baek, J.H.; Kim, H.R. The cooperation of CREB and NFAT is required for PTHrP-induced RANKL expression in mouse osteoblastic cells. J. Cell Physiol. 2015, 230, 667–679. [Google Scholar] [CrossRef]
- Kvissel, A.K.; Orstavik, S.; Oistad, P.; Rootwelt, T.; Jahnsen, T.; Skalhegg, B.S. Induction of Cbeta splice variants and formation of novel forms of protein kinase A type II holoenzymes during retinoic acid-induced differentiation of human NT2 cells. Cell Signal 2004, 16, 577–587. [Google Scholar] [CrossRef]
- Jilka, R.L. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 2007, 40, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, E.M. Anabolic effects of intermittent PTH on osteoblasts. Curr. Mol. Pharm. 2012, 5, 127–134. [Google Scholar] [CrossRef]
- Sands, W.A.; Palmer, T.M. Regulating gene transcription in response to cyclic AMP elevation. Cell Signal 2008, 20, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Tascau, L.; Gardner, T.; Anan, H.; Yongpravat, C.; Cardozo, C.P.; Bauman, W.A.; Lee, F.Y.; Oh, D.S.; Tawfeek, H.A. Activation of Protein Kinase A in Mature Osteoblasts Promotes a Major Bone Anabolic Response. Endocrinology 2016, 157, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Manolagas, S.C.; O’Brien, C.A. Parathyroid hormone controls receptor activator of NF-kappaB ligand gene expression via a distant transcriptional enhancer. Mol. Cell Biol. 2006, 26, 6453–6468. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yamazaki, M.; Shevde, N.K.; Pike, J.W. Transcriptional control of receptor activator of nuclear factor-kappaB ligand by the protein kinase A activator forskolin and the transmembrane glycoprotein 130-activating cytokine, oncostatin M, is exerted through multiple distal enhancers. Mol. Endocrinol. 2007, 21, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Bae, O.Y.; Baek, K.H.; Kwon, A.; Hwang, H.R.; Qadir, A.S.; Park, H.J.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. High extracellular calcium-induced NFATc3 regulates the expression of receptor activator of NF-kappaB ligand in osteoblasts. Bone 2011, 49, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Rao, A. Signaling to gene expression: Calcium, calcineurin and NFAT. Nat. Immunol. 2009, 10, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Musson, R.E.; Cobbaert, C.M.; Smit, N.P. Molecular diagnostics of calcineurin-related pathologies. Clin. Chem. 2012, 58, 511–522. [Google Scholar] [CrossRef]
- Takami, M.; Takahashi, N.; Udagawa, N.; Miyaura, C.; Suda, K.; Woo, J.T.; Martin, T.J.; Nagai, K.; Suda, T. Intracellular calcium and protein kinase C mediate expression of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in osteoblasts. Endocrinology 2000, 141, 4711–4719. [Google Scholar] [CrossRef]
- Philbrick, W.M.; Wysolmerski, J.J.; Galbraith, S.; Holt, E.; Orloff, J.J.; Yang, K.H.; Vasavada, R.C.; Weir, E.C.; Broadus, A.E.; Stewart, A.F. Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol. Rev. 1996, 76, 127–173. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, R.; Kitazawa, S.; Maeda, S.; Kobayashi, A. Expression of parathyroid hormone-related protein (PTHrP) in parathyroid tissue under normal and pathological conditions. Histol. Histopathol. 2002, 17, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Karaplis, A.C.; Huang, D.C.; Siegel, P.M.; Camirand, A.; Yang, X.F.; Muller, W.J.; Kremer, R. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J. Clin. Investig. 2011, 121, 4655–4669. [Google Scholar] [CrossRef] [PubMed]
- Camirand, A.; Goltzman, D.; Gupta, A.; Kaouass, M.; Panda, D.; Karaplis, A. The Role of Parathyroid Hormone-Related Protein (PTHrP) in Osteoblast Response to Microgravity: Mechanistic Implications for Osteoporosis Development. PLoS ONE 2016, 11, e0160034. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.L. Expression of mRNAs for the alpha 1 subunit of voltage-gated calcium channels in human osteoblast-like cell lines and in normal human osteoblasts. Calcif Tissue Int. 2000, 66, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.J.; Lees, R.L.; Trujillo, E.; Martin-Vasallo, P.; Heersche, J.N.; Mobasheri, A. ATPase pumps in osteoclasts and osteoblasts. Int. J. Biochem. Cell Biol. 2002, 34, 459–476. [Google Scholar] [CrossRef]
- Zayzafoon, M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J. Cell Biochem. 2006, 97, 56–70. [Google Scholar] [CrossRef]
- Koch, F.P.; Merkel, C.; Ziebart, T.; Smeets, R.; Walter, C.; Al-Nawas, B. Influence of bisphosphonates on the osteoblast RANKL and OPG gene expression in vitro. Clin. Oral Investig. 2012, 16, 79–86. [Google Scholar] [CrossRef]
- Soysa, N.S.; Alles, N.; Aoki, K.; Ohya, K. Osteoclast formation and differentiation: An overview. J. Med. Dent. Sci. 2012, 59, 65–74. [Google Scholar] [CrossRef]
- Peng, X.; Guo, W.; Ren, T.; Lou, Z.; Lu, X.; Zhang, S.; Lu, Q.; Sun, Y. Differential expression of the RANKL/RANK/OPG system is associated with bone metastasis in human non-small cell lung cancer. PLoS ONE 2013, 8, e58361. [Google Scholar] [CrossRef]
- Streicher, C.; Heyny, A.; Andrukhova, O.; Haigl, B.; Slavic, S.; Schuler, C.; Kollmann, K.; Kantner, I.; Sexl, V.; Kleiter, M.; et al. Estrogen Regulates Bone Turnover by Targeting RANKL Expression in Bone Lining Cells. Sci. Rep. 2017, 7, 6460. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Kikkawa, U.; Nishizuka, Y. The family of protein kinase C and membrane lipid mediators. J. Diabetes Complicat. 2002, 16, 4–8. [Google Scholar] [CrossRef]
- Shin, J.; Jang, H.; Lin, J.; Lee, S.Y. PKCbeta positively regulates RANKL-induced osteoclastogenesis by inactivating GSK-3beta. Mol. Cells 2014, 37, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Nardone, V.; D’Asta, F.; Brandi, M.L. Pharmacological management of osteogenesis. Clinics 2014, 69, 438–446. [Google Scholar] [CrossRef]
- Hanley, D.A.; Adachi, J.D.; Bell, A.; Brown, V. Denosumab: Mechanism of action and clinical outcomes. Int. J. Clin. Pr. 2012, 66, 1139–1146. [Google Scholar] [CrossRef]
- Cummings, S.R.; San Martin, J.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Lewiecki, E.M.; Cohen, S.B.; Bolognese, M.A.; Woodson, G.C.; Moffett, A.H.; Peacock, M.; Miller, P.D.; Lederman, S.N.; Chesnut, C.H.; et al. Denosumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2006, 354, 821–831. [Google Scholar] [CrossRef]
- Kashiwagi, M.; Hojo, H.; Kitaura, Y.; Maeda, Y.; Aini, H.; Takato, T.; Chung, U.I.; Ohba, S. Local administration of a hedgehog agonist accelerates fracture healing in a mouse model. Biochem. Biophys. Res. Commun. 2016, 479, 772–778. [Google Scholar] [CrossRef]
- Lee, S.; Wang, C.; Pan, H.C.; Shrestha, S.; Meyers, C.; Ding, C.; Shen, J.; Chen, E.; Lee, M.; Soo, C.; et al. Combining Smoothened Agonist and NEL-Like Protein-1 Enhances Bone Healing. Plast Reconstr. Surg. 2017, 139, 1385–1396. [Google Scholar] [CrossRef]
- Kha, H.T.; Basseri, B.; Shouhed, D.; Richardson, J.; Tetradis, S.; Hahn, T.J.; Parhami, F. Oxysterols regulate differentiation of mesenchymal stem cells: Pro-bone and anti-fat. J. Bone Min. Res. 2004, 19, 830–840. [Google Scholar] [CrossRef]
- Hokugo, A.; Sorice, S.; Parhami, F.; Yalom, A.; Li, A.; Zuk, P.; Jarrahy, R. A novel oxysterol promotes bone regeneration in rabbit cranial bone defects. J. Tissue Eng. Regen. Med. 2016, 10, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Hokugo, A.; Segovia, L.A.; Yalom, A.; Rezzadeh, K.; Zhou, S.; Zhang, Z.; Parhami, F.; Stappenbeck, F.; Jarrahy, R. Oxy133, a novel osteogenic agent, promotes bone regeneration in an intramembranous bone-healing model. J. Tissue Eng. Regen. Med. 2017, 11, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Castrogiovanni, P.; Trovato, F.M.; Szychlinska, M.A.; Nsir, H.; Imbesi, R.; Musumeci, G. The importance of physical activity in osteoporosis. From the molecular pathways to the clinical evidence. Histol. Histopathol. 2016, 31, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef]
- Wen, X.; Lai, C.K.; Evangelista, M.; Hongo, J.A.; de Sauvage, F.J.; Scales, S.J. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell Biol. 2010, 30, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Liu, A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 14751–14756. [Google Scholar] [CrossRef]
- Hilton, M.J.; Tu, X.; Cook, J.; Hu, H.; Long, F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development 2005, 132, 4339–4351. [Google Scholar] [CrossRef] [PubMed]
- Alman, B.A. The role of hedgehog signalling in skeletal health and disease. Nat. Rev. Rheumatol. 2015, 11, 552–560. [Google Scholar] [CrossRef]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef]
- Ali, S.A.; Al-Jazrawe, M.; Ma, H.; Whetstone, H.; Poon, R.; Farr, S.; Naples, M.; Adeli, K.; Alman, B.A. Regulation of Cholesterol Homeostasis by Hedgehog Signaling in Osteoarthritic Cartilage. Arthritis Rheumatol. 2016, 68, 127–137. [Google Scholar] [CrossRef]
- Tanabe, S. Signaling involved in stem cell reprogramming and differentiation. World J. Stem Cells 2015, 7, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Kazmers, N.H.; McKenzie, J.A.; Shen, T.S.; Long, F.; Silva, M.J. Hedgehog signaling mediates woven bone formation and vascularization during stress fracture healing. Bone 2015, 81, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Dohle, E.; Fuchs, S.; Kolbe, M.; Hofmann, A.; Schmidt, H.; Kirkpatrick, C.J. Sonic hedgehog promotes angiogenesis and osteogenesis in a coculture system consisting of primary osteoblasts and outgrowth endothelial cells. Tissue Eng. Part A 2010, 16, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.A.; Maschhoff, C.; Liu, X.; Migotsky, N.; Silva, M.J.; Gardner, M.J. Activation of hedgehog signaling by systemic agonist improves fracture healing in aged mice. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2019, 37, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Tomimori, Y.; Mori, K.; Koide, M.; Nakamichi, Y.; Ninomiya, T.; Udagawa, N.; Yasuda, H. Evaluation of pharmaceuticals with a novel 50-h animal model of bone loss. J. Bone Min. Res. 2009, 24, 1194–1205. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, W.-T.; Du, D.-H.; Gao, R.-J.; Yu, C.-W.; Jia, Y.; Jia, Z.-F.; Wang, C.-J. Regulation of Hedgehog signaling Offers A Novel Perspective for Bone Homeostasis Disorder Treatment. Int. J. Mol. Sci. 2019, 20, 3981. https://doi.org/10.3390/ijms20163981
Lv W-T, Du D-H, Gao R-J, Yu C-W, Jia Y, Jia Z-F, Wang C-J. Regulation of Hedgehog signaling Offers A Novel Perspective for Bone Homeostasis Disorder Treatment. International Journal of Molecular Sciences. 2019; 20(16):3981. https://doi.org/10.3390/ijms20163981
Chicago/Turabian StyleLv, Wen-Ting, Dong-Hua Du, Rui-Juan Gao, Chun-Wei Yu, Yan Jia, Zhi-Feng Jia, and Chun-Jie Wang. 2019. "Regulation of Hedgehog signaling Offers A Novel Perspective for Bone Homeostasis Disorder Treatment" International Journal of Molecular Sciences 20, no. 16: 3981. https://doi.org/10.3390/ijms20163981
APA StyleLv, W.-T., Du, D.-H., Gao, R.-J., Yu, C.-W., Jia, Y., Jia, Z.-F., & Wang, C.-J. (2019). Regulation of Hedgehog signaling Offers A Novel Perspective for Bone Homeostasis Disorder Treatment. International Journal of Molecular Sciences, 20(16), 3981. https://doi.org/10.3390/ijms20163981