Hydrogen Sulfide Attenuates Hydrogen Peroxide-Induced Injury in Human Lung Epithelial A549 Cells


Abstract
1. Introduction
2. Results
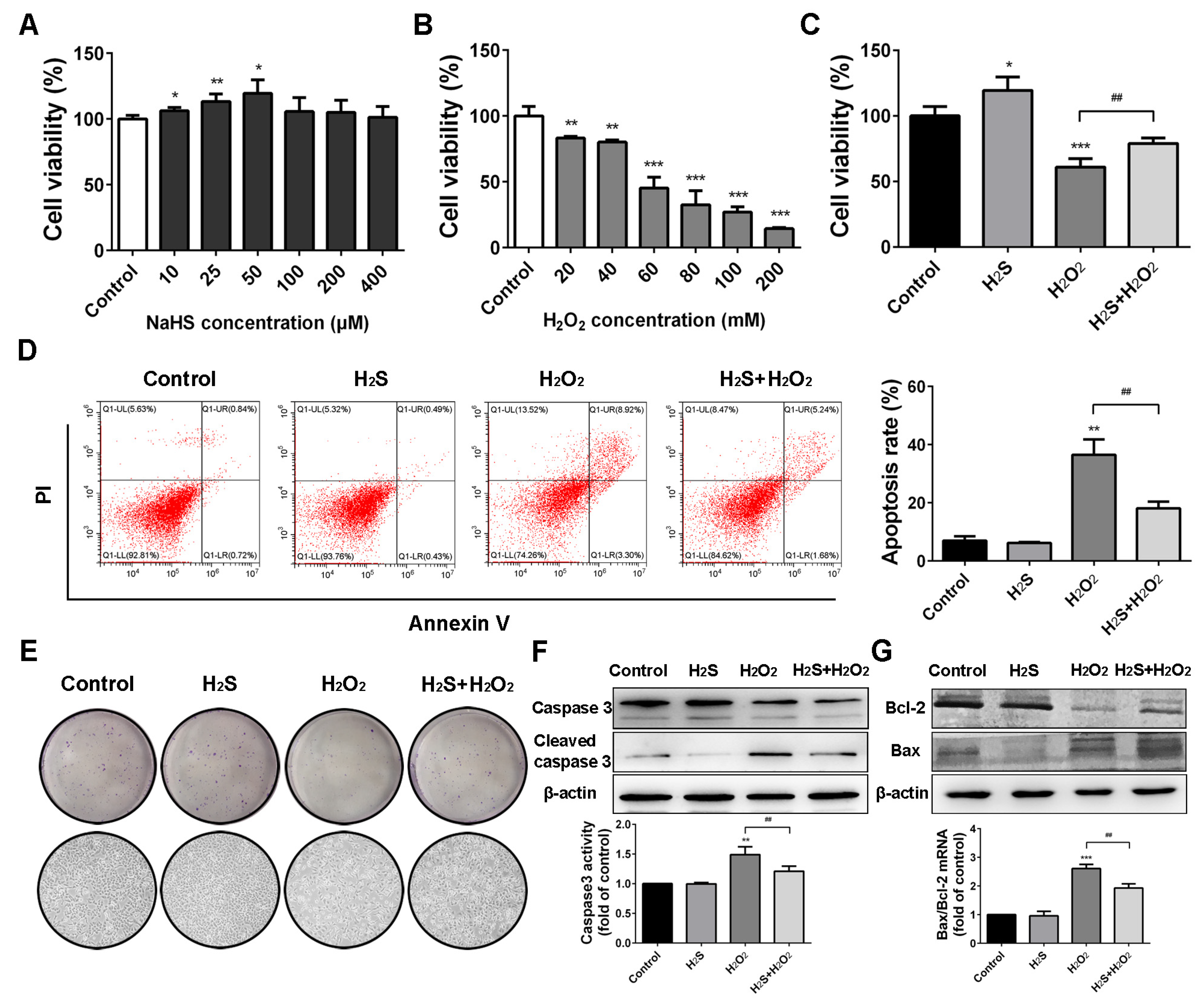
2.1. Protective Effect of H2S on Human Lung Epithelial Cells against H2O2-Induced Apoptosis
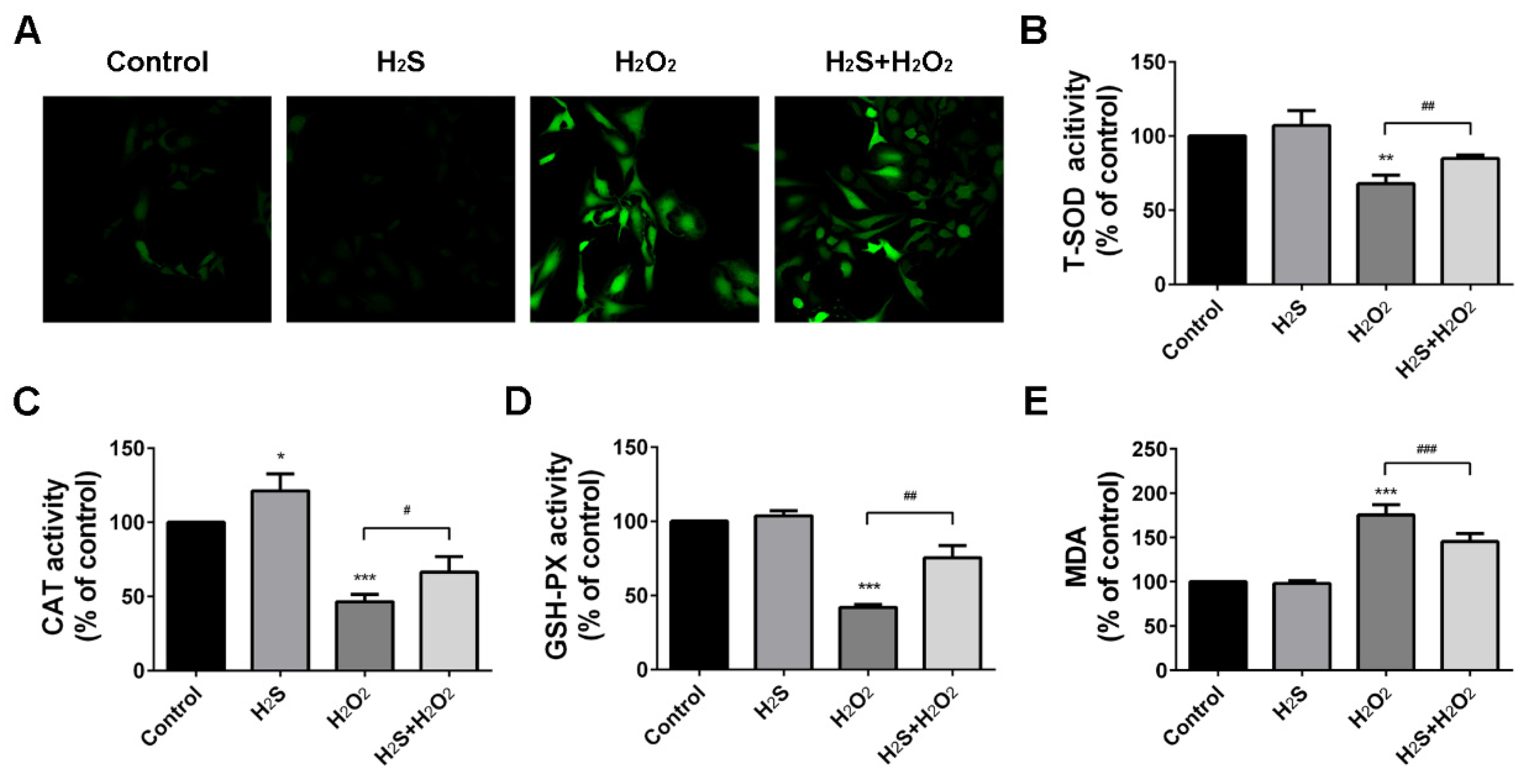
2.2. Protection Effect of H2S on Reactive Oxygen Species Injury
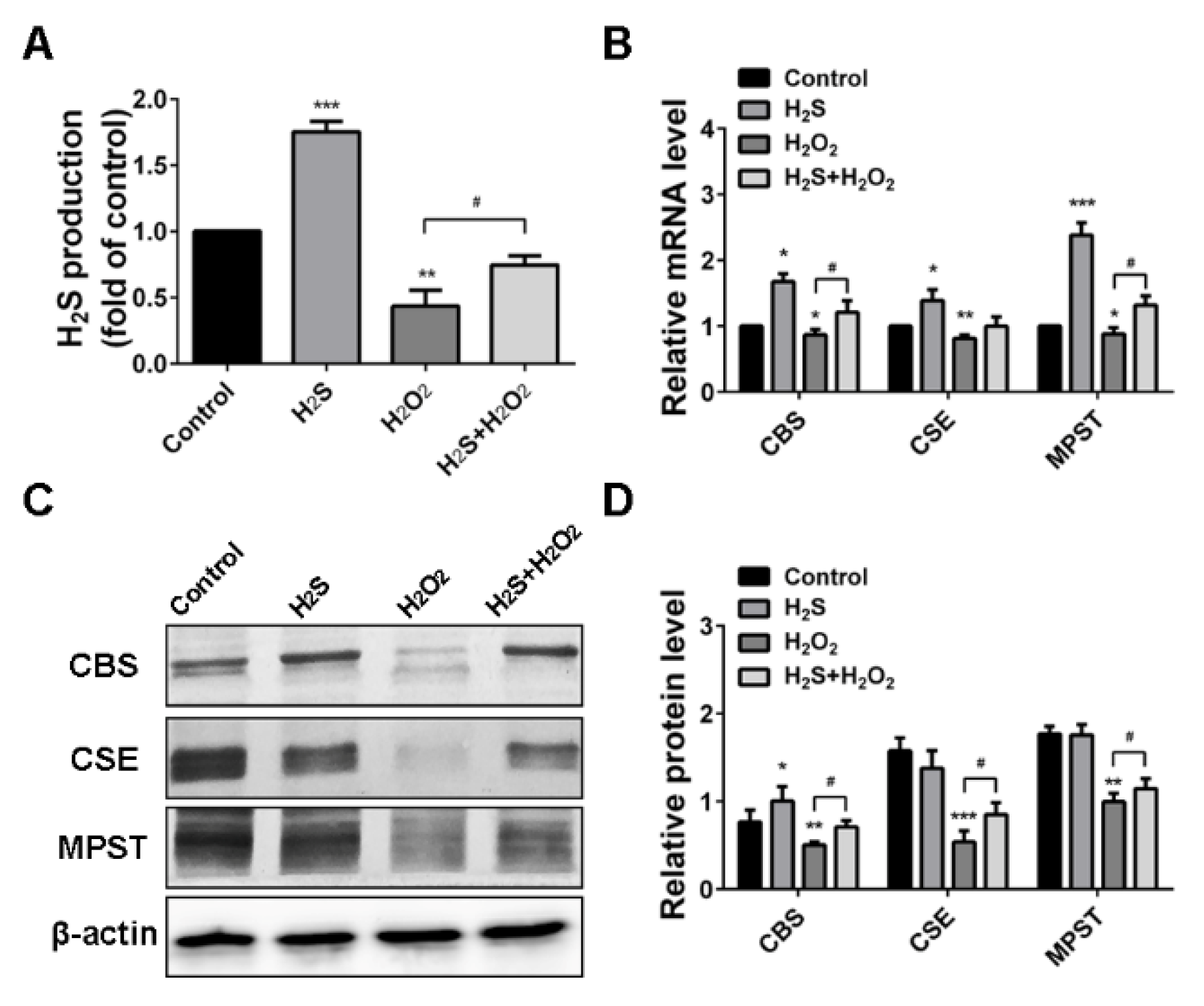
2.3. H2O2 Suppressed Endogenous H2S Production in A549 Cells
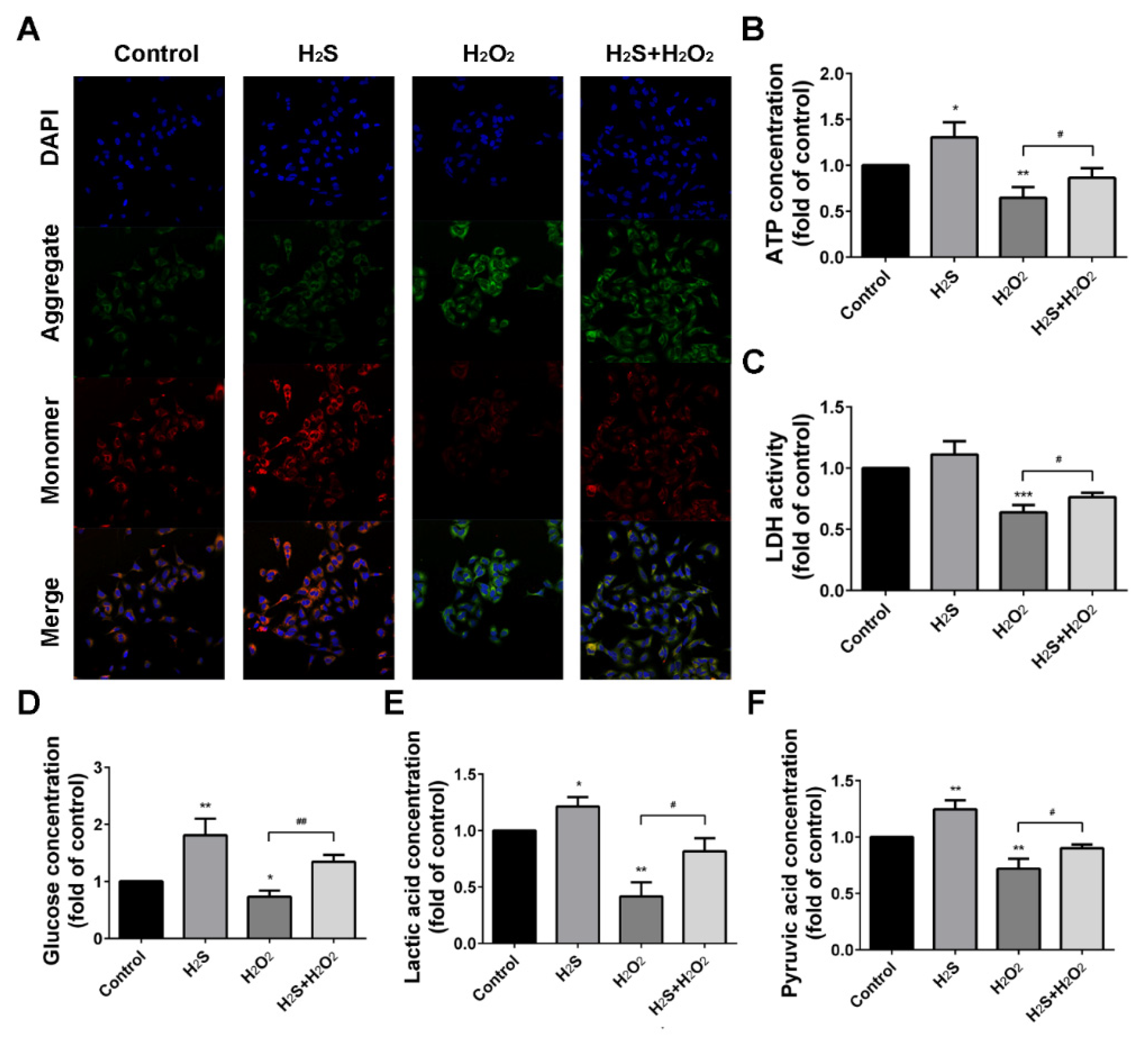
2.4. Effect of H2S on H2O2 Injury A549 Cells in Mitochondrial Membrane Potential (Δψ) and Energy Metabolism
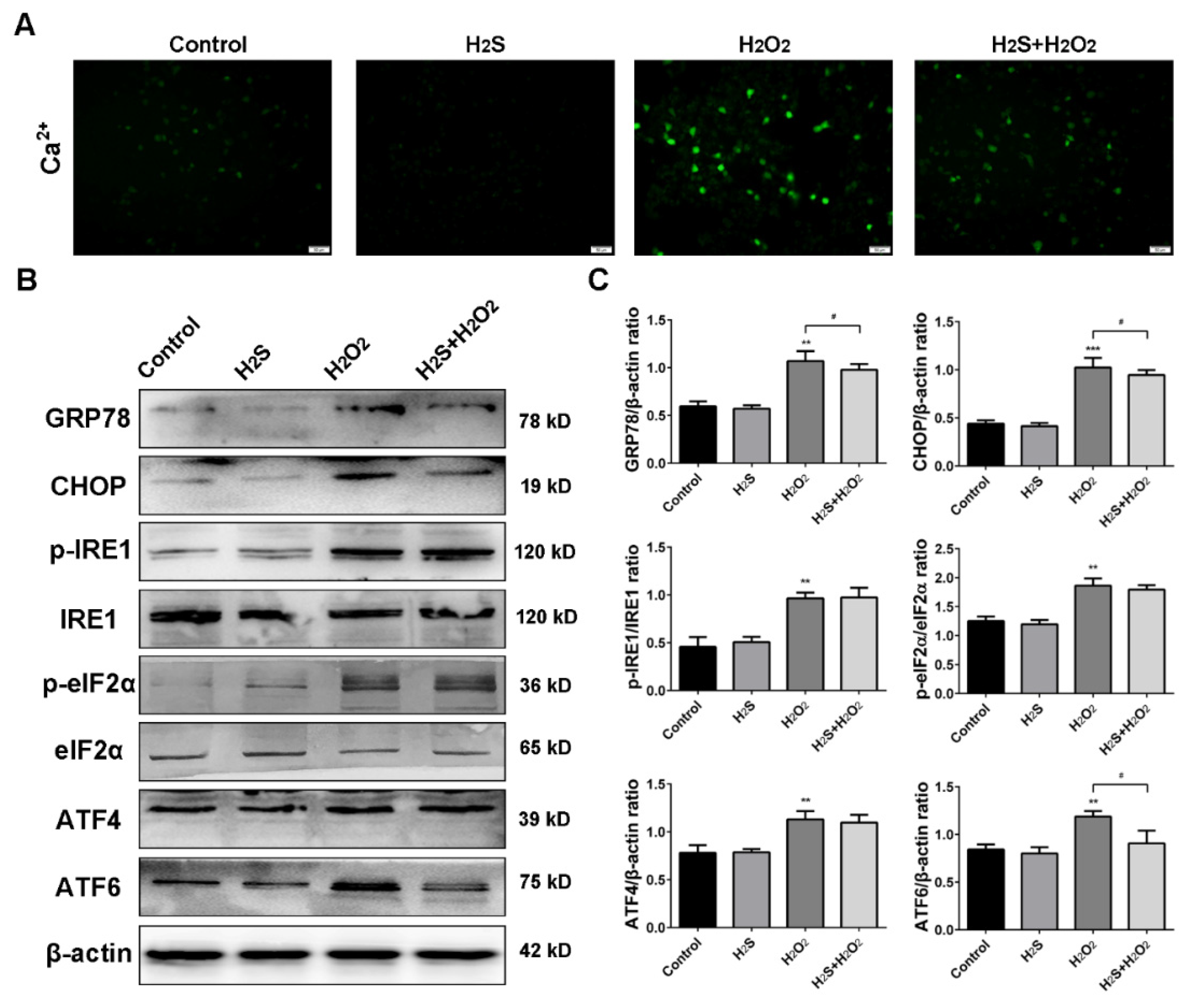
2.5. Effect of H2S on ROS-Induced Intracellular [Ca2+] and Endoplasmic Reticulum Stress
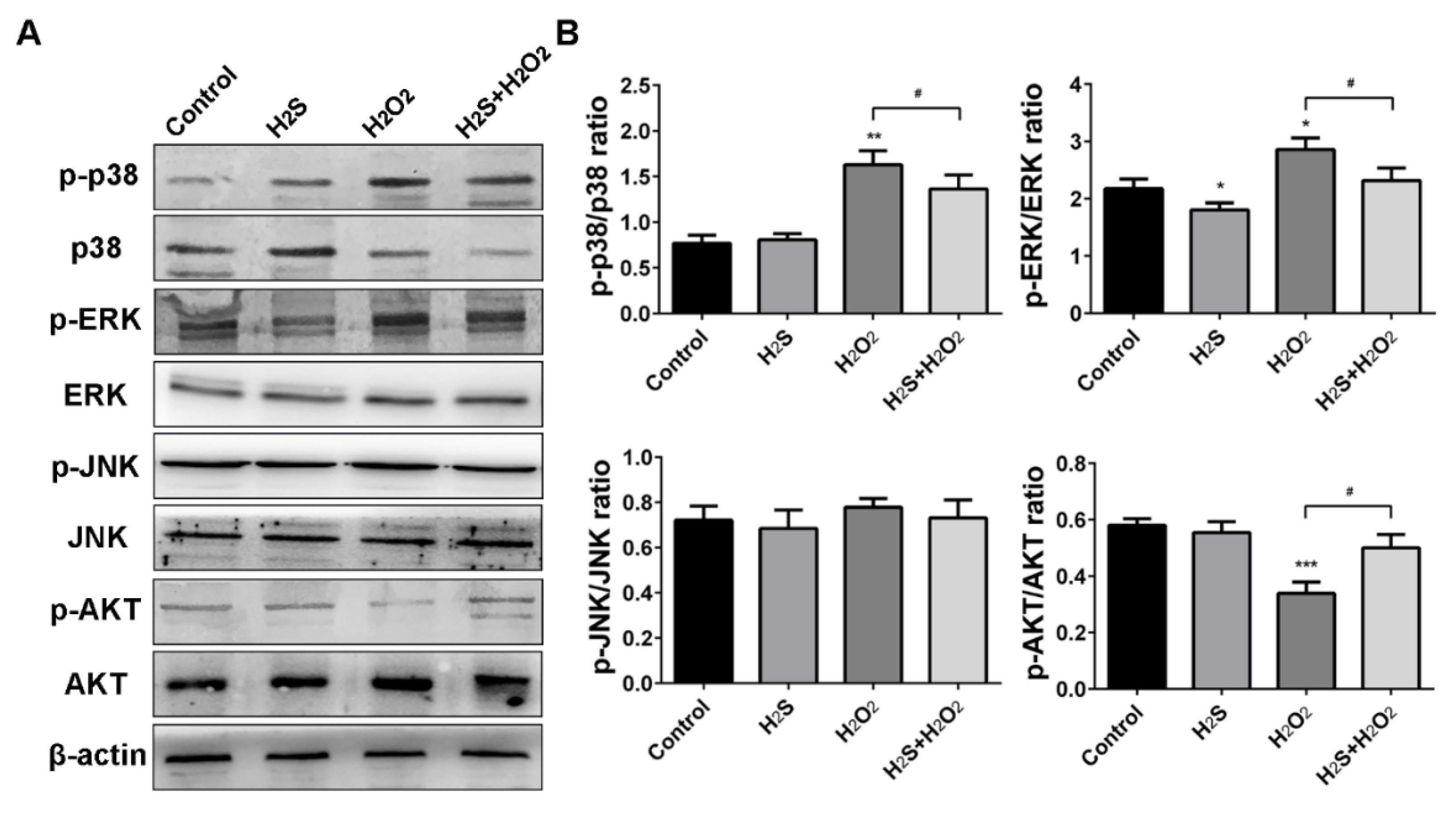
2.6. Effect of H2S on the Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway in H2O2-Treated A549 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability and Colony Formation
4.3. Analysis of Cell Apoptosis
4.4. Quantitative Real-Time PCR (qRT-PCR)
4.5. Western Blot
4.6. Measurement of H2S in Cell Culture Supernatants
4.7. Measurement of [Ca2+]
4.8. Measurement of Mitochondrial Membrane Potential (Δψ)
4.9. Reactive Oxygen Species (ROS), Malondialdehyde (MDA), Superoxide Dismutase (SOD), Glutathione (GSH), and Catalase-Peroxidase (CAT) Assays
4.10. Metabolic Assays
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| H2S | hydrogen sulfide |
| H2O2 | hydrogen peroxide |
| CBS | cystathionine-beta-synthase |
| CSE | cystathionine-gamma-lyase |
| MPST | 3-mercapto-pyruvate sulfurtransferase |
| ER | endoplasmic reticulum |
| MAPK | mitogen-activated protein kinase |
| ROS | reactive oxygen species |
| NaHS | sodium hydrosulfide |
| IC50 | half maximal inhibitory concentration |
| MTT | 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide |
| DCFH-DA | 2′,7′-dichloroflurescein-diacetate |
| SOD | superoxide dismutase |
| GSH-PX | glutathione peroxidase |
| CAT | catalase |
| MDA | malondialdehyde |
| Δψ | mitochondrial membrane potential |
| ATP | adenosine triphosphate |
| LDH | lactate dehydrogenase |
| IRE1 | inositol requiring enzyme 1 |
| PERK | protein kinase RNA-like ER kinase |
| ATF6 | activating transcription factor 6 |
| UPR | unfold protein response |
| FBS | fetal bovine serum |
| qRT-PCR | quantitative real-time PCR |
| PBS | phosphate buffer saline |
| NSAIDS | nonsteroidal anti-inflammatory drugs |
References
- Di Masi, A.; Ascenzi, P. H2S: a “double face” molecule in health and disease. Biofactors 2013, 39, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Bai, Y.; Yin, C.; Qian, H.; Xing, G.; Wang, S.; Li, F.; Bian, J.; Aschner, M.; Lu, R. Activation of autophagic flux and the Nrf2/ARE signaling pathway by hydrogen sulfide protects against acrylonitrile-induced neurotoxicity in primary rat astrocytes. Arch. Toxicol. 2018, 92, 2093–2108. [Google Scholar] [CrossRef]
- Wang, R. Hydrogen sulfide: the third gasotransmitter in biology and medicine. Antioxid. Redox Signal. 2010, 12, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Szabo, C. Hydrogen Sulfide and Cancer. Handb. Exp. Pharmacol. 2015, 230, 233–241. [Google Scholar] [PubMed]
- Szabo, C.; Coletta, C.; Chao, C.; Modis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Ivanciuc, T.; Sbrana, E.; Ansar, M.; Bazhanov, N.; Szabo, C.; Casola, A.; Garofalo, R.P. Hydrogen Sulfide Is an Antiviral and Antiinflammatory Endogenous Gasotransmitter in the Airways. Role in Respiratory Syncytial Virus Infection. Am. J. Respir. Cell Mol. Biol. 2016, 55, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide: its production, release and functions. Amino Acids 2011, 41, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Gargallo, C.J.; Lanas, A. Is NSAIDs-related gastrointestinal damage preventable? J. Dig. Dis. 2013, 14, 55–61. [Google Scholar] [CrossRef]
- Gemici, B.; Elsheikh, W.; Feitosa, K.B.; Costa, S.K.; Muscara, M.N.; Wallace, J.L. H2S-releasing drugs: anti-inflammatory, cytoprotective and chemopreventative potential. Nitric Oxide 2015, 46, 25–31. [Google Scholar] [CrossRef]
- Hsia, C.C.; Hyde, D.M.; Weibel, E.R. Lung Structure and the Intrinsic Challenges of Gas Exchange. Compr. Physiol. 2016, 6, 827–895. [Google Scholar] [PubMed]
- Groneberg-Kloft, B.; Kraus, T.; Mark, A.; Wagner, U.; Fischer, A. Analysing the causes of chronic cough: relation to diesel exhaust, ozone, nitrogen oxides, sulphur oxides and other environmental factors. J. Occup. Med. Toxicol. 2006, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Kallet, R.H.; Matthay, M.A. Hyperoxic acute lung injury. Respir. Care 2013, 58, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Escaffre, O.; Saito, T.B.; Juelich, T.L.; Ikegami, T.; Smith, J.K.; Perez, D.D.; Atkins, C.; Levine, C.B.; Huante, M.B.; Nusbaum, R.J.; et al. Contribution of Human Lung Parenchyma and Leukocyte Influx to Oxidative Stress and Immune System-Mediated Pathology following Nipah Virus Infection. J. Virol. 2017, 91, e00275-17. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef]
- Fudulu, D.; Angelini, G. Oxidative Stress after Surgery on the Immature Heart. Oxid. Med. Cell Longev. 2016, 2016, 1971452. [Google Scholar] [CrossRef]
- Liao, P.H.; Hsu, H.H.; Chen, T.S.; Chen, M.C.; Day, C.H.; Tu, C.C.; Lin, Y.M.; Tsai, F.J.; Kuo, W.W.; Huang, C.Y. Phosphorylation of cofilin-1 by ERK confers HDAC inhibitor resistance in hepatocellular carcinoma cells via decreased ROS-mediated mitochondria injury. Oncogene 2017, 36, 1978–1990. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, K.; Jin, Y.; Sheng, X. Endoplasmic reticulum proteostasis control and gastric cancer. Cancer Lett. 2019, 449, 263–271. [Google Scholar] [CrossRef]
- Marciniak, S.J. Endoplasmic reticulum stress in lung disease. Eur. Respir. Rev. 2017, 26, 170018. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, S.R.; Cho, S.H.; Lee, Y.C. Endoplasmic Reticulum Stress and Allergic Diseases. Curr. Allergy Asthma Rep. 2017, 17, 82. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Mozzini, C.; Cominacini, L.; Garbin, U.; Fratta Pasini, A.M. Endoplasmic Reticulum Stress, NRF2 Signalling and Cardiovascular Diseases in a Nutshell. Curr. Atheroscler. Rep. 2017, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E. Endoplasmic Reticulum Stress and Obesity. Adv. Exp. Med. Biol. 2017, 960, 261–276. [Google Scholar] [PubMed]
- Wei, J.; Rahman, S.; Ayaub, E.A.; Dickhout, J.G.; Ask, K. Protein misfolding and endoplasmic reticulum stress in chronic lung disease. Chest 2013, 143, 1098–1105. [Google Scholar] [CrossRef]
- Herken, H.; Uz, E.; Ozyurt, H.; Sogut, S.; Virit, O.; Akyol, O. Evidence that the activities of erythrocyte free radical scavenging enzymes and the products of lipid peroxidation are increased in different forms of schizophrenia. Mol. Psychiatry 2001, 6, 66–73. [Google Scholar] [CrossRef]
- Fujimura, N.; Sumita, S.; Aimono, M.; Masuda, Y.; Shichinohe, Y.; Narimatsu, E.; Namiki, A. Effect of free radical scavengers on diaphragmatic contractility in septic peritonitis. Am. J. Respir. Crit. Care Med. 2000, 162, 2159–2165. [Google Scholar] [CrossRef]
- Sun, C.K.; Zhang, X.Y.; Sheard, P.W.; Mabuchi, A.; Wheatley, A.M. Change in mitochondrial membrane potential is the key mechanism in early warm hepatic ischemia-reperfusion injury. Microvasc. Res. 2005, 70, 102–110. [Google Scholar] [CrossRef]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M.; Ma, Y.; Mao, Z.; Song, H.; Chen, F. beta-Thujaplicin induces autophagic cell death, apoptosis, and cell cycle arrest through ROS-mediated Akt and p38/ERK MAPK signaling in human hepatocellular carcinoma. Cell Death Dis. 2019, 10, 255. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Wallace, J.L.; Blackler, R.W.; Chan, M.V.; Da Silva, G.J.; Elsheikh, W.; Flannigan, K.L.; Gamaniek, I.; Manko, A.; Wang, L.; Motta, J.P.; et al. Anti-inflammatory and cytoprotective actions of hydrogen sulfide: translation to therapeutics. Antioxid Redox Signal. 2015, 22, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Siddiqui, N.; Alharbi, N.O.; Alharbi, M.M. Airway and systemic oxidant-antioxidant dysregulation in asthma: a possible scenario of oxidants spill over from lung into blood. Pulm. Pharmacol. Ther. 2014, 29, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Yuan, X.; Yu, L.; Gao, C.; Sun, X.; Wang, D.; Zheng, Q. Licochalcone A-induced human gastric cancer BGC-823 cells apoptosis by regulating ROS-mediated MAPKs and PI3K/AKT signaling pathways. Sci. Rep. 2015, 5, 10336. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Addabbo, F.; Montagnani, M.; Goligorsky, M.S. Mitochondria and reactive oxygen species. Hypertension 2009, 53, 885–892. [Google Scholar] [CrossRef]
- Feng, A.; Ling, C.; Xin-Duo, L.; Bing, W.; San-Wu, W.; Yu, Z.; Yu-Lan, H.; You-En, Z. Hydrogen Sulfide Protects Human Cardiac Fibroblasts Against H2O2-induced Injury Through Regulating Autophagy-Related Proteins. Cell Transplant. 2018, 27, 1222–1234. [Google Scholar] [CrossRef]
- Su, Y.B.; Cheng, Y.K.; Wang, D.D.; Zhang, F.R.; Su, Y.W.; Li, Z.Q. Effects of hydrogen sulfide (H2S) on respiration control of state 3/4 in mitochondria from bovine heart. Afr. J. Biotechnol. 2012, 11, 4876–4883. [Google Scholar]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef]
- Tajiri, S.; Oyadomari, S.; Yano, S.; Morioka, M.; Gotoh, T.; Hamada, J.I.; Ushio, Y.; Mori, M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004, 11, 403–415. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Hammadi, M.; Oulidi, A.; Gackiere, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.W.; Zhu, H.T.; Chen, K.L.; Dong, X.; Wei, J.; Qiu, C.; Xue, J.H. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc. Diabetol. 2013, 12, 158. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Aspects Med. 2018, 63, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yin, F.; Xu, J.; Zhang, T.; Wang, G.; Mao, M.; Wang, Z.; Sun, W.; Han, J.; Yang, M.; et al. CYT997(Lexibulin) induces apoptosis and autophagy through the activation of mutually reinforced ER stress and ROS in osteosarcoma. J. Exp. Clin. Cancer Res. 2019, 38, 44. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Chen, W.; Li, P.; Liu, Y.; Yang, Y.; Ye, X.; Zhang, F.; Huang, H. Isoalantolactone induces apoptosis through ROS-mediated ER stress and inhibition of STAT3 in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 309. [Google Scholar] [CrossRef]
- Hayashi, T.; Saito, A.; Okuno, S.; Ferrand-Drake, M.; Dodd, R.L.; Chan, P.H. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J. Cereb. Blood Flow Metab. 2005, 25, 41–53. [Google Scholar] [CrossRef]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| CBS | AATGGTGACGCTTGGGAA | TGAGGCGGATCTGTTTGA |
| CSE | AAGACGCCTCCTCACAAGGT | ATATTCAAAACCCGAGTGCTGG |
| MPST | GACCCCGCCTTCATCAAG | CATGTACCACTCCACCCA |
| Bax | TGGCAGCTGACATGTTTTCTGA | TCACCCAACCACCCTGGTCTT |
| Bcl-2 | CAGTTGGGCAACAGAGAACCAT | AGCCCTTGTCCCCAATTTGGAA |
| β-actin | CTGGAACGGTGAAGGTGACA | AAGGGACTTCCTGTAACAATGCA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Cao, X.; Luan, C.; Li, Z. Hydrogen Sulfide Attenuates Hydrogen Peroxide-Induced Injury in Human Lung Epithelial A549 Cells. Int. J. Mol. Sci. 2019, 20, 3975. https://doi.org/10.3390/ijms20163975
Wang M, Cao X, Luan C, Li Z. Hydrogen Sulfide Attenuates Hydrogen Peroxide-Induced Injury in Human Lung Epithelial A549 Cells. International Journal of Molecular Sciences. 2019; 20(16):3975. https://doi.org/10.3390/ijms20163975
Chicago/Turabian StyleWang, Mingqi, Xinyu Cao, Chang Luan, and Zhengqiang Li. 2019. "Hydrogen Sulfide Attenuates Hydrogen Peroxide-Induced Injury in Human Lung Epithelial A549 Cells" International Journal of Molecular Sciences 20, no. 16: 3975. https://doi.org/10.3390/ijms20163975
APA StyleWang, M., Cao, X., Luan, C., & Li, Z. (2019). Hydrogen Sulfide Attenuates Hydrogen Peroxide-Induced Injury in Human Lung Epithelial A549 Cells. International Journal of Molecular Sciences, 20(16), 3975. https://doi.org/10.3390/ijms20163975