Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges


Abstract
1. Introduction
2. RTT and RTT-Like Syndrome
3. New Technologies for a Rare Genetic Diagnosis
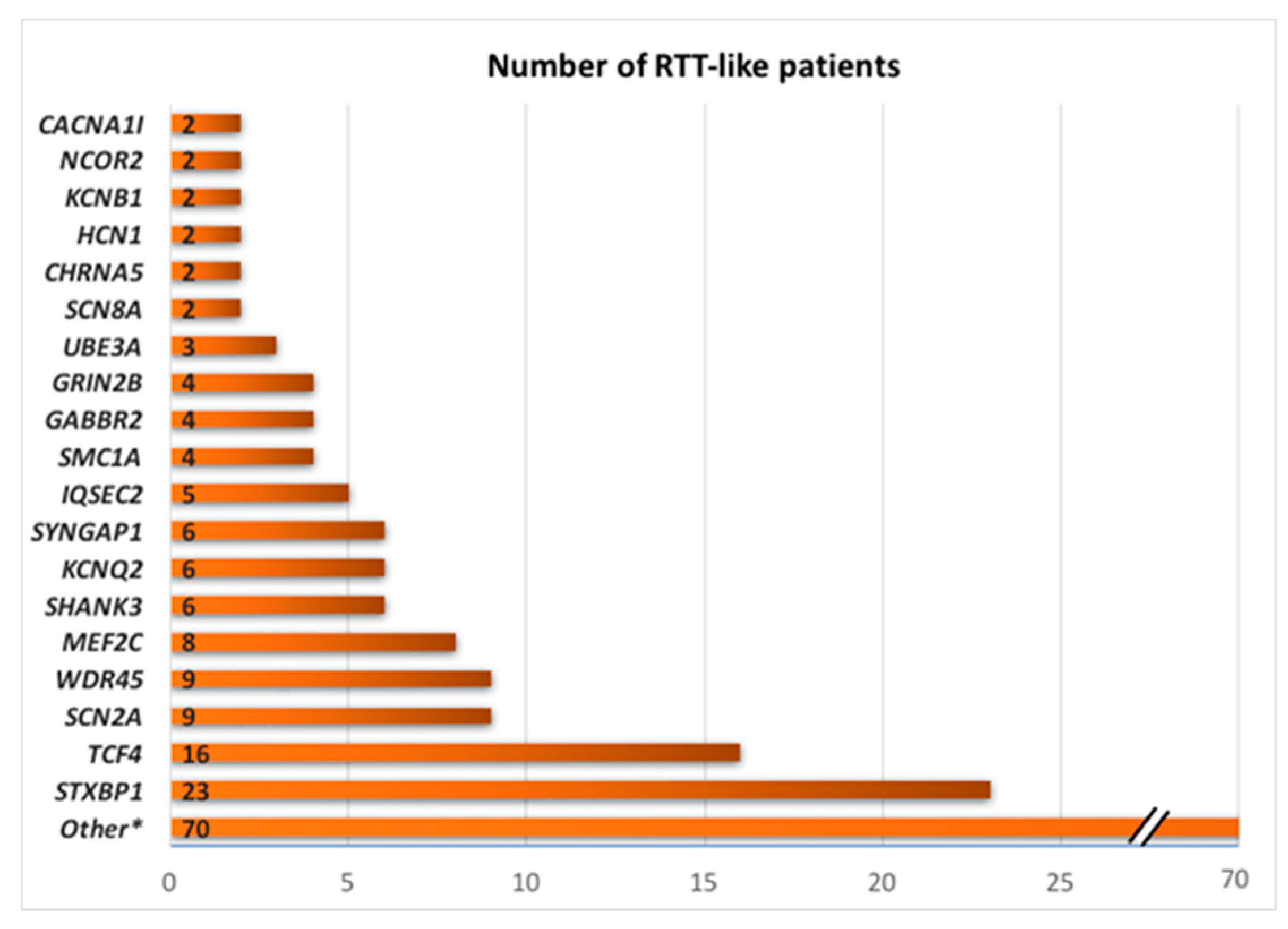
4. NGS Results: Many Genes, Many Disorders
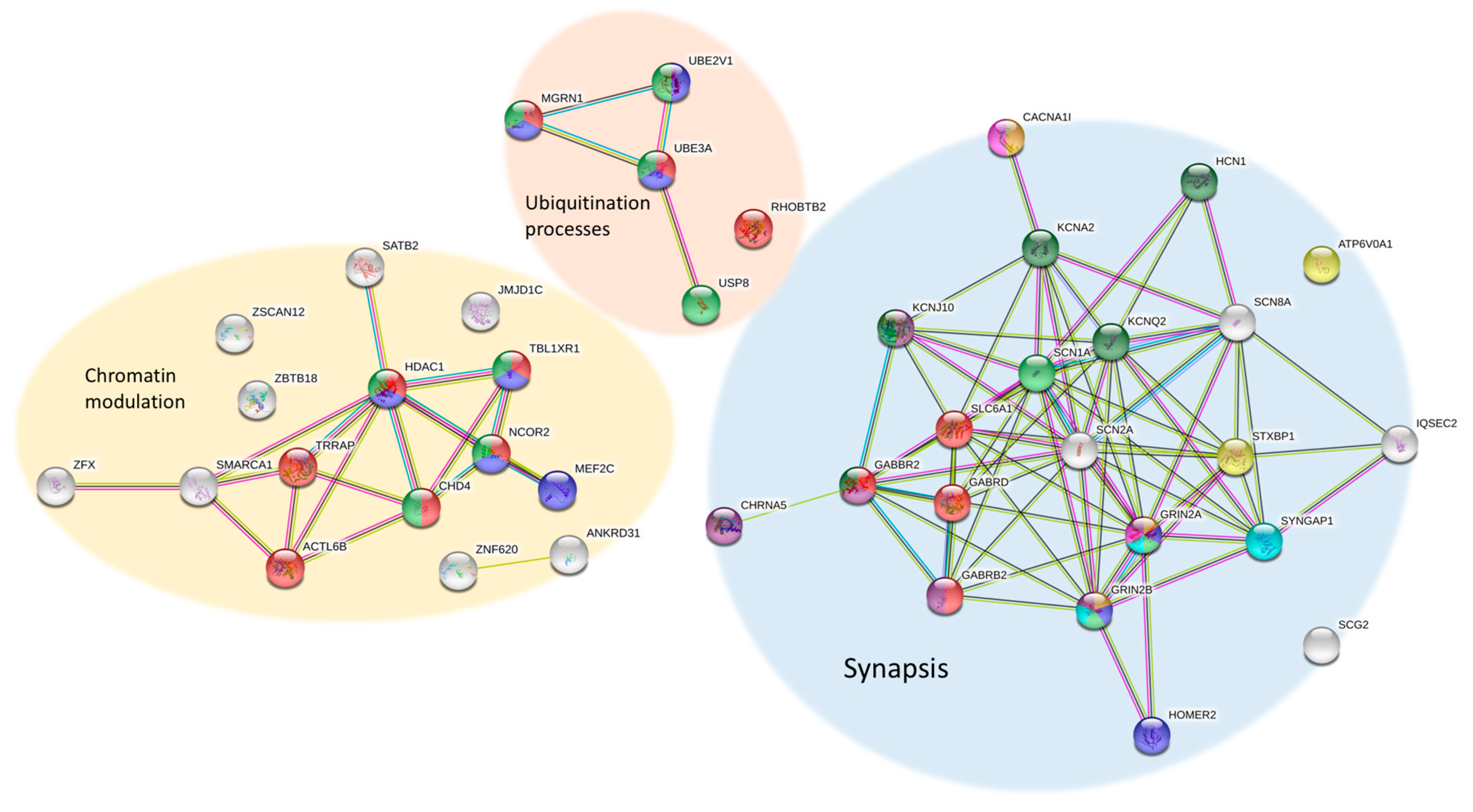
5. Functions and Pathways around RTT
6. Future Perspectives and Treatment Options
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Bird, A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004, 32, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004, 36, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Yasui, D.H.; Gonzales, M.L.; Aflatooni, J.O.; Crary, F.K.; Hu, D.J.; Gavino, B.J.; Golub, M.S.; Vincent, J.B.; Carolyn Schanen, N.; Olson, C.O.; et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mol. Genet. 2014, 23, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, J.; Grimm, A.; Maher, T.; Bennetts, B. RettBASE: The IRSA MECP2 variation database-a new mutation database in evolution. Hum. Mutat. 2003, 21, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; Freude, K.; Musante, L.; Jensen, L.R.; Yntema, H.G.; Gecz, J.; Sefiani, A.; Hoffmann, K.; Moser, B.; Haas, S.; et al. Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 2003, 35, 313–315. [Google Scholar] [CrossRef]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Hagberg, B.; Hanefeld, F.; Percy, A.; Skjeldal, O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur. J. Paediatr. Neurol. 2002, 6, 293–297. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Chen, Y.; Yu, S.; Wu, X.; Bao, X.; Wen, Y. Novel MEF2C point mutations in Chinese patients with Rett (-like) syndrome or non-syndromic intellectual disability: Insights into genotype-phenotype correlation. BMC Med. Genet. 2018, 19, 191. [Google Scholar] [CrossRef]
- Vidal, S.; Brandi, N.; Pacheco, P.; Gerotina, E.; Blasco, L.; Trotta, J.R.; Derdak, S.; Del Mar O’Callaghan, M.; Garcia-Cazorla, A.; Pineda, M.; et al. The utility of Next Generation Sequencing for molecular diagnostics in Rett syndrome. Sci. Rep. 2017, 7, 12288. [Google Scholar] [CrossRef]
- Schonewolf-Greulich, B.; Bisgaard, A.M.; Moller, R.S.; Duno, M.; Brondum-Nielsen, K.; Kaur, S.; Van Bergen, N.J.; Lunke, S.; Eggers, S.; Jespersgaard, C.; et al. Clinician’s guide to genes associated with Rett-like phenotypes-Investigation of a Danish cohort and review of the literature. Clin. Genet. 2019, 95, 221–230. [Google Scholar] [CrossRef]
- Di Resta, C.; Galbiati, S.; Carrera, P.; Ferrari, M. Next-generation sequencing approach for the diagnosis of human diseases: Open challenges and new opportunities. EJIFCC 2018, 29, 4–14. [Google Scholar]
- Srivastava, S.; Desai, S.; Cohen, J.; Smith-Hicks, C.; Baranano, K.; Fatemi, A.; Naidu, S. Monogenic disorders that mimic the phenotype of Rett syndrome. Neurogenetics 2018, 19, 41–47. [Google Scholar] [CrossRef]
- Lucariello, M.; Vidal, E.; Vidal, S.; Saez, M.; Roa, L.; Huertas, D.; Pineda, M.; Dalfo, E.; Dopazo, J.; Jurado, P.; et al. Whole exome sequencing of Rett syndrome-like patients reveals the mutational diversity of the clinical phenotype. Hum. Genet. 2016, 135, 1343–1354. [Google Scholar] [CrossRef]
- Mak, C.C.; Leung, G.K.; Mok, G.T.; Yeung, K.S.; Yang, W.; Fung, C.W.; Chan, S.H.; Lee, S.L.; Lee, N.C.; Pfundt, R.; et al. Exome sequencing for paediatric-onset diseases: Impact of the extensive involvement of medical geneticists in the diagnostic odyssey. NPJ Genom. Med. 2018, 3, 19. [Google Scholar] [CrossRef]
- Lelieveld, S.H.; Veltman, J.A.; Gilissen, C. Novel bioinformatic developments for exome sequencing. Hum. Genet. 2016, 135, 603–614. [Google Scholar] [CrossRef]
- Lupski, J.R.; Reid, J.G.; Gonzaga-Jauregui, C.; Rio Deiros, D.; Chen, D.C.; Nazareth, L.; Bainbridge, M.; Dinh, H.; Jing, C.; Wheeler, D.A.; et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N. Engl. J. Med. 2010, 362, 1181–1191. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Halvorsen, M.; Petrovski, S.; Shellhaas, R.; Tang, Y.; Crandall, L.; Goldstein, D.; Devinsky, O. Mosaic mutations in early-onset genetic diseases. Genet. Med. 2016, 18, 746–749. [Google Scholar] [CrossRef]
- de Lange, I.M.; Koudijs, M.J.; van ’t Slot, R.; Sonsma, A.C.M.; Mulder, F.; Carbo, E.C.; van Kempen, M.J.A.; Nijman, I.J.; Ernst, R.F.; Savelberg, S.M.C.; et al. Assessment of parental mosaicism in SCN1A-related epilepsy by single-molecule molecular inversion probes and next-generation sequencing. J. Med. Genet. 2019, 56, 75–80. [Google Scholar] [CrossRef]
- Uddin, M.; Woodbury-Smith, M.; Chan, A.; Brunga, L.; Lamoureux, S.; Pellecchia, G.; Yuen, R.K.C.; Faheem, M.; Stavropoulos, D.J.; Drake, J.; et al. Germline and somatic mutations in STXBP1 with diverse neurodevelopmental phenotypes. Neurol. Genet. 2017, 3, e199. [Google Scholar] [CrossRef]
- D’Gama, A.M.; Walsh, C.A. Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 2018, 21, 1504–1514. [Google Scholar] [CrossRef]
- de Lange, I.M.; Koudijs, M.J.; van ’t Slot, R.; Gunning, B.; Sonsma, A.C.M.; van Gemert, L.; Mulder, F.; Carbo, E.C.; van Kempen, M.J.A.; Verbeek, N.E.; et al. Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia 2018, 59, 690–703. [Google Scholar] [CrossRef]
- Baasch, A.L.; Huning, I.; Gilissen, C.; Klepper, J.; Veltman, J.A.; Gillessen-Kaesbach, G.; Hoischen, A.; Lohmann, K. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia 2014, 55, e25–e29. [Google Scholar] [CrossRef]
- Saitsu, H.; Tohyama, J.; Walsh, T.; Kato, M.; Kobayashi, Y.; Lee, M.; Tsurusaki, Y.; Miyake, N.; Goto, Y.; Nishino, I.; et al. A girl with West syndrome and autistic features harboring a de novo TBL1XR1 mutation. J. Hum. Genet. 2014, 59, 581–583. [Google Scholar] [CrossRef]
- Okamoto, N.; Miya, F.; Tsunoda, T.; Kato, M.; Saitoh, S.; Yamasaki, M.; Shimizu, A.; Torii, C.; Kanemura, Y.; Kosaki, K. Targeted next-generation sequencing in the diagnosis of neurodevelopmental disorders. Clin. Genet. 2015, 88, 288–292. [Google Scholar] [CrossRef]
- Hara, M.; Ohba, C.; Yamashita, Y.; Saitsu, H.; Matsumoto, N.; Matsuishi, T. De novo SHANK3 mutation causes Rett syndrome-like phenotype in a female patient. Am. J. Med. Genet. A 2015, 167, 1593–1596. [Google Scholar] [CrossRef]
- Olson, H.E.; Tambunan, D.; LaCoursiere, C.; Goldenberg, M.; Pinsky, R.; Martin, E.; Ho, E.; Khwaja, O.; Kaufmann, W.E.; Poduri, A. Mutations in epilepsy and intellectual disability genes in patients with features of Rett syndrome. Am. J. Med. Genet. A 2015, 167A, 2017–2025. [Google Scholar] [CrossRef]
- Hoffjan, S.; Ibisler, A.; Tschentscher, A.; Dekomien, G.; Bidinost, C.; Rosa, A.L. WDR45 mutations in Rett (-like) syndrome and developmental delay: Case report and an appraisal of the literature. Mol. Cell Probes 2016, 30, 44–49. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J.H. SATB2-associated syndrome presenting with Rett-like phenotypes. Clin. Genet. 2016, 89, 728–732. [Google Scholar] [CrossRef]
- Saez, M.A.; Fernandez-Rodriguez, J.; Moutinho, C.; Sanchez-Mut, J.V.; Gomez, A.; Vidal, E.; Petazzi, P.; Szczesna, K.; Lopez-Serra, P.; Lucariello, M.; et al. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet. Med. 2016, 18, 378–385. [Google Scholar] [CrossRef]
- Rocha, H.; Sampaio, M.; Rocha, R.; Fernandes, S.; Leao, M. MEF2C haploinsufficiency syndrome: Report of a new MEF2C mutation and review. Eur. J. Med. Genet. 2016, 59, 478–482. [Google Scholar] [CrossRef]
- Lopes, F.; Barbosa, M.; Ameur, A.; Soares, G.; de Sa, J.; Dias, A.I.; Oliveira, G.; Cabral, P.; Temudo, T.; Calado, E.; et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016, 53, 190–199. [Google Scholar] [CrossRef]
- Sajan, S.A.; Jhangiani, S.N.; Muzny, D.M.; Gibbs, R.A.; Lupski, J.R.; Glaze, D.G.; Kaufmann, W.E.; Skinner, S.A.; Annese, F.; Friez, M.J.; et al. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet. Med. 2017, 19, 13–19. [Google Scholar] [CrossRef]
- Allou, L.; Julia, S.; Amsallem, D.; El Chehadeh, S.; Lambert, L.; Thevenon, J.; Duffourd, Y.; Saunier, A.; Bouquet, P.; Pere, S.; et al. Rett-like phenotypes: Expanding the genetic heterogeneity to the KCNA2 gene and first familial case of CDKL5-related disease. Clin. Genet. 2017, 91, 431–440. [Google Scholar] [CrossRef]
- Yoo, Y.; Jung, J.; Lee, Y.N.; Lee, Y.; Cho, H.; Na, E.; Hong, J.; Kim, E.; Lee, J.S.; Lee, J.S.; et al. GABBR2 mutations determine phenotype in rett syndrome and epileptic encephalopathy. Ann. Neurol. 2017, 82, 466–478. [Google Scholar] [CrossRef]
- Vuillaume, M.L.; Jeanne, M.; Xue, L.; Blesson, S.; Denomme-Pichon, A.S.; Alirol, S.; Brulard, C.; Colin, E.; Isidor, B.; Gilbert-Dussardier, B.; et al. A novel mutation in the transmembrane 6 domain of GABBR2 leads to a Rett-like phenotype. Ann. Neurol. 2018, 83, 437–439. [Google Scholar] [CrossRef]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. A 2017, 173, 2108–2125. [Google Scholar] [CrossRef]
- Percy, A.K.; Lane, J.; Annese, F.; Warren, H.; Skinner, S.A.; Neul, J.L. When Rett syndrome is due to genes other than MECP2. Transl. Sci. Rare Dis. 2018, 3, 49–53. [Google Scholar] [CrossRef]
- Iwama, K.; Mizuguchi, T.; Takeshita, E.; Nakagawa, E.; Okazaki, T.; Nomura, Y.; Iijima, Y.; Kajiura, I.; Sugai, K.; Saito, T.; et al. Genetic landscape of Rett syndrome-like phenotypes revealed by whole exome sequencing. J. Med. Genet. 2019. [Google Scholar] [CrossRef]
- Vidal, S.; Brandi, N.; Pacheco, P.; Maynou, J.; Fernandez, G.; Xiol, C.; Pascual-Alonso, A.; Pineda, M.; Rett Working, G.; Armstrong, J. The most recurrent monogenic disorders that overlap with the phenotype of Rett syndrome. Eur. J. Paediatr. Neurol. 2019. [Google Scholar] [CrossRef]
- Swanson, D.A.; Steel, J.M.; Valle, D. Identification and characterization of the human ortholog of rat STXBP1, a protein implicated in vesicle trafficking and neurotransmitter release. Genomics 1998, 48, 373–376. [Google Scholar] [CrossRef]
- Toonen, R.F.; Wierda, K.; Sons, M.S.; de Wit, H.; Cornelisse, L.N.; Brussaard, A.; Plomp, J.J.; Verhage, M. Munc18-1 expression levels control synapse recovery by regulating readily releasable pool size. Proc. Natl. Acad. Sci. USA 2006, 103, 18332–18337. [Google Scholar] [CrossRef]
- Medrihan, L.; Tantalaki, E.; Aramuni, G.; Sargsyan, V.; Dudanova, I.; Missler, M.; Zhang, W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 2008, 99, 112–121. [Google Scholar] [CrossRef]
- Sepp, M.; Pruunsild, P.; Timmusk, T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum. Mol. Genet. 2012, 21, 2873–2888. [Google Scholar] [CrossRef]
- de Winter, C.F.; Baas, M.; Bijlsma, E.K.; van Heukelingen, J.; Routledge, S.; Hennekam, R.C. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J. Rare Dis. 2016, 11, 37. [Google Scholar] [CrossRef]
- Sanders, S.J.; Campbell, A.J.; Cottrell, J.R.; Moller, R.S.; Wagner, F.F.; Auldridge, A.L.; Bernier, R.A.; Catterall, W.A.; Chung, W.K.; Empfield, J.R.; et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018, 41, 442–456. [Google Scholar] [CrossRef]
- Proikas-Cezanne, T.; Waddell, S.; Gaugel, A.; Frickey, T.; Lupas, A.; Nordheim, A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004, 23, 9314–9325. [Google Scholar] [CrossRef]
- Haack, T.B.; Hogarth, P.; Kruer, M.C.; Gregory, A.; Wieland, T.; Schwarzmayr, T.; Graf, E.; Sanford, L.; Meyer, E.; Kara, E.; et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012, 91, 1144–1149. [Google Scholar] [CrossRef]
- Ohba, C.; Nabatame, S.; Iijima, Y.; Nishiyama, K.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Tanaka, F.; Ozono, K.; Saitsu, H.; et al. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J. Hum. Genet. 2014, 59, 292–295. [Google Scholar] [CrossRef]
- Zweier, M.; Gregor, A.; Zweier, C.; Engels, H.; Sticht, H.; Wohlleber, E.; Bijlsma, E.K.; Holder, S.E.; Zenker, M.; Rossier, E.; et al. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum. Mutat. 2010, 31, 722–733. [Google Scholar] [CrossRef]
- Bienvenu, T.; Diebold, B.; Chelly, J.; Isidor, B. Refining the phenotype associated with MEF2C point mutations. Neurogenetics 2013, 14, 71–75. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Chapleau, C.A.; Calfa, G.D.; Lane, M.C.; Albertson, A.J.; Larimore, J.L.; Kudo, S.; Armstrong, D.L.; Percy, A.K.; Pozzo-Miller, L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 2009, 35, 219–233. [Google Scholar] [CrossRef]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef]
- Moretti, P.; Levenson, J.M.; Battaglia, F.; Atkinson, R.; Teague, R.; Antalffy, B.; Armstrong, D.; Arancio, O.; Sweatt, J.D.; Zoghbi, H.Y. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 2006, 26, 319–327. [Google Scholar] [CrossRef]
- Samaco, R.C.; Mandel-Brehm, C.; Chao, H.T.; Ward, C.S.; Fyffe-Maricich, S.L.; Ren, J.; Hyland, K.; Thaller, C.; Maricich, S.M.; Humphreys, P.; et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2009, 106, 21966–21971. [Google Scholar] [CrossRef]
- Harrington, A.J.; Raissi, A.; Rajkovich, K.; Berto, S.; Kumar, J.; Molinaro, G.; Raduazzo, J.; Guo, Y.; Loerwald, K.; Konopka, G.; et al. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife 2016, 5. [Google Scholar] [CrossRef]
- Berret, E.; Barron, T.; Xu, J.; Debner, E.; Kim, E.J.; Kim, J.H. Oligodendroglial excitability mediated by glutamatergic inputs and Nav1.2 activation. Nat. Commun. 2017, 8, 557. [Google Scholar] [CrossRef]
- Shin, W.; Kweon, H.; Kang, R.; Kim, D.; Kim, K.; Kang, M.; Kim, S.Y.; Hwang, S.N.; Kim, J.Y.; Yang, E.; et al. Scn2a Haploinsufficiency in Mice Suppresses Hippocampal Neuronal Excitability, Excitatory Synaptic Drive, and Long-Term Potentiation, and Spatial Learning and Memory. Front. Mol. Neurosci. 2019, 12, 145. [Google Scholar] [CrossRef]
- Santos, T.C.; Wierda, K.; Broeke, J.H.; Toonen, R.F.; Verhage, M. Early Golgi Abnormalities and Neurodegeneration upon Loss of Presynaptic Proteins Munc18-1, Syntaxin-1, or SNAP-25. J. Neurosci. 2017, 37, 4525–4539. [Google Scholar] [CrossRef]
- Li, H.; Zhu, Y.; Morozov, Y.M.; Chen, X.; Page, S.C.; Rannals, M.D.; Maher, B.J.; Rakic, P. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol. Psychiatry 2019, 24, 1235–1246. [Google Scholar] [CrossRef]
- Wan, H.; Wang, Q.; Chen, X.; Zeng, Q.; Shao, Y.; Fang, H.; Liao, X.; Li, H.S.; Liu, M.G.; Xu, T.L.; et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy 2019. [Google Scholar] [CrossRef]
- Ehrhart, F.; Coort, S.L.; Cirillo, E.; Smeets, E.; Evelo, C.T.; Curfs, L.M. Rett syndrome - biological pathways leading from MECP2 to disorder phenotypes. Orphanet J. Rare Dis. 2016, 11, 158. [Google Scholar] [CrossRef]
- Ehrhart, F.; Coort, S.L.; Eijssen, L.; Cirillo, E.; Smeets, E.E.; Bahram Sangani, N.; Evelo, C.T.; Curfs, L.M.G. Integrated analysis of human transcriptome data for Rett syndrome finds a network of involved genes. World J. Biol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Lekman, A.Y.; Hagberg, B.A.; Svennerholm, L.T. Membrane cerebral lipids in Rett syndrome. Pediatr. Neurol. 1991, 7, 186–190. [Google Scholar] [CrossRef]
- Kishi, N.; MacDonald, J.L.; Ye, J.; Molyneaux, B.J.; Azim, E.; Macklis, J.D. Reduction of aberrant NF-kappaB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice. Nat. Commun. 2016, 7, 10520. [Google Scholar] [CrossRef]
- di Michele, F.; Luchetti, S.; Bernardi, G.; Romeo, E.; Longone, P. Neurosteroid and neurotransmitter alterations in Parkinson’s disease. Front. Neuroendocrinol. 2013, 34, 132–142. [Google Scholar] [CrossRef]
- Goffin, D.; Brodkin, E.S.; Blendy, J.A.; Siegel, S.J.; Zhou, Z. Cellular origins of auditory event-related potential deficits in Rett syndrome. Nat. Neurosci. 2014, 17, 804–806. [Google Scholar] [CrossRef]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Banerjee, A.; Rikhye, R.V.; Breton-Provencher, V.; Tang, X.; Li, C.; Li, K.; Runyan, C.A.; Fu, Z.; Jaenisch, R.; Sur, M. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E7287–E7296. [Google Scholar] [CrossRef]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef]
- Cosentino, L.; Vigli, D.; Franchi, F.; Laviola, G.; De Filippis, B. Rett syndrome before regression: A time window of overlooked opportunities for diagnosis and intervention. Neurosci. Biobehav. Rev. 2019. [Google Scholar] [CrossRef]
- Bedogni, F.; Cobolli Gigli, C.; Pozzi, D.; Rossi, R.L.; Scaramuzza, L.; Rossetti, G.; Pagani, M.; Kilstrup-Nielsen, C.; Matteoli, M.; Landsberger, N. Defects During Mecp2 Null Embryonic Cortex Development Precede the Onset of Overt Neurological Symptoms. Cereb Cortex 2016, 26, 2517–2529. [Google Scholar] [CrossRef]
- Smith-Hicks, C.L.; Gupta, S.; Ewen, J.B.; Hong, M.; Kratz, L.; Kelley, R.; Tierney, E.; Vaurio, R.; Bibat, G.; Sanyal, A.; et al. Randomized open-label trial of dextromethorphan in Rett syndrome. Neurology 2017, 89, 1684–1690. [Google Scholar] [CrossRef]
- Mancini, J.; Dubus, J.C.; Jouve, E.; Roux, J.C.; Franco, P.; Lagrue, E.; Castelnau, P.; Cances, C.; Chaix, Y.; Rougeot-Jung, C.; et al. Effect of desipramine on patients with breathing disorders in RETT syndrome. Ann. Clin. Transl. Neurol. 2018, 5, 118–127. [Google Scholar] [CrossRef]
- O’Leary, H.M.; Kaufmann, W.E.; Barnes, K.V.; Rakesh, K.; Kapur, K.; Tarquinio, D.C.; Cantwell, N.G.; Roche, K.J.; Rose, S.A.; Walco, A.C.; et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann. Clin. Transl. Neurol. 2018, 5, 323–332. [Google Scholar] [CrossRef]
- Stamberger, H.; Weckhuysen, S.; De Jonghe, P. STXBP1 as a therapeutic target for epileptic encephalopathy. Expert. Opin. Ther. Targets 2017, 21, 1027–1036. [Google Scholar] [CrossRef]
- Gerber, S.H.; Rah, J.C.; Min, S.W.; Liu, X.; de Wit, H.; Dulubova, I.; Meyer, A.C.; Rizo, J.; Arancillo, M.; Hammer, R.E.; et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 2008, 321, 1507–1510. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Kavalali, E.T. Rett syndrome and the impact of MeCP2 associated transcriptional mechanisms on neurotransmission. Biol. Psychiatry 2009, 65, 204–210. [Google Scholar] [CrossRef]
- Dani, V.S.; Nelson, S.B. Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J. Neurosci. 2009, 29, 11263–11270. [Google Scholar] [CrossRef]
- Chamma, I.; Chevy, Q.; Poncer, J.C.; Levi, S. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front. Cell Neurosci. 2012, 6, 5. [Google Scholar] [CrossRef]
- Duarte, S.T.; Armstrong, J.; Roche, A.; Ortez, C.; Perez, A.; O’Callaghan Mdel, M.; Pereira, A.; Sanmarti, F.; Ormazabal, A.; Artuch, R.; et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS ONE 2013, 8, e68851. [Google Scholar] [CrossRef]
- Lozovaya, N.; Nardou, R.; Tyzio, R.; Chiesa, M.; Pons-Bennaceur, A.; Eftekhari, S.; Bui, T.T.; Billon-Grand, M.; Raserom, J.; Bonifazi, P.; et al. Early alterations in a mouse model of Rett syndrome: The GABA developmental shift is abolished at birth. Sci. Rep. 2019, 9, 9276. [Google Scholar] [CrossRef]
- Tang, X.; Drotar, J.; Li, K.; Clairmont, C.D.; Brumm, A.S.; Sullins, A.J.; Wu, H.; Liu, X.S.; Wang, J.; Gray, N.S.; et al. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 2019, 11, eaau0164. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Data Bases | Description | Website |
| Human mutation database (HGMD) | Database that represents an attempt to collate all known (published) gene lesions responsible for human inherited disease. | www.hgmd.cf.ac.uk/ |
| Varsome | The human genomic variant search engine. | https://varsome.com/ |
| GnomAD | Data from exome and genome sequencing from a variety of large-scale sequencing projects. | https://gnomad.broadinstitute.org/ |
| dbSNP | Public-domain archive for a broad collection of simple genetic polymorphisms. | www.ncbi.nlm.nih.gov/snp/ |
| ClinVar | Public archive of reports of the relationships among human variations and phenotypes, with supporting evidence. | www.ncbi.nlm.nih.gov/clinvar/ |
| Specific disease databases | Databases such as RettBASE that are freely-available resources for mutation and polymorphism data pertaining to Rett syndrome and other related clinical disorders. | mecp2.chw.edu.au |
| Software Tools | Description | Website |
| Mutation Taster | An in silico prediction tool for the pathogenicity of a variant based on evolutionary conservation, splice-site, mRNA, protein and regulatory features. | www.mutationtaster.org/ |
| SIFT | An in silico prediction tool for nonsynonymous variants based on sequence homology derived from closely related sequences collected through PSI-BLAST. | https://sift.bii.a-star.edu.sg/ |
| Polyphen-2 | Tool which predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations. | genetics.bwh.harvard.edu/pph2/ |
| Provean | An in silico tool that predicts how nonsynonymous or in-frame indel variant will affect a protein’s biological function. | provean.jcvi.org/ |
| Humans Splicing Finder | This tool is aimed to help studying the pre-mRNA splicing. | http://www.umd.be/HSF/ |
| Publications | Genes |
|---|---|
| Gilissen et al. 2014 [23] | SMC1A |
| Baasch et al. 2014 [29] | CN2A |
| Saitsu et al. 2014 [30] | TBL1XR1 |
| Okamoto et al. 2015 [31] | GABRD |
| Hara et al. 2015 [32] | SHANK3 |
| Olson et al. 2015 [33] | STXBP1, SCN8A, IQSEC2 |
| Hoffjan et al. 2016 [34] | WDR45 |
| Lee et al. 2016 [35] | SATB2 |
| Saez et al. 2016 [36] | JMJD1C |
| Rocha et al. 2016 [37] | MEF2C |
| Lucariello et al. 2016 [19] | ANKRD31, CHRNA5, HCN1, SCN1A, TCF4, GRIN2B, SLC6A1, MGRN1, BTBD9, SEMA6B, AGAP6, MGRN1,VASH2, ZNF620, GRAMD1A, GABBR2, ATP8B1, HAP1, PDLIM7, SRRM3, CACNA1I |
| Lopes et al. 2016 [38] | TCF4, EEF1A2, STXBP1, ZNF238, SLC35A2, ZFX, SHROOM4, EIF2B2, RHOBTB2, SMARCA1, GABBR2, EIF4G1, HTT |
| Vidal et al. 2017 [15] | GRIN2B, GABBR2, MEF2C, STXBP1, KCNQ2, SLC2A1, TCF4, SCN2A, SYNGAP1, CACNA1I, CHRNA5, HCN1 |
| Sajan et al. 2017 [39] | PWP2, SCG2, IZUMO4, XAB2, ZSCAN12, IQSEC2, FAM151A, SYNE2, SMC1A, ARHGEF10L, HDAC1, TAF1B, KCNJ10, CHD4, LRRC40, LAMB2, GRIN2B, IMPDH2, SAFB2, ACTL6B, STXBP1, TRRAP, WDR45, SLC39A13, FAT3, IQGAP3, NCOR2, GABRB2, TCF4, GRIN2A |
| Allou et al. 2017 [40] | IQSEC2, KCNA2 |
| Yoo et al. 2017 [41] | GABBR2 |
| Vuillaume et al. 2018 [42] | GABBR2 |
| Huisman et al. 2017 [43] | SMC1A |
| Wang et al. 2018 [14] | MEF2C |
| Percy et al. 2018 [44] | CTNNB1, WDR45 |
| Srivastava et al. 2018 [18] | KCNB1, IQSEC2, MEIS2, TCF4, WDR45 |
| Iwama et al. 2019 [45] | ATP6V0A1, USP8, MAST3, NCOR2, WDR45, STXBP1, SHANK3, UBE3A, GABRA1, SCN2A, SCN8A, GRIN2B, IQSEC2, CAMK2B, CUX2, CACNA1D, CACNA1G, ITPR1, KIF1A, SYNGAP1, NALCN, NR2F1, IRF2BPL, MAST1, COL4A1, HDAC8, TCF4, PDHA1, PPT1, DNMT3A, MEF2C |
| Schönewolf-Greulich et al. 2019 [16] | STXBP1, SCN2A, KCNB1, TCF4, SHANK3, SMC1A |
| Vidal et al. 2019 [46] | STXBP1, TCF4, SCN2A, KCNQ2, MEF2C, SYNGAP1 |
| RTT Genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MECP2 | CDKL5 | FOXG1 | STXBP1 | TCF4 | SCN2A | WDR45 | MEF2C | |||
| Disorder | Rett syndrome | EEP 2 | RTT, congenital variant | EEP 4 | Pitt- Hopkins syndrome | EEP 11 | Neurodegeneration with brain iron accumulation 5 | MEF2C haploinsufficiency syndrome | ||
| OMIM# | 312750 | 300672 | 613454 | 612164 | 610954 | 613721 | 300894 | 613443 | ||
| Inheritance | XLD | XLD | AD | AD | AD | AD | XLD | AD | ||
| Present in RTT | Required | Developmental regression | + | + | + | + | - | + | + | - |
| Four main criteria | Purposeful hand movements lost/absent | + | + | + | + | + | + | + | + | |
| Speech severe deficit/loss | + | + | + | + | + | + | + | + | ||
| Gait abnormality | + | + | + | + | + | + | + | + | ||
| Stereotypic hand movements | + | + | + | + | + | + | + | + | ||
| Other common symptoms | Breathing abnormality | + | + | - | - | + | - | - | - | |
| ID | + | + | + | + | + | + | + | + | ||
| Epilepsy | + | + | + | + | + | + | + | + | ||
| Microcephaly | + | + | + | + | + | + | + | - | ||
| Not present in RTT | Exclusion criteria | CNS abnormality | - | - | + | - | + | - | + | + |
| Other symptoms | Dysmorphic facial features | - | + | - | - | + | - | + | + | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidal, S.; Xiol, C.; Pascual-Alonso, A.; O’Callaghan, M.; Pineda, M.; Armstrong, J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int. J. Mol. Sci. 2019, 20, 3925. https://doi.org/10.3390/ijms20163925
Vidal S, Xiol C, Pascual-Alonso A, O’Callaghan M, Pineda M, Armstrong J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. International Journal of Molecular Sciences. 2019; 20(16):3925. https://doi.org/10.3390/ijms20163925
Chicago/Turabian StyleVidal, Silvia, Clara Xiol, Ainhoa Pascual-Alonso, M. O’Callaghan, Mercè Pineda, and Judith Armstrong. 2019. "Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges" International Journal of Molecular Sciences 20, no. 16: 3925. https://doi.org/10.3390/ijms20163925
APA StyleVidal, S., Xiol, C., Pascual-Alonso, A., O’Callaghan, M., Pineda, M., & Armstrong, J. (2019). Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. International Journal of Molecular Sciences, 20(16), 3925. https://doi.org/10.3390/ijms20163925