Normalizing Plasma Renin Activity in Experimental Dilated Cardiomyopathy: Effects on Edema, Cachexia, and Survival

 ,
,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
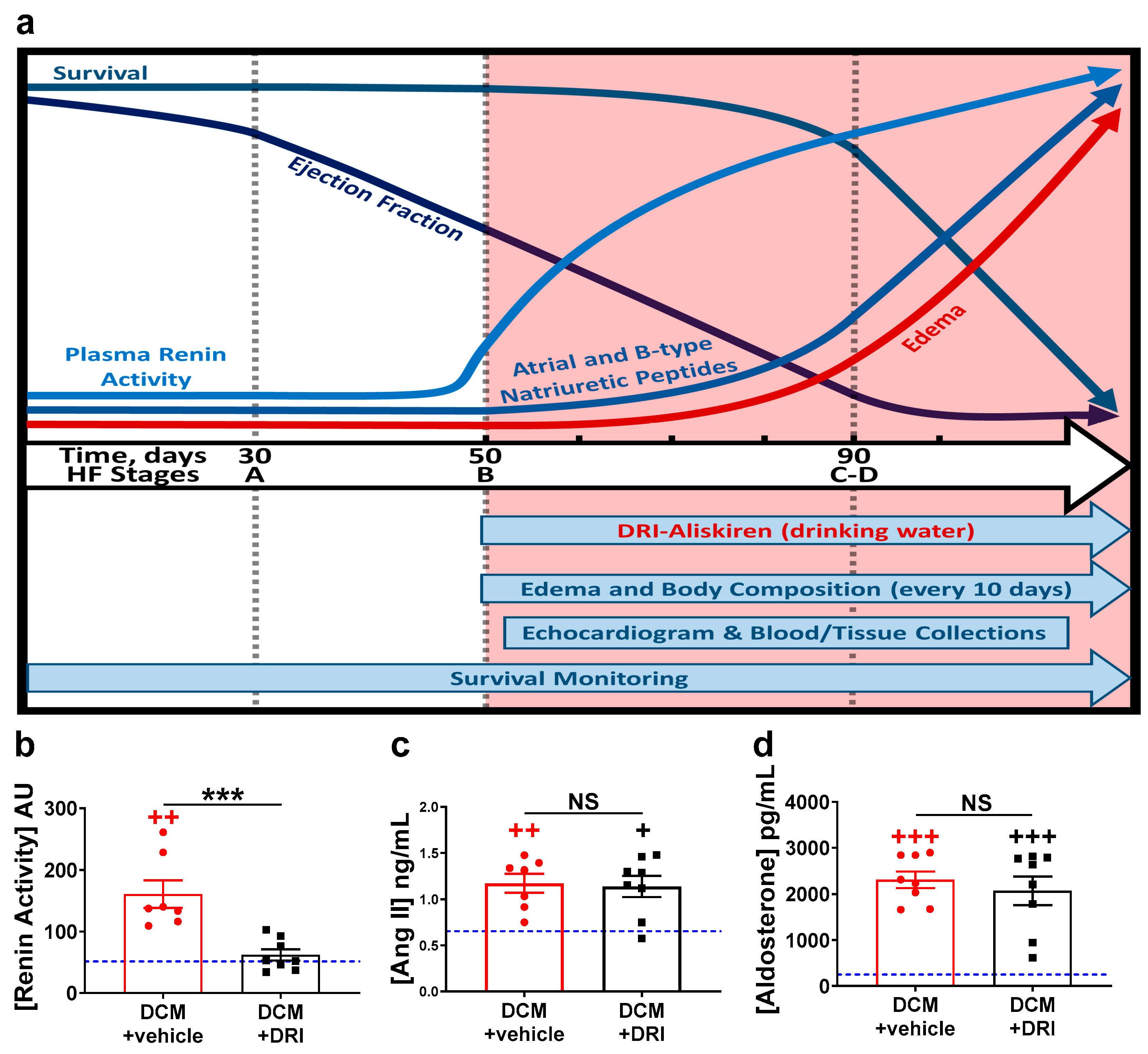
2.1. Normalization of Elevated Plasma Renin Activity
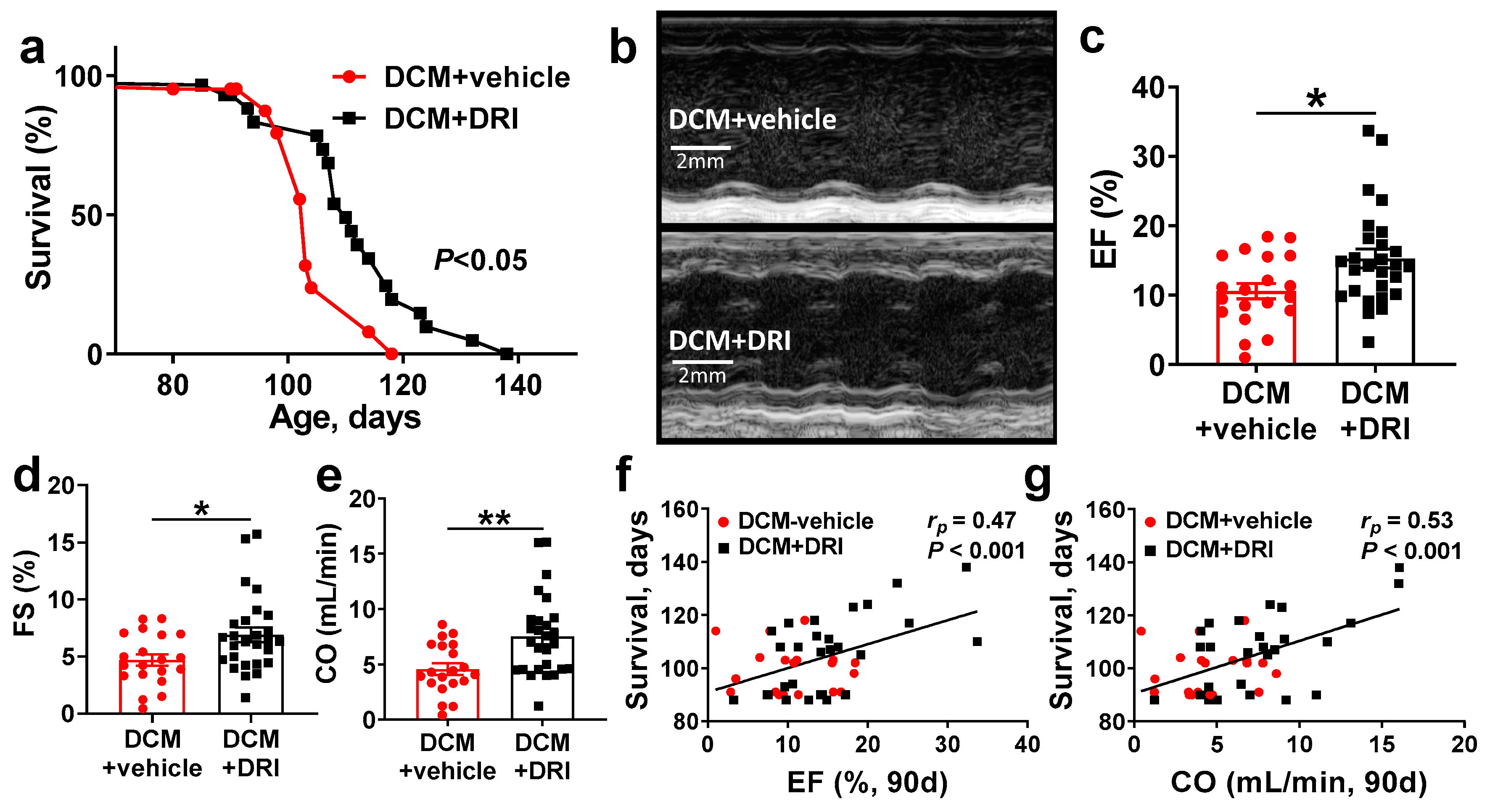
2.2. Normalization of Plasma Renin Activity Prolongs Survival and Delays Progression of Left Ventricular Systolic Dysfunction
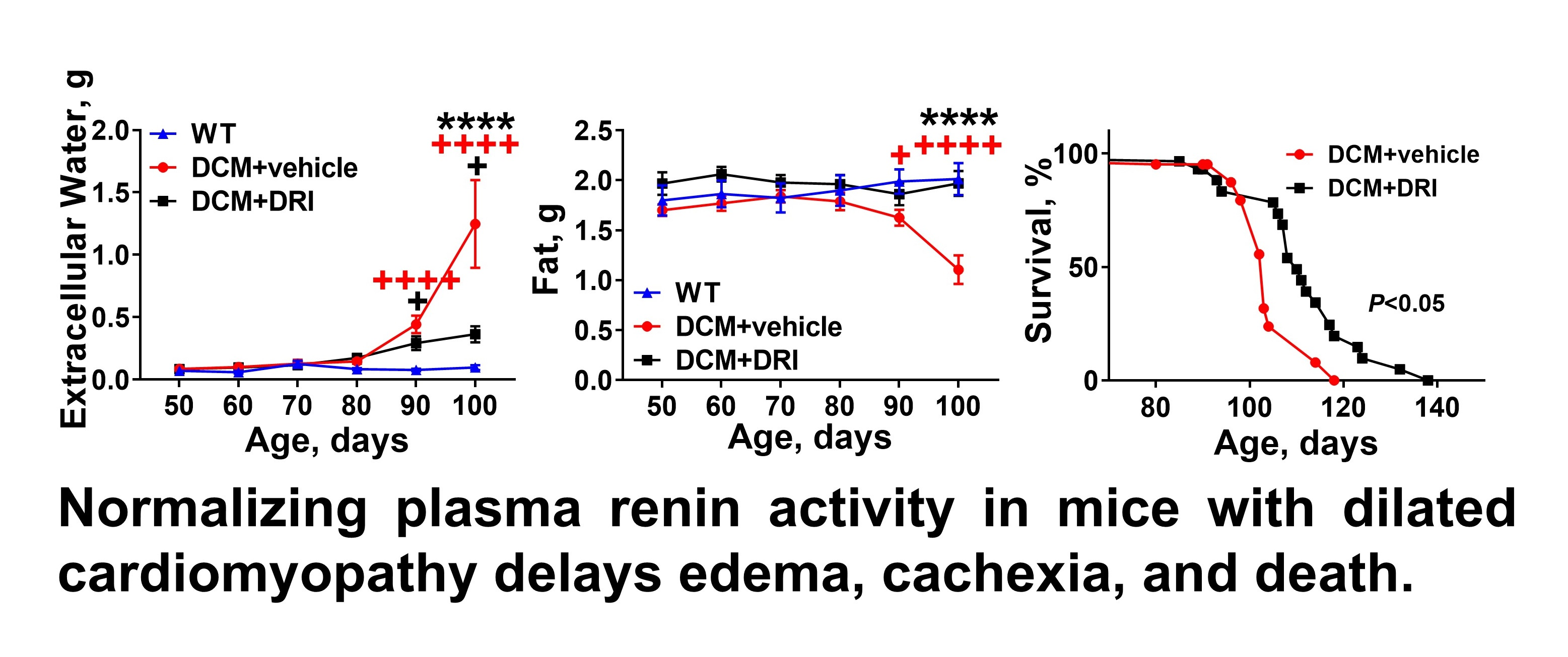
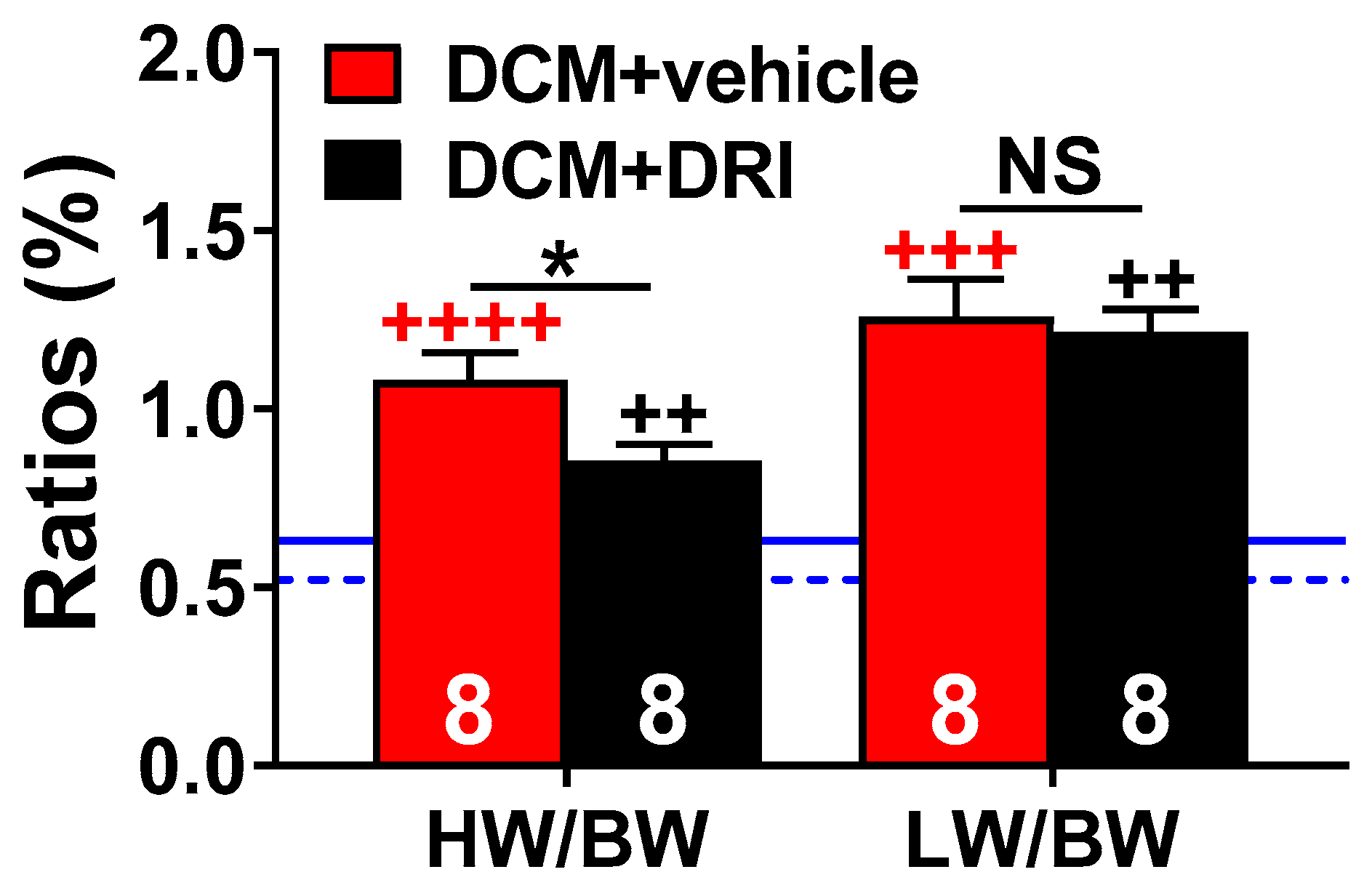
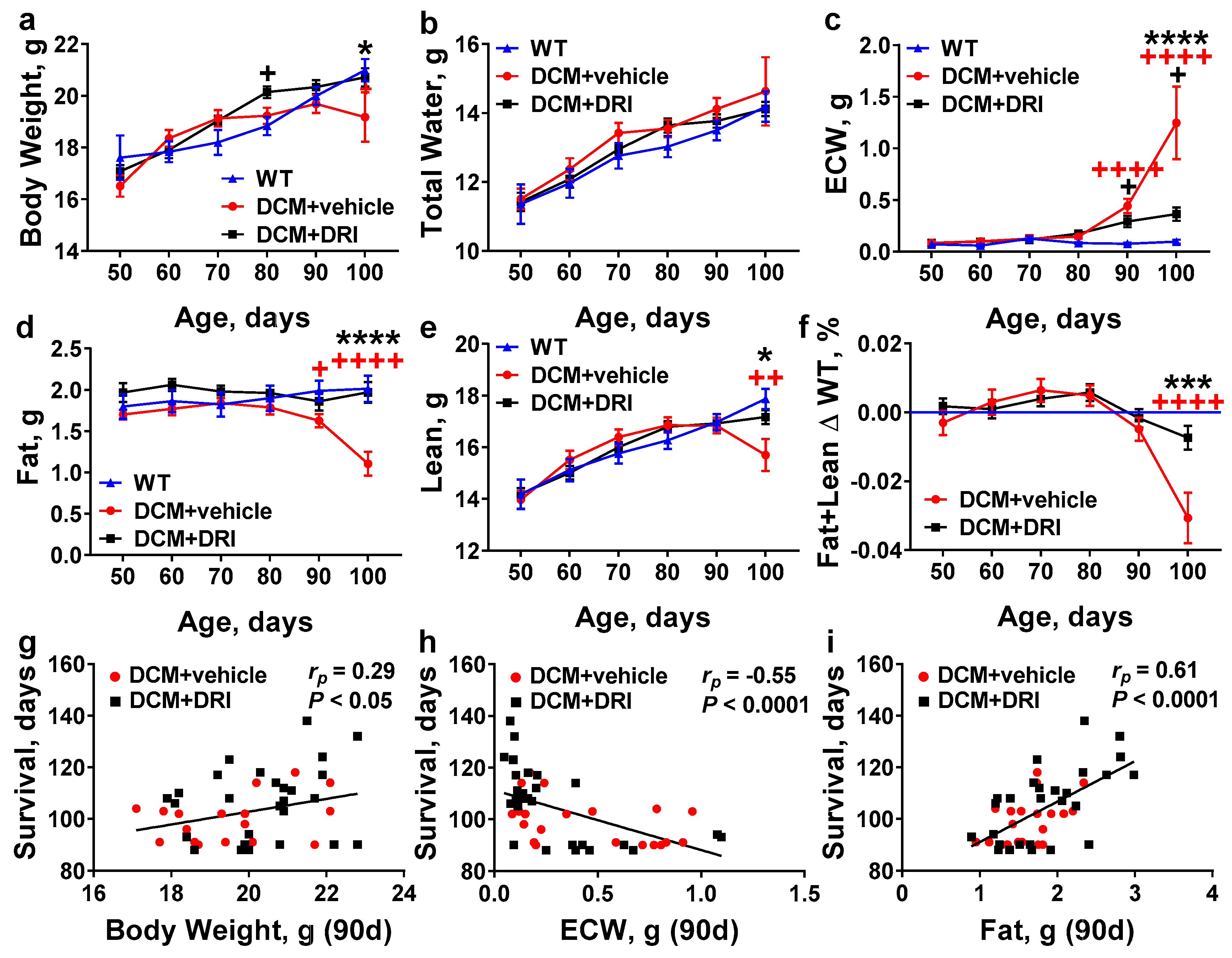
2.3. Normalization of Plasma Renin Activity Delays Development of Systemic Edema and Cachexia/Sarcopenia
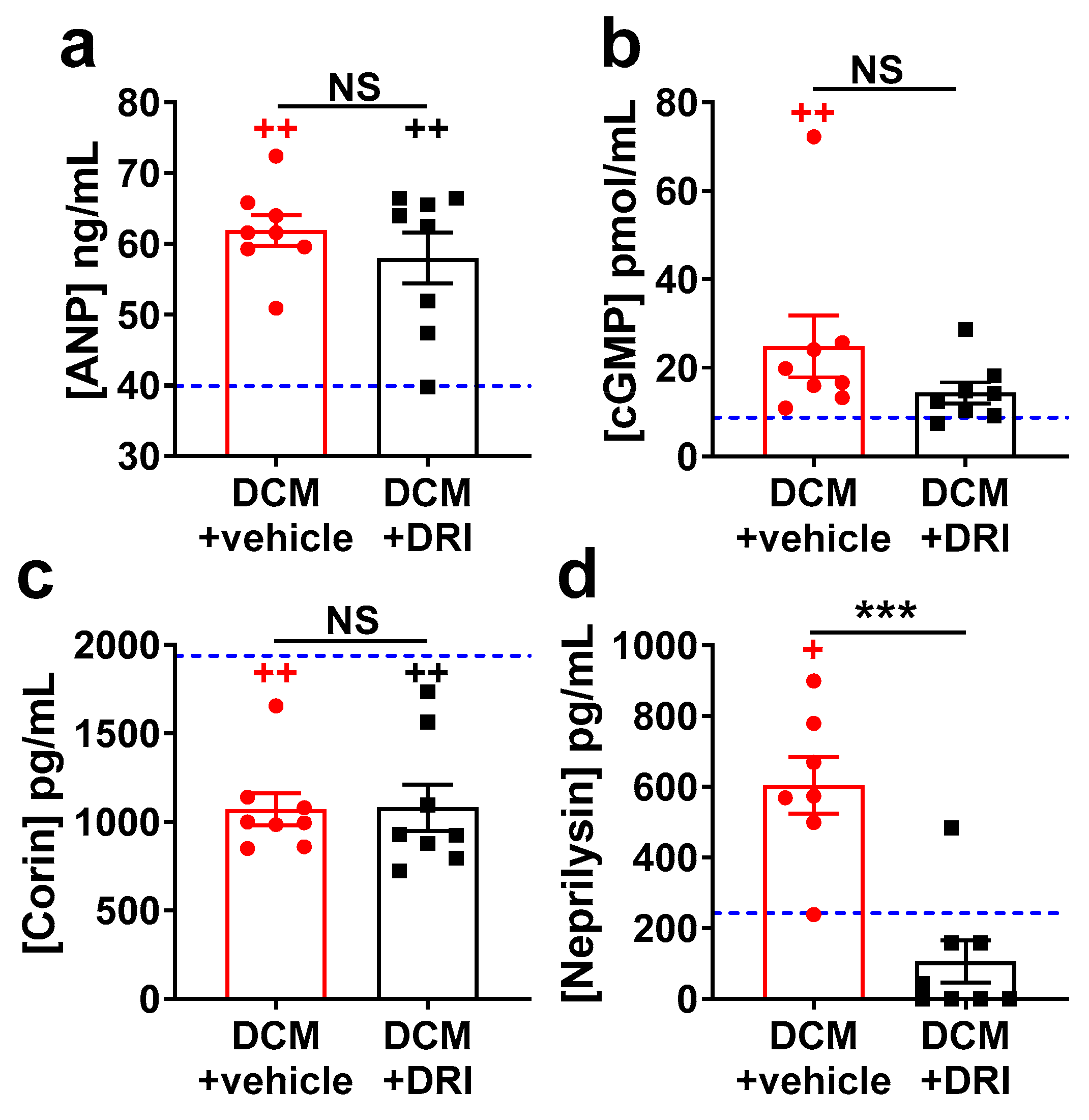
2.4. Heart Failure Plasma Biomarkers Independently Respond to Direct Renin Inhibition.
3. Discussion
4. Materials and Methods
4.1. Institution and Environment
4.2. Mice
4.3. Direct Renin-Inhibitor Treatment
4.4. Body Composition
4.5. Echocardiography
4.6. Enzyme Immunoassay
4.7. Plasma Renin Activity Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HF | heart failure |
| rEF | reduced ejection fraction |
| DCM | renin-angiotensin-aldosterone-system |
| RAAS | linear dichroism |
| NP | natriuretic peptide |
| ANP | atrial natriuretic peptide |
| BNP | b-type natriuretic peptide |
| Ang II | angiotensin II |
| Ang (1-7) | angiotensin (1-7) |
| DRI | direct renin inhibitor |
| NEP | neprilysin |
| cGMP | cyclic guanosine monophosphate |
| ARC | active renin concentration |
| ELISA | enzyme-linked immunosorbent assay |
| AU | arbitrary units |
| SEM | standard error of the mean |
| WT | wild type |
| HW/BW | heart weight to bodyweight ratio |
| LW/BW | lung weight to bodyweight ratio |
| QMR | quantitative magnetic resonance |
| ECW | extracellular water |
| LV | left ventricle |
| LVMc | left ventricular mass corrected |
| PRA | plasma renin activity |
| ARC | active renin concentration |
| APRC | active plasma renin concentration |
References
- Schrier, R.W.; Abraham, W.T. Hormones and hemodynamics in heart failure. N. Engl. J. Med. 1999, 341, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Carnovali, M.; Mastromarino, V. The natriuretic peptides system in the pathophysiology of heart failure: From molecular basis to treatment. Clin. Sci. (Lond.) 2016, 130, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; van der Meer, P.; van Tintelen, J.P.; van den Berg, M.P. Sex differences in cardiomyopathies. Eur. J. Heart Fail. 2014, 16, 238–247. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Struthers, A.D.; Lang, C.C. Modulation of the renin-angiotensin-aldosterone system in heart failure. Curr. Atheroscler Rep. 2014, 16, 403. [Google Scholar] [CrossRef] [PubMed]
- Sartiani, L.; Spinelli, V.; Laurino, A.; Blescia, S.; Raimondi, L.; Cerbai, E.; Mugelli, A. Pharmacological perspectives in sarcopenia: A potential role for renin-angiotensin system blockers? Clin. Cases. Min. Bone Metab. 2015, 12, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Di Palo, K.E.; Pina, I.L. Sex differences in heart failure. Clin. Cardiol. 2018, 41, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.; Fonarow, G.C.; Givertz, M.M.; et al. 2016 ACC/AHA/HFSA Focused Update on New Pharmacological Therapy for Heart Failure: An Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J. Am. Coll. Cardiol. 2016, 68, 1476–1488. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef] [PubMed]
- Akwo, E.A.; Kabagambe, E.K.; Wang, T.J.; Harrell, F.E., Jr.; Blot, W.J.; Mumma, M.; Gupta, D.K.; Lipworth, L. Heart Failure Incidence and Mortality in the Southern Community Cohort Study. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.F.; Claggett, B.; Shah, A.M.; Liu, J.; Shah, S.J.; Anand, I.; O’Meara, E.; Sweitzer, N.K.; Rouleau, J.L.; Fang, J.C.; et al. Racial Differences in Characteristics and Outcomes of Patients With Heart Failure and Preserved Ejection Fraction in the Treatment of Preserved Cardiac Function Heart Failure Trial. Circ. Heart Fail. 2018, 11, e004457. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Severino, P.; Calcagno, S.; Mancone, M. Heart failure: TNM-like classification. J. Am. Coll. Cardiol. 2014, 63, 1959–1960. [Google Scholar] [CrossRef] [PubMed]
- Severino, P.; Mestrini, V.; Mariani, M.V.; Birtolo, L.I.; Scarpati, R.; Mancone, M.; Fedele, F. Structural and myocardial dysfunction in heart failure beyond ejection fraction. Heart Fail. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef]
- Fedele, F.; Mancone, M.; Adamo, F.; Severino, P. Heart Failure With Preserved, Mid-Range, and Reduced Ejection Fraction: The Misleading Definition of the New Guidelines. Cardiol. Rev. 2017, 25, 4–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orsborne, C.; Chaggar, P.S.; Shaw, S.M.; Williams, S.G. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: The past, the present and the future. Postgrad. Med. J. 2017, 93, 29–37. [Google Scholar] [CrossRef]
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Seferovic, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef]
- Antman, E.M.; Loscalzo, J. Precision medicine in cardiology. Nat. Rev. Cardiol. 2016, 13, 591–602. [Google Scholar] [CrossRef]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Fentzke, R.C.; Korcarz, C.E.; Lang, R.M.; Lin, H.; Leiden, J.M. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J. Clin. Invest. 1998, 101, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Birdsey, N.; Huggins, G.S.; Svensson, E.; Heppe, D.; Knaub, L. Cardiac-specific overexpression of dominant-negative CREB leads to increased mortality and mitochondrial dysfunction in female mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2056–H2068. [Google Scholar] [CrossRef] [PubMed]
- Gladysheva, I.P.; Wang, D.; McNamee, R.A.; Houng, A.K.; Mohamad, A.A.; Fan, T.M.; Reed, G.L. Corin overexpression improves cardiac function, heart failure, and survival in mice with dilated cardiomyopathy. Hypertension 2013, 61, 327–332. [Google Scholar] [CrossRef]
- Wang, D.; Gladysheva, I.P.; Fan, T.H.; Sullivan, R.; Houng, A.K.; Reed, G.L. Atrial natriuretic peptide affects cardiac remodeling, function, heart failure, and survival in a mouse model of dilated cardiomyopathy. Hypertension 2014, 63, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Wang, D.; Sullivan, R.; Fan, T.H.; Gladysheva, I.P.; Reed, G.L. Depressed Corin Levels Indicate Early Systolic Dysfunction Before Increases of Atrial Natriuretic Peptide/B-Type Natriuretic Peptide and Heart Failure Development. Hypertension 2016, 67, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Sullivan, R.; Fan, T.M.; Wang, D.; Sun, Y.; Reed, G.L.; Gladysheva, I.P. Enhanced heart failure, mortality and renin activation in female mice with experimental dilated cardiomyopathy. PLoS ONE 2017, 12, e0189315. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Skeggs, L.T., Jr.; Kahn, J.R.; Lentz, K.; Shumway, N.P. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J. Exp. Med. 1957, 106, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T. Aldosterone in congestive heart failure. N. Engl. J. Med. 2001, 345, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Seto, S.W.; Golledge, J. Angiotensin II, sympathetic nerve activity and chronic heart failure. Heart Fail. Rev. 2014, 19, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Reed, G.L.; Gladysheva, I.P. Renin Activity in Heart Failure with Reduced Systolic Function—New Insights. Int. J. Mol. Sci. 2019, 20, 3182. [Google Scholar] [CrossRef] [PubMed]
- Mentz, R.J.; Stevens, S.R.; DeVore, A.D.; Lala, A.; Vader, J.M.; AbouEzzeddine, O.F.; Khazanie, P.; Redfield, M.M.; Stevenson, L.W.; O’Connor, C.M.; et al. Decongestion strategies and renin-angiotensin-aldosterone system activation in acute heart failure. JACC Heart Fail. 2015, 3, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Seed, A.; Gardner, R.; McMurray, J.; Hillier, C.; Murdoch, D.; MacFadyen, R.; Bobillier, A.; Mann, J.; McDonagh, T. Neurohumoral effects of the new orally active renin inhibitor, aliskiren, in chronic heart failure. Eur. J. Heart Fail. 2007, 9, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Bohm, M.; Greene, S.J.; Fonarow, G.C.; Lewis, E.F.; Zannad, F.; Solomon, S.D.; Baschiera, F.; Botha, J.; Hua, T.A.; et al. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: The ASTRONAUT randomized trial. JAMA 2013, 309, 1125–1135. [Google Scholar] [CrossRef]
- Schroten, N.F.; Damman, K.; Hemmelder, M.H.; Voors, A.A.; Navis, G.; Gaillard, C.A.; van Veldhuisen, D.J.; Van Gilst, W.H.; Hillege, H.L. Effect of additive renin inhibition with aliskiren on renal blood flow in patients with Chronic Heart Failure and Renal Dysfunction (Additive Renin Inhibition with Aliskiren on renal blood flow and Neurohormonal Activation in patients with Chronic Heart Failure and Renal Dysfunction). Am. Heart J. 2015, 169, 693–701. [Google Scholar]
- McMurray, J.J.; Krum, H.; Abraham, W.T.; Dickstein, K.; Kober, L.V.; Desai, A.S.; Solomon, S.D.; Greenlaw, N.; Ali, M.A.; Chiang, Y.; et al. Aliskiren, Enalapril, or Aliskiren and Enalapril in Heart Failure. N Engl J. Med. 2016, 374, 1521–1532. [Google Scholar] [CrossRef]
- Wal, P.; Wal, A.; Rai, A.K.; Dixit, A. Aliskiren: An orally active renin inhibitor. J. Pharm. Bioallied. Sci. 2011, 3, 189–193. [Google Scholar] [CrossRef]
- Liu, H.; Luo, H.; Wang, S.; Zhang, C.; Hao, J.; Gao, C. Aliskiren for heart failure: A systematic review and meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 88189–88198. [Google Scholar] [CrossRef]
- Yamada, C.; Kuwahara, K.; Yamazaki, M.; Nakagawa, Y.; Nishikimi, T.; Kinoshita, H.; Kuwabara, Y.; Minami, T.; Yamada, Y.; Shibata, J.; et al. The renin-angiotensin system promotes arrhythmogenic substrates and lethal arrhythmias in mice with non-ischaemic cardiomyopathy. Cardiovasc. Res. 2016, 109, 162–173. [Google Scholar] [CrossRef]
- Anker, S.D.; Sharma, R. The syndrome of cardiac cachexia. Int. J. Cardiol. 2002, 85, 51–66. [Google Scholar] [CrossRef]
- Fischer, M.; Baessler, A.; Schunkert, H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc. Res. 2002, 53, 672–677. [Google Scholar] [CrossRef]
- Muller, D.N.; Derer, W.; Dechend, R. Aliskiren--mode of action and preclinical data. J. Mol. Med. (Berl) 2008, 86, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Shin, S.H.; Shah, A.; Skali, H.; Desai, A.; Kober, L.; Maggioni, A.P.; Rouleau, J.L.; Kelly, R.Y.; Hester, A.; et al. Aliskiren Study in Post, M.I.P. t. R. R. I., Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur. Heart J. 2011, 32, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Goliasch, G.; Wurm, R.; Novak, J.; Strunk, G.; Gyongyosi, M.; Poglitsch, M.; Saemann, M.D.; Hulsmann, M. Low- and High-renin Heart Failure Phenotypes with Clinical Implications. Clin. Chem. 2018, 64, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Riad, A.; Lettau, O.; Roks, A.; Savvatis, K.; Becher, P.M.; Escher, F.; Jan Danser, A.H.; Schultheiss, H.P.; Tschope, C. Renin inhibition improves cardiac function and remodeling after myocardial infarction independent of blood pressure. Hypertension 2008, 52, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Yong, Q.C.; Seqqat, R.; Chandel, N.; Feldman, D.L.; Baker, K.M.; Kumar, R. Direct renin inhibition prevents cardiac dysfunction in a diabetic mouse model: Comparison with an angiotensin receptor antagonist and angiotensin-converting enzyme inhibitor. Clin. Sci. (Lond) 2013, 124, 529–541. [Google Scholar] [CrossRef]
- Connelly, K.A.; Advani, A.; Advani, S.; Zhang, Y.; Thai, K.; Thomas, S.; Krum, H.; Kelly, D.J.; Gilbert, R.E. Combination angiotensin converting enzyme and direct renin inhibition in heart failure following experimental myocardial infarction. Cardiovasc. Ther. 2013, 31, 84–91. [Google Scholar] [CrossRef]
- Yang, N.I.; Liao, C.C.; Hung, M.J.; Cherng, W.J. Direct Renin Inhibitor Attenuates Left Ventricular Remodeling in Post-Myocardial Infarction Heart Failure Mice. Acta Cardiol. Sin. 2013, 29, 160–167. [Google Scholar]
- Koid, S.S.; Ziogas, J.; Campbell, D.J. Aliskiren reduces myocardial ischemia-reperfusion injury by a bradykinin B2 receptor- and angiotensin AT2 receptor-mediated mechanism. Hypertension 2014, 63, 768–773. [Google Scholar] [CrossRef]
- Desjarlais, M.; Dussault, S.; Dhahri, W.; Mathieu, R.; Rivard, A. Direct renin inhibition with aliskiren improves ischemia-induced neovascularization: Blood pressure-independent effect. Atherosclerosis 2015, 242, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Toma, M.; Ezekowitz, J.A.; Bakal, J.A.; O’Connor, C.M.; Hernandez, A.F.; Sardar, M.R.; Zolty, R.; Massie, B.M.; Swedberg, K.; Armstrong, P.W.; et al. The relationship between left ventricular ejection fraction and mortality in patients with acute heart failure: Insights from the ASCEND-HF Trial. Eur. J. Heart Fail. 2014, 16, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Henriquez-Palop, F.; Anton-Perez, G.; Marrero-Robayna, S.; Gonzalez-Cabrera, F.; Rodriguez-Perez, J.C. Water overload as a biomarker for heart failure and acute renal failure. Nefrologia 2013, 33, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Choi, M.J.; Lee, J.H.; Oh, J.E.; Seo, J.W.; Lee, Y.K.; Yoon, J.W.; Kim, H.J.; Noh, J.W.; Koo, J.R. Extracellular Fluid/Intracellular Fluid Volume Ratio as a Novel Risk Indicator for All-Cause Mortality and Cardiovascular Disease in Hemodialysis Patients. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Pellicori, P.; Kaur, K.; Clark, A.L. Fluid Management in Patients with Chronic Heart Failure. Card. Fail. Rev. 2015, 1, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Okoshi, M.P.; Capalbo, R.V.; Romeiro, F.G.; Okoshi, K. Cardiac Cachexia: Perspectives for Prevention and Treatment. Arq. Bras. Cardiol. 2017, 108, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Jaffrin, M.Y.; Morel, H. Body fluid volumes measurements by impedance: A review of bioimpedance spectroscopy (BIS) and bioimpedance analysis (BIA) methods. Med. Eng. Phys. 2008, 30, 1257–1269. [Google Scholar] [CrossRef]
- Kovner, I.; Taicher, G.Z.; Mitchell, A.D. Calibration and validation of EchoMRI whole body composition analysis based on chemical analysis of piglets, in comparison with the same for DXA. Int. J. Body Compos. Res. 2010, 8, 17–29. [Google Scholar]
- Gallagher, D.; Thornton, J.C.; He, Q.; Wang, J.; Yu, W.; Bradstreet, T.E.; Burke, J.; Heymsfield, S.B.; Rivas, V.M.; Kaufman, R. Quantitative magnetic resonance fat measurements in humans correlate with established methods but are biased. Obes. (Silver Spring) 2010, 18, 2047–2054. [Google Scholar] [CrossRef]
- Metzinger, M.N.; Miramontes, B.; Zhou, P.; Liu, Y.; Chapman, S.; Sun, L.; Sasser, T.A.; Duffield, G.E.; Stack, M.S.; Leevy, W.M. Correlation of X-ray computed tomography with quantitative nuclear magnetic resonance methods for pre-clinical measurement of adipose and lean tissues in living mice. Sensors 2014, 14, 18526–18542. [Google Scholar] [CrossRef]
- Toro-Ramos, T.; Paley, C.; Wong, W.W.; Pi-Sunyer, F.X.; Yu, W.W.; Thornton, J.; Gallagher, D. Reliability of the EchoMRI Infants System for Water and Fat Measurements in Newborns. Obes. (Silver Spring) 2017, 25, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.S.; Ward, R.D.; Ramanathan, K.; Yu, X.; Gladysheva, I.P.; Reed, G.L. Possible Enzymatic Downregulation of the Natriuretic Peptide System in Patients with Reduced Systolic Function and Heart Failure: A Pilot Study. BioMed. Res. Int. 2018, 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Ibebuogu, U.N.; Gladysheva, I.P.; Houng, A.K.; Reed, G.L. Decompensated heart failure is associated with reduced corin levels and decreased cleavage of pro-atrial natriuretic peptide. Circ. Heart Fail. 2011, 4, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef] [PubMed]
- McKee, P.A.; Castelli, W.P.; McNamara, P.M.; Kannel, W.B. The natural history of congestive heart failure: The Framingham study. N. Engl. J. Med. 1971, 285, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Barallat, J.; Galan, A.; de Antonio, M.; Domingo, M.; Zamora, E.; Urrutia, A.; Lupon, J. Soluble neprilysin is predictive of cardiovascular death and heart failure hospitalization in heart failure patients. J. Am. Coll. Cardiol. 2015, 65, 657–665. [Google Scholar] [CrossRef]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef]
- Von Lueder, T.G.; Sangaralingham, S.J.; Wang, B.H.; Kompa, A.R.; Atar, D.; Burnett, J.C., Jr.; Krum, H. Renin-angiotensin blockade combined with natriuretic peptide system augmentation: Novel therapeutic concepts to combat heart failure. Circ. Heart Fail. 2013, 6, 594–605. [Google Scholar] [CrossRef]
- Vodovar, N.; Seronde, M.F.; Laribi, S.; Gayat, E.; Lassus, J.; Januzzi, J.L., Jr.; Boukef, R.; Nouira, S.; Manivet, P.; Samuel, J.L.; et al. Elevated Plasma B-Type Natriuretic Peptide Concentrations Directly Inhibit Circulating Neprilysin Activity in Heart Failure. JACC Heart Fail. 2015, 3, 629–636. [Google Scholar] [CrossRef]
- Campbell, D.J.; Zhang, Y.; Kelly, D.J.; Gilbert, R.E.; McCarthy, D.J.; Shi, W.; Smyth, G.K. Aliskiren increases bradykinin and tissue kallikrein mRNA levels in the heart. Clin. Exp. Pharm. Physiol. 2011, 38, 623–631. [Google Scholar] [CrossRef]
- Hirose, T.; Mori, N.; Totsune, K.; Morimoto, R.; Maejima, T.; Kawamura, T.; Metoki, H.; Asayama, K.; Kikuya, M.; Ohkubo, T.; et al. Gene expression of (pro)renin receptor is upregulated in hearts and kidneys of rats with congestive heart failure. Peptides 2009, 30, 2316–2322. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, P.; Masson, S.; Barlera, S.; Girerd, N.; Castelnovo, A.; Zannad, F.; Clemenza, F.; Tognoni, G.; Anand, I.S.; Cohn, J.N.; et al. Loss in body weight is an independent prognostic factor for mortality in chronic heart failure: Insights from the GISSI-HF and Val-HeFT trials. Eur. J. Heart Fail. 2015, 17, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Groban, L. Role of the renin-angiotensin system in age-related sarcopenia and diastolic dysfunction. Aging Health 2008, 4, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Sukhanov, S.; Semprun-Prieto, L.; Yoshida, T.; Michael Tabony, A.; Higashi, Y.; Galvez, S.; Delafontaine, P. Angiotensin II, oxidative stress and skeletal muscle wasting. Am. J. Med. Sci. 2011, 342, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Huggins, G.S.; Lepore, J.J.; Greytak, S.; Patten, R.; McNamee, R.; Aronovitz, M.; Wang, P.J.; Reed, G.L. The CREB leucine zipper regulates CREB phosphorylation, cardiomyopathy, and lethality in a transgenic model of heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1877–H1882. [Google Scholar] [CrossRef]
- Poss, J.; Werner, C.; Lorenz, D.; Gensch, C.; Bohm, M.; Laufs, U. The renin inhibitor aliskiren upregulates pro-angiogenic cells and reduces atherogenesis in mice. Basic. Res. Cardiol. 2010, 105, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Sullivan, R.D.; You, D.; Zafar, N.; He Yang, C.; Thiyagarajan, T.; Johnson, D.L.; Barrett, M.L.; Koehler, N.J.; Star, M.; et al. Androgen receptor agonists increase lean mass, improve cardiopulmonary functions and extend survival in preclinical models of Duchenne muscular dystrophy. Hum. Mol. Genet. 2017, 26, 2526–2540. [Google Scholar] [CrossRef]
- Schmiedt, C.W.; Hurley, K.A.; Tong, X.; Rakhmanova, V.A.; Po, C.L.; Hurley, D.J. Measurement of plasma renin concentration in cats by use of a fluorescence resonance energy transfer peptide substrate of renin. Am. J. Vet. Res. 2009, 70, 1315–1322. [Google Scholar] [CrossRef]
- Prysyazhna, O.; Rudyk, O.; Eaton, P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 2012, 18, 286–290. [Google Scholar] [CrossRef]
- Kakoki, M.; Pochynyuk, O.M.; Hathaway, C.M.; Tomita, H.; Hagaman, J.R.; Kim, H.S.; Zaika, O.L.; Mamenko, M.; Kayashima, Y.; Matsuki, K.; et al. Primary aldosteronism and impaired natriuresis in mice underexpressing TGFbeta1. Proc. Natl. Acad. Sci. USA 2013, 110, 5600–5605. [Google Scholar] [CrossRef]
- Takada, S.; Kinugawa, S.; Hirabayashi, K.; Suga, T.; Yokota, T.; Takahashi, M.; Fukushima, A.; Homma, T.; Ono, T.; Sobirin, M.A.; et al. Angiotensin II receptor blocker improves the lowered exercise capacity and impaired mitochondrial function of the skeletal muscle in type 2 diabetic mice. J. Appl. Physiol. 2013, 114, 844–857. [Google Scholar] [CrossRef] [PubMed][Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Gladysheva, I.P.; Reed, G.L. Normalizing Plasma Renin Activity in Experimental Dilated Cardiomyopathy: Effects on Edema, Cachexia, and Survival. Int. J. Mol. Sci. 2019, 20, 3886. https://doi.org/10.3390/ijms20163886
Sullivan RD, Mehta RM, Tripathi R, Gladysheva IP, Reed GL. Normalizing Plasma Renin Activity in Experimental Dilated Cardiomyopathy: Effects on Edema, Cachexia, and Survival. International Journal of Molecular Sciences. 2019; 20(16):3886. https://doi.org/10.3390/ijms20163886
Chicago/Turabian StyleSullivan, Ryan D., Radhika M. Mehta, Ranjana Tripathi, Inna P. Gladysheva, and Guy L. Reed. 2019. "Normalizing Plasma Renin Activity in Experimental Dilated Cardiomyopathy: Effects on Edema, Cachexia, and Survival" International Journal of Molecular Sciences 20, no. 16: 3886. https://doi.org/10.3390/ijms20163886
APA StyleSullivan, R. D., Mehta, R. M., Tripathi, R., Gladysheva, I. P., & Reed, G. L. (2019). Normalizing Plasma Renin Activity in Experimental Dilated Cardiomyopathy: Effects on Edema, Cachexia, and Survival. International Journal of Molecular Sciences, 20(16), 3886. https://doi.org/10.3390/ijms20163886