Retrotransposons in Plant Genomes: Structure, Identification, and Classification through Bioinformatics and Machine Learning



Abstract
1. Introduction
2. Structure, Diversity, Dynamics, and Function of Retrotransposons in Host Genomes
2.1. Retrotransposons Structure
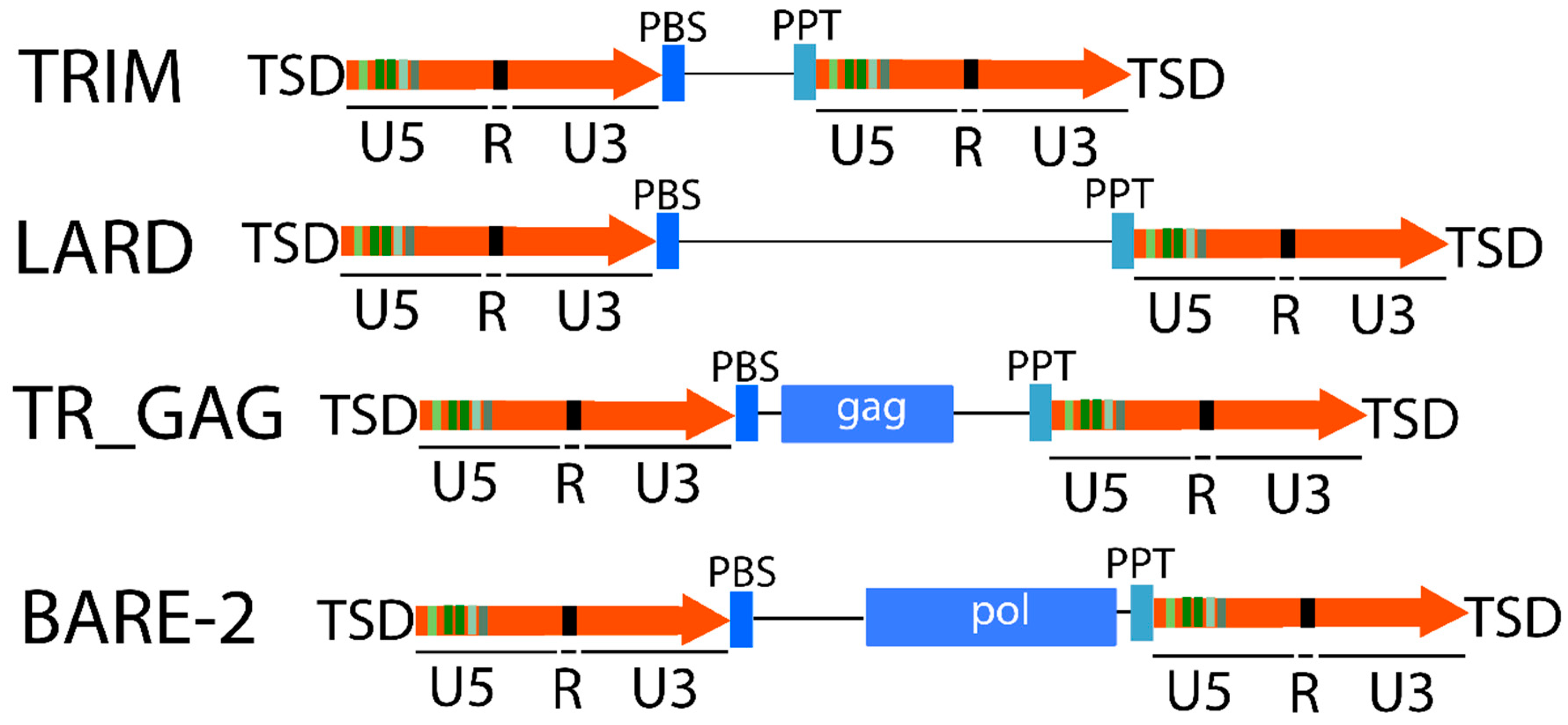
2.1.1. LTR Retrotransposons
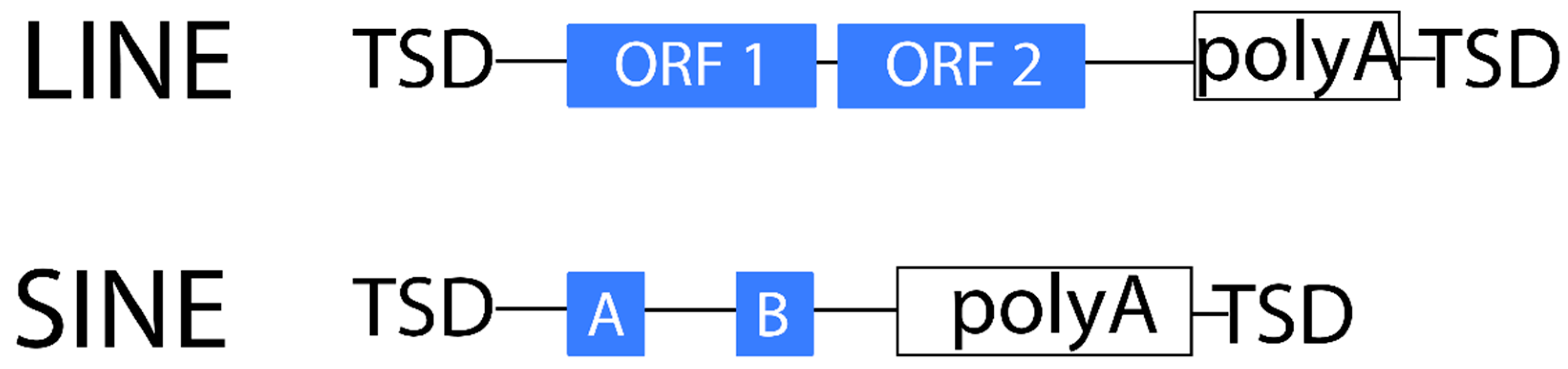
2.1.2. Non-LTR Retrotransposons
2.1.3. PLEs or Penelope-Like Elements
2.1.4. DIRS
2.2. Retrotransposon Dynamics
2.2.1. How are Retrotransposons Activated
2.2.2. How Are Retrotransposons Silenced
2.2.3. Horizontal Transfer of TEs
2.3. Function of Retrotransposons in a Chromosome’s Structure
2.3.1. Chromosomal Distribution of Retrotransposons
2.3.2. Sex-Specific Chromosomes
2.4. Interaction of Retrotransposons with Genes
3. Why Is It Important to Classify Retrotransposons (into Superfamilies and Lineages)?
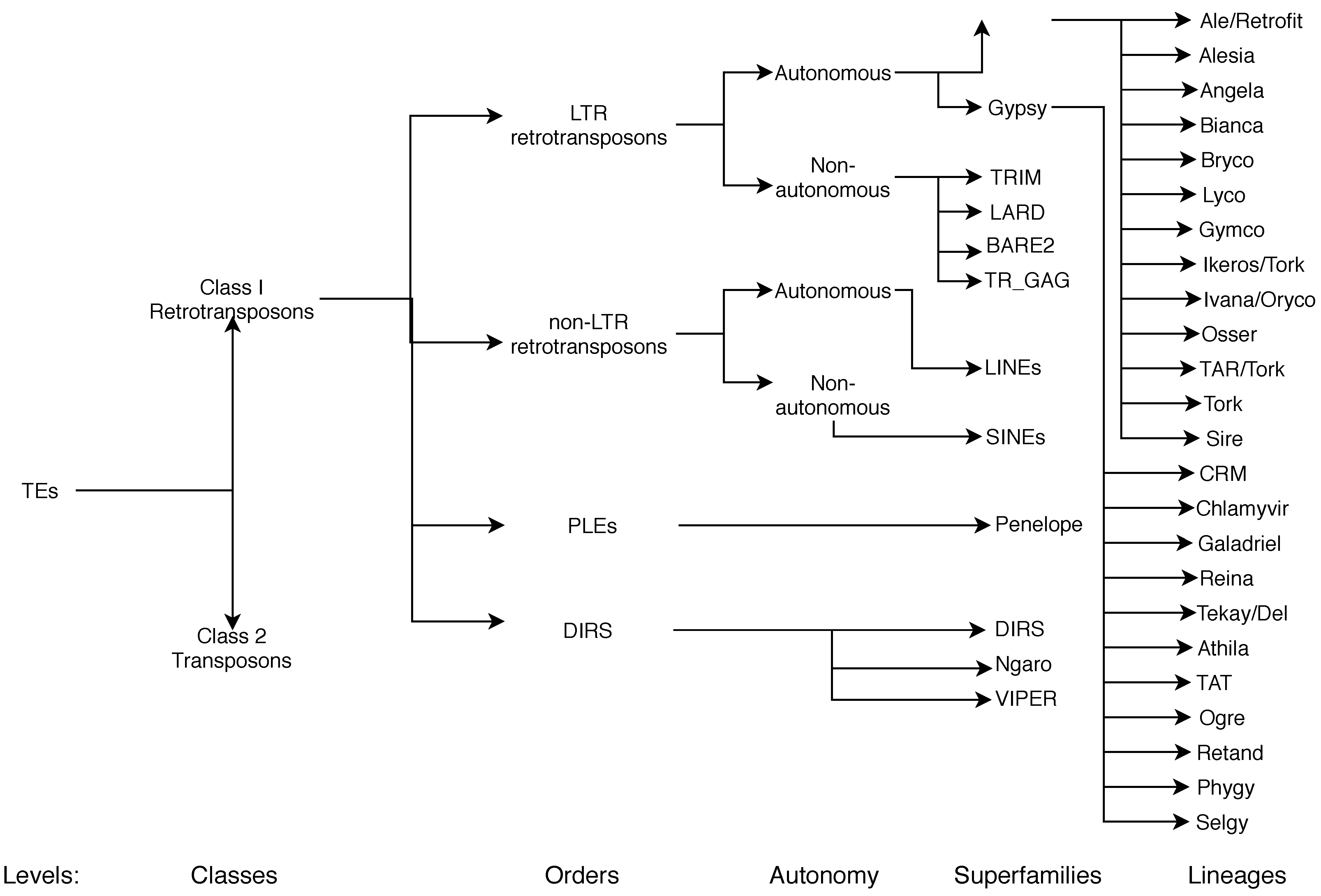
3.1. Current Classifications
3.2. Current Nomenclature
4. How to Identify and Classify Retrotransposons
4.1. Current Problems for Retrotransposon Identification and Classification
- The difficulties in constructing a representative and comprehensive library of TE sequences, since it depends on the sensibility and specificity of the bioinformatics programs used.
- Nested elements.
- The false identification of TEs (for example, large gene families).
- The difficulties in classifying non-autonomous elements.
4.2. Current Strategies and Methodologies
4.2.1. Structure-Based Methods
4.2.2. Homology-Based Methods
4.2.3. De Novo
4.2.4. Comparative Genomics
4.3. Most Popular Bioinformatics Resources
5. How can Machine Learning and Deep Learning Techniques Improve the Identification and Classification of Retrotransposons?
5.1. Current Machine Learning Techniques for Genomics and Transposable Elements
5.2. Current Deep Neural Networks Techniques for Genomics and Transposable Elements
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Aspartic protease |
| CR | centromeric retrotransposons |
| DIRS | Dictyostelium intermediate repeat sequence |
| DL | Deep Learning |
| DNN | Deep Neural Networks |
| ENV | Enveloppe |
| FISH | Fluorescent In Situ Hybridization |
| GAG | Group Specific Antigen |
| HC | Hierarchical Classification |
| HMM | Hidden Markov Models |
| HT | Horizontal Transfer |
| HTT | Horizontal Transfer of Transposable element |
| indel | Insersion-Deletion |
| INT | Integrase |
| LINE | Long Interspersed Nuclear Element |
| LTR | Long Terminal Repeat |
| LTR-RT | Long Terminal Repeat retrotransposon |
| ML | Machine learning |
| NGS | Next Generation Sequencing |
| ORF | Open Reading Frame |
| PBS | primer binding site |
| PPT | Poly-Purine Tract |
| PLEs | Penelope-like elements |
| RT | Reverse transcriptase |
| SINE | Short Interspersed Nuclear Element |
| SVM | Support Vector Machine |
| TE | transposable elements |
| TIR | Terminal Inverted Repeat |
| TSD | Target Site Duplication |
| UTR | Untranslated Regions |
References
- Mita, P.; Boeke, J.D. How Retrotransposons shape genome regulation. Curr. Opin. Genet. Dev. 2016, 37, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Keidar, D.; Doron, C.; Kashkush, K. Genome-wide analysis of a recently active retrotransposon, Au SINE, in wheat: Content, distribution within subgenomes and chromosomes, and gene associations. Plant Cell Rep. 2018, 37, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Chen, J.; Jiang, N. Assessing genome assembly quality using the LTR assembly index (LAI). Nucleic Acids Res. 2018, 46, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mustafin, R.N.; Khusnutdinova, E.K. The role of transposons in epigenetic regulation of ontogenesis. Russ. J. Dev. Biol. 2018, 49, 61–78. [Google Scholar] [CrossRef]
- Muszewska, A.; Hoffman-Sommer, M.; Grynberg, M. LTR Retrotransposons in Fungi. PLoS ONE 2011, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Arango-López, J.; Orozco-Arias, S.; Salazar, J.A.; Guyot, R. Application of data mining algorithms to classify biological data: The Coffea canephora genome case. In Colombian Conference on Computing; Springer: Cham, Switzerland, 2017; pp. 156–170. [Google Scholar] [CrossRef]
- Chaparro, C.; Gayraud, T.; de Souza, R.F.; Domingues, D.S.; Akaffou, S.; Laforga Vanzela, A.L.; de Kochko, A.; Rigoreau, M.; Crouzillat, D.; Hamon, S.; et al. Terminal-repeat retrotransposons with GAG domain in plant genomes: A new testimony on the complex world of transposable elements. Genome Biol. Evol. 2015, 7, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Grandbastien, M.-A. LTR Retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim. Biophys. Acta 2015, 1849, 403–416. [Google Scholar] [CrossRef]
- Gao, D.; Jimenez-Lopez, J.C.; Iwata, A.; Gill, N.; Jackson, S.A. Functional and structural divergence of an unusual LTR retrotransposon family in plants. PLoS ONE 2012, 7, e48595. [Google Scholar] [CrossRef]
- Rahman, A.Y.A.; Usharraj, A.O.; Misra, B.B.; Thottathil, G.P.; Jayasekaran, K.; Feng, Y.; Hou, S.; Ong, S.Y.; Ng, F.L.; Lee, L.S.; et al. Draft genome sequence of the rubber tree hevea brasiliensis. BMC Genomics 2013, 14, 75. [Google Scholar] [CrossRef]
- Gao, D.; Chen, J.; Chen, M.; Meyers, B.C.; Jackson, S. A Highly conserved, small LTR retrotransposon that preferentially targets genes in grass genomes. PLoS ONE 2012, 7, e32010. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Muñoz-Pomer, A.; Bernad, L.; Botella, H.; Moya, A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct 2009, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Futami, R.; Covelli, L.; Domínguez-Escribá, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodríguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2010, gkq1061. [Google Scholar] [CrossRef] [PubMed]
- De Castro Nunes, R.; Orozco-Arias, S.; Crouzillat, D.; Mueller, L.A.; Strickler, S.R.; Descombes, P.; Fournier, C.; Moine, D.; de Kochko, A.; Yuyama, P.M.; et al. Structure and distribution of centromeric retrotransposons at diploid and allotetraploid Coffea centromeric and pericentromeric regions. Front. Plant Sci. 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Novák, P.; Hoštáková, N.; MacAs, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, T.; Fonseca, N.; Camacho, R. Application of Machine Learning Techniques on the Discovery and Annotation of Transposons in Genomes. Master’s Thesis, Faculdade de Engenharia, Universidade Do Porto, Porto, Portugal, 2012. [Google Scholar]
- Girgis, H.Z. Red: An intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinform. 2015, 16, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zytnicki, M.; Akhunov, E.; Quesneville, H. Tedna: A transposable element de novo assembler. Bioinformatics 2014, 30, 2656–2658. [Google Scholar] [CrossRef] [PubMed]
- Staton, S.E.; Burke, J.M. Transposome: A toolkit for annotation of transposable element families from unassembled sequence reads. Bioinformatics 2015, 31, 1827–1829. [Google Scholar] [CrossRef]
- Chu, C.; Nielsen, R.; Wu, Y. REPdenovo: Inferring de novo repeat motifs from short sequence reads. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Monat, C.; Tando, N.; Tranchant-Dubreuil, C.; Sabot, F. LTRclassifier: A website for fast structural LTR retrotransposons classification in plants. Mob. Genet. Elem. 2016, 6, e1241050. [Google Scholar] [CrossRef] [PubMed]
- Orozco-arias, S.; Liu, J.; Id, R.T.; Ceballos, D.; Silva, D.; Id, D.; Ming, R.; Guyot, R. Inpactor, integrated and parallel analyzer and classifier of LTR retrotransposons and its application for pineapple LTR retrotransposons diversity and dynamics. Biology 2018. [Google Scholar] [CrossRef] [PubMed]
- Larrañaga, P.; Calvo, B.; Santana, R.; Bielza, C.; Galdiano, J.; Inza, I.; Lozano, J.A.; Armañanzas, R.; Santafé, G.; Pérez, A.; et al. Machine Learning in Bioinformatics. Brief. Bioinform. 2006, 7, 86–112. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Schietgat, L.; Vens, C.; Cerri, R.; Fischer, C.N.; Costa, E.; Ramon, J.; Carareto, C.M.A.; Blockeel, H. A machine learning based framework to identify and classify long terminal repeat retrotransposons. PLoS Comput. Biol. 2018, 14, e1006097. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Garzón, J.A.; Díaz, F. Boosting the detection of transposable elements using machine learning. Adv. Intell. Syst. Comput. 2013, 222, 15–22. [Google Scholar] [CrossRef]
- Santos, B.Z.; Cerri, R.; Lu, R.W. A new machine learning dataset for hierarchical classification of transposable elements. In Proceedings of the XIII Encontro Nacional de Inteligência Artificial-ENIAC, Sao Paulo, Brazil, 9–12 October 2016; pp. 661–672. [Google Scholar]
- Makałowski, W.; Pande, A.; Gotea, V.; Makałowska, I. Transposable elements and their identification. In Evolutionary Genomics Statistical and Computational Methods; Anisimova, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 337–359. [Google Scholar] [CrossRef]
- Dashti, T.H.; Masoudi-Nejad, A. Mining biological repetitive sequences using support vector machines and fuzzy SVM. Iran. J. Chem. Chem. Eng. 2010, 29, 1–17. [Google Scholar]
- Schulman, A.H. Retrotransposon replication in plants. Curr. Opin. Virol. 2013, 3, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Y.; Zhang, Z.; Li, X.; Yao, D.; Wang, Y.; Ouyang, X.; Li, Y.; Song, W.; Xiao, Y. A D-genome-originated Ty1/copia-type retrotransposon family expanded significantly in tetraploid cottons. Mol. Genet. Genomics 2018, 293, 33–43. [Google Scholar] [CrossRef]
- Negi, P.; Rai, A.N.; Suprasanna, P. Moving through the stressed genome: Emerging regulatory roles for transposons in plant stress response. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Casacuberta, E.; González, J. The impact of transposable elements in environmental adaptation. Mol. Ecol. 2013, 22, 1503–1517. [Google Scholar] [CrossRef]
- Kejnovsky, E.; Tokan, V.; Lexa, M. Transposable elements and G-Quadruplexes. Chromosome Res. 2015, 23, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-J.; Gao, L.-Z. Rapid and recent evolution of LTR retrotransposons drives rice genome evolution during the speciation of AA-genome oryza species. G3 Genes Genomes Genet. 2017, 7, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Eickbush, T.H.; Jamburuthugoda, V.K. The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res. 2008, 134, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.; Paulo, O.S.; Morais-Cecilio, L.; Rocheta, M. A new gypsy-like retroelement family in Vitis Vinifera. VITIS 2012, 51, 65–72. [Google Scholar]
- Mascagni, F.; Giordani, T.; Ceccarelli, M.; Cavallini, A.; Natali, L. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genomics 2017, 18, 634. [Google Scholar] [CrossRef]
- Cossu, R.M.; Buti, M.; Giordani, T.; Natali, L.; Cavallini, A. A computational study of the dynamics of LTR retrotransposons in the Populus Trichocarpa genome. TREE Genet. Genomes 2012, 8, 61–75. [Google Scholar] [CrossRef]
- Kubat, Z.; Zluvova, J.; Vogel, I.; Kovacova, V.; Cermak, T.; Cegan, R.; Hobza, R.; Vyskot, B.; Kejnovsky, E. Possible mechanisms responsible for absence of a retrotransposon family on a plant Y chromosome. New Phytol. 2014, 202, 662–678. [Google Scholar] [CrossRef]
- Bento, M.; Tomás, D.; Viegas, W.; Silva, M. Retrotransposons represent the most labile fraction for genomic rearrangements in polyploid plant species. Cytogenet. Genome Res. 2013, 140, 286–294. [Google Scholar] [CrossRef]
- Gao, D.; Li, Y.; Do Kim, K.; Abernathy, B.; Jackson, S.A. Landscape and evolutionary dynamics of terminal repeat retrotransposons in miniature in plant genomes. Genome Biol. 2016, 17, 7. [Google Scholar] [CrossRef]
- Du, D.; Du, X.; Mattia, M.R.; Wang, Y.; Yu, Q.; Huang, M.; Yu, Y.; Grosser, J.W.; Gmitter, F.G., Jr. LTR retrotransposons from the citrus X clementina genome: Characterization and application. Tree Genet. Genomes 2018, 14, 43. [Google Scholar] [CrossRef]
- Chang, W.; Jääskeläinen, M.; Li, S.; Schulman, A.H. BARE Retrotransposons are translated and replicated via distinct RNA pools. PLoS ONE 2013, 8, e72270. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Cavallini, A.; Giordani, T.; Natali, L. Different histories of two highly variable LTR retrotransposons in sunflower species. Gene 2017, 634, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Joly-Lopez, Z.; Bureau, T.E. Exaptation of transposable element coding sequences. Curr. Opin. Genet. Dev. 2018, 49, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ustyantsev, K.; Novikova, O.; Blinov, A.; Smyshlyaev, G. Convergent evolution of ribonuclease H in LTR retrotransposons and retroviruses. Mol. Biol. Evol. 2015, 32, 1197–1207. [Google Scholar] [CrossRef]
- Zhao, D.; Ferguson, A.A.; Jiang, N. What makes up plant genomes: The vanishing line between transposable elements and genes. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Ustyantsev, K.; Blinov, A.; Smyshlyaev, G. Convergence of retrotransposons in oomycetes and plants. Mob. DNA 2017, 8, 4. [Google Scholar] [CrossRef]
- Piednoël, M.; Carrete-Vega, G.; Renner, S.S. Characterization of the LTR retrotransposon repertoire of a plant clade of six diploid and one tetraploid species. Plant J. 2013, 75, 699–709. [Google Scholar] [CrossRef]
- Usai, G.; Mascagni, F.; Natali, L.; Giordani, T.; Cavallini, A. Comparative genome-wide analysis of repetitive DNA in the Genus Populus L. Tree Genet. Genomes 2017, 13, 96. [Google Scholar] [CrossRef]
- Paz, R.C.; Kozaczek, M.E.; Rosli, H.G.; Andino, N.P.; Sanchez-Puerta, M.V. Diversity, distribution and dynamics of full-length copia and gypsy LTR retroelements in solanum Lycopersicum. Genetica 2017, 145, 417–430. [Google Scholar] [CrossRef]
- Sanchez, D.H.; Gaubert, H.; Drost, H.-G.; Zabet, N.R.; Paszkowski, J. High-frequency recombination between members of an LTR retrotransposon family during transposition bursts. Nat. Commun. 2017, 8, 1283. [Google Scholar] [CrossRef]
- Novikov, A.; Smyshlyaev, G.; Novikova, O. Evolutionary history of LTR retrotransposon chromodomains in plants. Int. J. Plant Genomics 2012, 2012, 874743. [Google Scholar] [CrossRef] [PubMed]
- Bousios, A.; Minga, E.; Kalitsou, N.; Pantermali, M.; Tsaballa, A.; Darzentas, N. MASiVEdb: The Sirevirus plant retrotransposon database. BMC Genomics 2012, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Alzohairy, A.M.; Sabir, J.S.M.; Gyulai, G.; Younis, R.A.A.; Jansen, R.K.; Bahieldin, A. Environmental stress activation of plant long-terminal repeat retrotransposons. Funct. Plant Biol. 2014, 41, 557–567. [Google Scholar] [CrossRef]
- Grover, A.; Sharma, P.C. Repetitive sequences in the potato and related genomes. In The Potato Genome; Chakrabarti, S.K., Xie, C., Tiwari, J.K., Eds.; Springer: Cham, Switzerland, 2017; pp. 143–160. [Google Scholar] [CrossRef]
- Zhou, M.; Liang, L.; Hänninen, H. A transposition-active phyllostachys edulis long terminal repeat (LTR) retrotransposon. J. Plant Res. 2018, 131, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Giordani, T.; Cossu, R.M.; Mascagni, F.; Marroni, F.; Morgante, M.; Cavallini, A.; Natali, L. Genome- wide analysis of LTR-retrotransposon expression in leaves of populus X Canadensis water-deprived plants. Tree Genet. Genomes 2016, 12, 75. [Google Scholar] [CrossRef][Green Version]
- Kriedt, R.A.; Cruz, G.M.Q.; Bonatto, S.L.; Freitas, L.B. Novel transposable elements in Solanaceae: Evolutionary relationships among Tnt1-related sequences in wild petunia species. Plant Mol. Biol. Rep. 2014, 32, 142–152. [Google Scholar] [CrossRef]
- Casacuberta, J.M.; Santiago, N. Plant LTR-Retrotransposons and MITEs: Control of transposition and impact on the evolution of plant genes and genomes. Gene 2003, 311, 1–11. [Google Scholar] [CrossRef]
- Benachenhou, F.; Sperber, G.O.; Bongcam-Rudloff, E.; Andersson, G.; Boeke, J.D.; Blomberg, J. Conserved structure and inferred evolutionary history of long terminal repeats (LTRs). Mob. DNA 2013, 4, 5. [Google Scholar] [CrossRef]
- Mascagni, F.; Usai, G.; Natali, L.; Cavallini, A.; Giordani, T. A Comparison of methods for LTR-retrotransposon insertion time profiling in the populus trichocarpa genome. Caryologia 2018, 71, 85–92. [Google Scholar] [CrossRef]
- Mascagni, F.; Barghini, E.; Giordani, T.; Rieseberg, L.H.; Cavallini, A.; Natali, L. Repetitive DNA and plant domestication: Variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus Annuus) genotypes. Genome Biol. Evol. 2015, 7, 3368–3382. [Google Scholar] [CrossRef]
- Yin, H.; Liu, J.; Xu, Y.; Liu, X.; Zhang, S.; Ma, J.; Du, J. TARE1, a mutated copia-like LTR retrotransposon followed by recent massive amplification in tomato. PLoS ONE 2013, 8, e68587. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wu, X.; Shi, D.; Chen, Y.; Qi, K.; Ma, Z.; Zhang, S. TGTT and AACA: Two transcriptionally active LTR retrotransposon subfamilies with a specific LTR structure and horizontal transfer in four Rosaceae species. Mob. DNA 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Monden, Y.; Fujii, N.; Yamaguchi, K.; Ikeo, K.; Nakazawa, Y.; Waki, T.; Hirashima, K.; Uchimura, Y.; Tahara, M. Efficient screening of long terminal repeat retrotransposons that show high insertion polymorphism via high-throughput sequencing of the primer binding site. Genome 2014, 57, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Choi, J.-Y.; Lee, S.-I.; Kim, N.-S. Marker utility of transposable elements for plant genetics, breeding, and ecology: A review. Genes Genomics 2015, 37, 141–151. [Google Scholar] [CrossRef]
- Gao, D.; Abernathy, B.; Rohksar, D.; Schmutz, J.; Jackson, S.A. Annotation and sequence diversity of transposable elements in common bean (Phaseolus Vulgaris). Front. Plant Sci. 2014, 5, 339. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Du, J.; Li, L.; Jin, C.; Fan, L.; Li, M.; Wu, J.; Zhang, S. Comparative genomic analysis reveals multiple long terminal repeats, lineage-specific amplification, and frequent interelement recombination for Cassandra retrotransposon in pear (Pyrus Bretschneideri Rehd.). Genome Biol. Evol. 2014, 6, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Witte, C.-P.; Le, Q.H.; Bureau, T.; Kumar, A. Terminal-Repeat Retrotransposons in Miniature (TRIM) are involved in restructuring plant genomes. Proc. Natl. Acad. Sci. USA 2001, 98, 13778–13783. [Google Scholar] [CrossRef] [PubMed]
- Sampath, P.; Yang, T.-J. Comparative analysis of Cassandra TRIMs in three Brassicaceae genomes. Plant Genet. Resour. Util. 2014, 12, S146–S150. [Google Scholar] [CrossRef]
- Kalendar, R.; Vicient, C.M.; Peleg, O.; Anamthawat-Jonsson, K.; Bolshoy, A.; Schulman, A.H. Large retrotransposon derivatives: Abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics 2004, 166, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- Schulman, A.H. Hitching a Ride: Nonautonomous retrotransposons and parasitism as a lifestyle. In Plant Transposable Elements; Grandbastien, M.-A., Casacuberta, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 71–88. [Google Scholar] [CrossRef]
- Huang, C.R.L.; Burns, K.H.; Boeke, J.D. Active transposition in genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N. Overview of repeat annotation and de novo repeat identification. In Plant Transposable Elements; Peterson, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 275–287. [Google Scholar] [CrossRef]
- Kim, N.-S. The genomes and transposable elements in plants: Are they friends or foes? Genes Genomics 2017, 39, 359–370. [Google Scholar] [CrossRef]
- Poulter, R.T.M.; Butler, M.I. Tyrosine recombinase retrotransposons and transposons. Microbiol. Spectr. 2015, 3, MDNA3-0036-2014. [Google Scholar] [CrossRef] [PubMed]
- Kralova, T.; Cegan, R.; Kubat, Z.; Vrana, J.; Vyskot, B.; Vogel, I.; Kejnovsky, E.; Hobza, R. Identification of a novel retrotransposon with sex chromosome-specific distribution in silene latifolia. Cytogenet. Genome Res. 2014, 143, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Bonchev, G.N. Useful parasites: The evolutionary biology and biotechnology applications of transposable elements. J. Genet. 2016, 95, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Vicient, C.M.; Casacuberta, J.M. Impact of transposable elements on polyploid plant genomes. Ann. Bot. 2017, 120, 195–207. [Google Scholar] [CrossRef]
- Bonchev, G.; Parisod, C. Transposable elements and microevolutionary changes in natural populations. Mol. Ecol. Resour. 2013, 13, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Todorovska, E. Retrotransposons and their role in plant—Genome Evolution. Biotechnol. Biotechnol. Equip. 2007, 21, 294–305. [Google Scholar] [CrossRef]
- Wessler, S.R.; Bureau, T.E.; White, S.E. LTR-retrotransposons and MITEs: Important players in the evolution of plant genomes. Curr. Opin. Genet. Dev. 1995, 5, 814–821. [Google Scholar] [CrossRef]
- Galindo-González, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.-A.M.-A.; Galindo-Gonzalez, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.-A.; Galindo-González, L.; Mhiri, C.; et al. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef]
- Fan, F.; Wen, X.; Ding, G.; Cui, B. Isolation, identification, and characterization of genomic LTR retrotransposon sequences from masson pine (Pinus Massoniana). Tree Genet. Genomes 2013, 9, 1237–1246. [Google Scholar] [CrossRef]
- Wang, L.; He, Y.; Qiu, H.; Guo, J.; Han, M.; Zhou, J.; Sun, Q.; Sun, J. Mdoryco1-1, a Bidirectionally transcriptional Ty1-Copia retrotransposon from Malus X Domestica. Sci. Hortic. 2017, 220, 283–290. [Google Scholar] [CrossRef]
- El baidouri, M.; Panaud, O. Genome-Wide Analysis of Transposition Using Next Generation Sequencing Technologies; Springer: Berlin/Heidelberg, Germany, 2012; pp. 59–70. [Google Scholar] [CrossRef]
- Cavrak, V.V.; Lettner, N.; Jamge, S.; Kosarewicz, A.; Bayer, L.M.; Mittelsten Scheid, O. How a retrotransposon exploits the plant’s heat stress response for its activation. PLoS Genet. 2014, 10, e1004115. [Google Scholar] [CrossRef] [PubMed]
- Paszkowski, J. Controlled activation of retrotransposition for plant breeding. Curr. Opin. Biotechnol. 2015, 32, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Pouteau, S.; Huttner, E.; Grandbastien, M.A.; Caboche, M. Specific expression of the tobacco Tnt1 retrotransposon in protoplasts. EMBO J. 1991, 10, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Hirochika, H.; Otsuki, H.; Yoshikawa, M.; Otsuki, Y.; Sugimoto, K.; Takeda, S. Autonomous transposition of the tobacco retrotransposon Tto1 in rice. Plant Cell 1996, 8, 725–734. [Google Scholar] [PubMed]
- Hirochika, H. Contribution of the Tos17 retrotransposon to rice functional genomics. Curr. Opin. Plant Biol. 2001, 4, 118–122. [Google Scholar] [CrossRef]
- Kimura, Y.; Tosa, Y.; Shimada, S.; Sogo, R.; Kusaba, M.; Sunaga, T.; Betsuyaku, S.; Eto, Y.; Nakayashiki, H.; Mayama, S. OARE-1, a Ty1-copia retrotransposon in Oat activated by abiotic and biotic stresses. Plant Cell Physiol. 2001, 42, 1345–1354. [Google Scholar] [CrossRef]
- Ramallo, E.; Kalendar, R.; Schulman, A.H.; Martínez-Izquierdo, J.A. Reme1, a copia retrotransposon in melon, is transcriptionally induced by UV Light. Plant Mol. Biol. 2008, 66, 137. [Google Scholar] [CrossRef]
- Matsunaga, W.; Kobayashi, A.; Kato, A.; Ito, H. The effects of heat induction and the siRNA biogenesis pathway on the transgenerational transposition of ONSEN, a copia-like retrotransposon in Arabidopsis Thaliana. Plant Cell Physiol. 2011, 53, 824–833. [Google Scholar] [CrossRef]
- Cao, Y.; Jiang, Y.; Ding, M.; He, S.; Zhang, H.; Lin, L.; Rong, J. Molecular characterization of a transcriptionally active Ty1/copia-like retrotransposon in gossypium. Plant Cell Rep. 2015, 34, 1037–1047. [Google Scholar] [CrossRef]
- He, P.; Ma, Y.; Zhao, G.; Dai, H.; Li, H.; Chang, L.; Zhang, Z. FaRE1: A transcriptionally active Ty1-copia retrotransposon in strawberry. J. Plant Res. 2010, 123, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Vicient, C.M.; Suoniemi, A.; Anamthawat-Jónsson, K.; Tanskanen, J.; Beharav, A.; Nevo, E.; Schulman, A.H. Retrotransposon BARE-1 and its role in genome evolution in the Genus Hordeum. Plant Cell 1999, 11, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Tapia, G.; Verdugo, I.; Yañez, M.; Ahumada, I.; Theoduloz, C.; Cordero, C.; Poblete, F.; González, E.; Ruiz-Lara, S. Involvement of ethylene in stress-induced expression of the TLC1. 1 retrotransposon from Lycopersicon Chilense Dun. Plant Physiol. 2005, 138, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.-K.; Bennetzen, J.L. Structure and coding properties of Bs1, a maize retrovirus-like transposon. Proc. Natl. Acad. Sci. USA 1989, 86, 6235–6239. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zhang, J. Plant genomic DNA methylation in response to stresses: Potential applications and challenges in plant breeding. Prog. Nat. Sci. USA 2009, 19, 1037–1045. [Google Scholar] [CrossRef]
- Farman, M.L.; Tosa, Y.; Nitta, N.; Leong, S.A. MAGGY, a retrotransposon in the genome of the rice blast Fungus Magnaporthe Grisea. Mol. Gen. Genet. MGG 1996, 251, 665–674. [Google Scholar]
- Madsen, L.H.; Fukai, E.; Radutoiu, S.; Yost, C.K.; Sandal, N.; Schauser, L.; Stougaard, J. LORE1, an active low-copy-number TY3-gypsy retrotransposon family in the model legume Lotus Japonicus. Plant J. 2005, 44, 372–381. [Google Scholar] [CrossRef]
- Zuccolo, A.; Scofield, D.G.; De Paoli, E.; Morgante, M. The Ty1-Copia LTR retroelement family PARTC is highly conserved in conifers over 200 MY of evolution. Gene 2015, 568, 89–99. [Google Scholar] [CrossRef]
- Sun, J.; Huang, Y.; Zhou, J.; Guo, J.; Sun, Q. LTR-retrotransposon diversity and transcriptional activation under phytoplasma stress in Ziziphus Jujuba. Tree Genet. Genomes 2013, 9, 423–431. [Google Scholar] [CrossRef]
- Jia, L.; Lou, Q.; Jiang, B.; Wang, D.; Chen, J. LTR retrotransposons cause expression changes of adjacent genes in early generations of the newly formed allotetraploid Cucumis Hytivus. Sci. Hortic. 2014, 174, 171–177. [Google Scholar] [CrossRef]
- Cui, X.; Cao, X. Epigenetic regulation and functional exaptation of transposable elements in higher plants. Curr. Opin. Plant Biol. 2014, 21, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramirez-Gonzalez, R.H.; De Oliveira, R.; Mayer, K.F.; Paux, E.; Choulet, F. Impact of Transposable Elements on Genome Structure and Evolution in Bread Wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef] [PubMed]
- Baruch, O.; Kashkush, K. Analysis of copy-number variation, insertional polymorphism, and methylation status of the tiniest class I (TRIM) and class II (MITE) transposable element families in various rice strains. Plant Cell Rep. 2012, 31, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yoshida, T.; Tsukahara, S.; Kawabe, A. Evolution of the ONSEN retrotransposon family activated upon heat stress in Brassicaceae. Gene 2013, 518, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; He, Z.; Wu, C.-I.; Shi, S. Convergent adaptive evolution in marginal environments: Unloading transposable elements as a common strategy among mangrove genomes. New Phytol. 2018, 217, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Cossu, R.M.; Casola, C.; Giacomello, S.; Vidalis, A.; Scofield, D.G.; Zuccolo, A. LTR retrotransposons show low levels of unequal recombination and high rates of intraelement gene conversion in large plant genomes. Genome Biol. Evol. 2017, 9, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Moisy, C.; Schulman, A.H.; Kalendar, R.; Buchmann, J.P.; Pelsy, F. The Tvv1 retrotransposon family is conserved between plant genomes separated by over 100 million years. Theor. Appl. Genet. 2014, 127, 1223–1235. [Google Scholar] [CrossRef][Green Version]
- Dias, E.S.; Hatt, C.; Hamon, S.; Hamon, P.; Rigoreau, M.; Crouzillat, D.; Carareto, C.M.A.; de Kochko, A.; Guyot, R. Large distribution and high sequence identity of a copia-type retrotransposon in angiosperm families. Plant Mol. Biol. 2015, 89, 83–97. [Google Scholar] [CrossRef]
- Woodrow, P.; Ciarmiello, L.F.; Fantaccione, S.; Annunziata, M.G.; Pontecorvo, G.; Carillo, P. Ty1-Copia group retrotransposons and the evolution of retroelements in several angiosperm plants: Evidence of horizontal transmission. Bioinformation 2012, 8, 267–271. [Google Scholar] [CrossRef][Green Version]
- Sharma, A.; Presting, G.G. Evolution of centromeric retrotransposons in grasses. Genome Biol. Evol. 2014, 6, 1335–1352. [Google Scholar] [CrossRef]
- El Baidouri, M.; Carpentier, M.-C.; Cooke, R.; Gao, D.; Lasserre, E.; Llauro, C.; Mirouze, M.; Picault, N.; Jackson, S.A.; Panaud, O. Widespread and frequent horizontal transfers of transposable elements in plants. Genome Res. 2014, 24, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Ma, B.; Xin, Y.; Kuang, L.; He, N. Horizontal transfers of LTR retrotransposons in seven species of rosales. Genome 2018, 61, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Hermann, D.; Egue, F.; Tastard, E.; Nguyen, D.-H.; Casse, N.; Caruso, A.; Hiard, S.; Marchand, J.; Chenais, B.; Morant-Manceau, A.; et al. An introduction to the vast world of transposable elements—What about the Diatoms? Diatom Res. 2014, 29, 91–104. [Google Scholar] [CrossRef]
- Parisod, C.; Alix, K.; Just, J.; Petit, M.; Sarilar, V.; Mhiri, C.; Ainouche, M.; Chalhoub, B.; Grandbastien, M.-A. Impact of transposable elements on the organization and function of allopolyploid genomes. New Phytol. 2010, 186, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. LINE-1 Elements and X chromosome inactivation: A Function for “junk” DNA? Proc. Natl. Acad. Sci. USA 2000, 97, 6248–6249. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-F.; Su, T.; Cheng, G.-Q.; Wang, B.-X.; Li, X.; Deng, C.-L.; Gao, W.-J. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Genes 2017, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Vergara, Z.; Sequeira-Mendes, J.; Morata, J.; Peiró, R.; Hénaff, E.; Costas, C.; Casacuberta, J.M.; Gutierrez, C. Retrotransposons are specified as DNA replication origins in the gene-poor regions of arabidopsis heterochromatin. Nucleic Acids Res. 2017, 45, 8358–8368. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Park, J.; Kim, S.; Kwon, J.-K.; Park, H.M.; Bae, I.H.; Yang, T.-J.; Lee, Y.-H.; Kang, B.-C.; Choi, D. Evolution of the large genome in capsicum annuum occurred through accumulation of single-type long terminal repeat retrotransposons and their derivatives. Plant J. 2012, 69, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Datema, E.; Guzman, M.O.; de Boer, J.M.; van Eck, H.J.; Bachem, C.W.B.; Visser, R.G.F.; de Jong, H. Chromosomal organizations of major repeat families on potato (Solanum Tuberosum) and further exploring in its sequenced genome. Mol. Genet. Genomics 2014, 289, 1307–1319. [Google Scholar] [CrossRef]
- de Setta, N.; Monteiro-Vitorello, C.; Metcalfe, C.; Cruz, G.M.; Del Bem, L.; Vicentini, R.; Nogueira, F.T.; Campos, R.; Nunes, S.; Turrini, P.C.; et al. Building the sugarcane genome for biotechnology and identifying evolutionary trends. BMC Genomics 2014, 15, 540. [Google Scholar] [CrossRef]
- Gao, D.; Jiang, N.; Wing, R.A.; Jiang, J.; Jackson, S.A. Transposons play an important role in the evolution and diversification of centromeres among closely related species. Front. Plant Sci. 2015, 6, 216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J. Co-evolution of plant LTR-retrotransposons and their host genomes. Protein Cell 2013, 4, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zuo, S.; Zhang, Z.; Li, Z.; Han, J.; Chu, Z.; Hasterok, R.; Wang, K. Centromeric DNA characterization in the model grass Brachypodium Distachyon provides insights on the evolution of the genus. Plant J. 2018, 93, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cao, Y.; Wang, K.; Zhao, T.; Chen, J.; Pan, M.; Wang, Q.; Feng, S.; Guo, W.; Zhou, B.; et al. Identification of centromeric regions on the linkage map of cotton using centromere-related repeats. Genomics 2014, 104, 587–593. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Cai, Z.; Hu, T.; Liu, H.; Bao, C.; Mao, W.; Jin, W. Repetitive sequence analysis and karyotyping reveals centromere-associated DNA sequences in Radish (Raphanus Sativus L.). BMC Plant Biol. 2015, 15, 105. [Google Scholar] [CrossRef] [PubMed]
- Divashuk, M.G.; Khuat, T.M.L.; Kroupin, P.Y.; Kirov, I.V.; Romanov, D.V.; Kiseleva, A.V.; Khrustaleva, L.I.; Alexeev, D.G.; Zelenin, A.S.; Klimushina, M.V.; et al. Variation in copy number of Ty3/Gypsy centromeric retrotransposons in the genomes of Thinopyrum Intermedium and its diploid progenitors. PLoS ONE 2016, 11, e0154241. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Choulet, F.; Heng, Y.; Hao, W.; Paux, E.; Liu, Z.; Yue, W.; Jin, W.; Feuillet, C.; Zhang, X. Wheat centromeric retrotransposons: The new ones take a major role in centromeric structure. Plant J. 2013, 73, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Cao, H.X.; Jovtchev, G.; Neumann, P.; Novák, P.; Fojtová, M.; Vu, G.T.H.; Macas, J.; Fajkus, J.; Schubert, I.; et al. Centromere and telomere sequence alterations reflect the rapid genome evolution within the carnivorous plant genus Genlisea. Plant J. 2015, 84, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Meštrović, N.; Mravinac, B. Centromere identity from the DNA point of view. Chromosoma 2014, 123, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Gao, Z.; Han, F. Plant centromeres. In Plant Cytogenetics; Springer: New York, NY, USA, 2012; pp. 133–142. [Google Scholar] [CrossRef]
- Qi, L.L.; Wu, J.J.; Friebe, B.; Qian, C.; Gu, Y.Q.; Fu, D.L.; Gill, B.S. Sequence organization and evolutionary dynamics of Brachypodium-specific centromere retrotransposons. Chromosome Res. 2013, 21, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-Y.; Ramachandran, S. Genome-wide survey and comparative analysis of LTR retrotransposons and their captured genes in rice and sorghum. PLoS ONE 2013, 8, e71118. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-R.; Liu, C.; Wei, P.; Song, X.-J.; Yang, Z.-J. Chromosomal distribution of a new centromeric Ty3-gypsy retrotransposon sequence in Dasypyrum and related Triticeae species. J. Genet. 2012, 91, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, J.P.; Keller, B.; Wicker, T. Transposons in cereals: Shaping genomes and driving their evolution. In Cereal Genomics II; Springer: Dordrecht, The Netherlands, 2013; pp. 127–154. [Google Scholar] [CrossRef]
- Luo, S.; Mach, J.; Abramson, B.; Ramirez, R.; Schurr, R.; Barone, P.; Copenhaver, G.; Folkerts, O. The cotton centromere contains a Ty3-Gypsy-like LTR retroelement. PLoS ONE 2012, 7, e35261. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Na, J.-K.; Yu, Q.; Gschwend, A.R.; Han, J.; Zeng, F.; Aryal, R.; VanBuren, R.; Murray, J.E.; Zhang, W.; et al. Sequencing Papaya X and Yh chromosomes reveals molecular basis of incipient sex chromosome evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 13710–13715. [Google Scholar] [CrossRef] [PubMed]
- Steflova, P.; Tokan, V.; Vogel, I.; Lexa, M.; Macas, J.; Novak, P.; Hobza, R.; Vyskot, B.; Kejnovsky, E. Contrasting patterns of transposable element and satellite distribution on sex chromosomes (XY1Y2) in the dioecious plant rumex acetosa. Genome Biol. Evol. 2013, 5, 769–782. [Google Scholar] [CrossRef]
- Gschwend, A.R.; Weingartner, L.A.; Moore, R.C.; Ming, R. The sex-specific region of sex chromosomes in animals and plants. Chromosom. Res. 2012, 20, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Puterova, J.; Kubat, Z.; Kejnovsky, E.; Jesionek, W.; Cizkova, J.; Vyskot, B.; Hobza, R. The slowdown of Y chromosome expansion in Dioecious Silene Latifolia due to DNA loss and male-specific silencing of retrotransposons. BMC Genomics 2018, 19, 153. [Google Scholar] [CrossRef]
- Hobza, R.; Cegan, R.; Jesionek, W.; Kejnovsky, E.; Vyskot, B.; Kubat, Z. Impact of repetitive elements on the Y chromosome formation in plants. Genes 2017, 8, 302. [Google Scholar] [CrossRef]
- Hobza, R.; Kubat, Z.; Cegan, R.; Jesionek, W.; Vyskot, B.; Kejnovsky, E. Impact of repetitive DNA on sex chromosome evolution in plants. Chromosom. Res. 2015, 23, 561–570. [Google Scholar] [CrossRef]
- Na, J.-K.; Wang, J.; Ming, R. Accumulation of interspersed and sex-specific repeats in the non-recombining region of papaya sex chromosomes. BMC Genomics 2014, 15, 335. [Google Scholar] [CrossRef]
- Testori, A.; Caizzi, L.; Cutrupi, S.; Friard, O.; De Bortoli, M.; Cora, D.; Caselle, M. The Role of transposable elements in shaping the combinatorial interaction of transcription factors. BMC Genomics 2012, 13, 400. [Google Scholar] [CrossRef] [PubMed]
- Krom, N.; Ramakrishna, W. Retrotransposon insertions in rice gene pairs associated with reduced conservation of gene pairs in grass genomes. Genomics 2012, 99, 308–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mascagni, F.; Vangelisti, A.; Giordani, T.; Cavallini, A.; Natali, L. Specific LTR-retrotransposons show copy number variations between wild and cultivated sunflowers. Genes 2018, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Waminal, N.E.; Choi, H.-I.; Perumal, S.; Lee, S.-C.; Nguyen, V.B.; Jang, W.; Kim, N.-H.; Gao, L.-Z.; Yang, T.-J. Rapid amplification of four retrotransposon families promoted speciation and genome size expansion in the genus panax. Sci. Rep. 2017, 7, 9045. [Google Scholar] [CrossRef]
- Dhadi, S.R.; Xu, Z.; Shaik, R.; Driscoll, K.; Ramakrishna, W. Differential regulation of genes by retrotransposons in rice promoters. Plant Mol. Biol. 2015, 87, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Natali, L.; Cossu, R.M.; Mascagni, F.; Giordani, T.; Cavallini, A. A survey of gypsy and copia LTR-retrotransposon superfamilies and lineages and their distinct dynamics in the Populus Trichocarpa (L.) genome. Tree Genet. Genomes 2015, 11, 107. [Google Scholar] [CrossRef]
- Yue, T.; Wang, H. Deep learning for genomics: A concise overview. arXiv 2018, arXiv:1802.00810. [Google Scholar]
- Wei, L.; Xiao, M.; An, Z.; Ma, B.; Mason, A.S.; Qian, W.; Li, J.; Fu, D. New insights into nested long terminal repeat retrotransposons in brassica species. Mol. Plant 2013, 6, 470–482. [Google Scholar] [CrossRef]
- Wicker, T.; Schulman, A.H.; Tanskanen, J.; Spannagl, M.; Twardziok, S.; Mascher, M.; Springer, N.M.; Li, Q.; Waugh, R.; Li, C.; et al. The repetitive landscape of the 5100 Mbp barley genome. Mob. DNA 2017, 8, 22. [Google Scholar] [CrossRef]
- Ferreira de Carvalho, J.; Chelaifa, H.; Boutte, J.; Poulain, J.; Couloux, A.; Wincker, P.; Bellec, A.; Fourment, J.; Bergès, H.; Salmon, A.; et al. Exploring the genome of the salt-marsh spartina maritima (Poaceae, Chloridoideae) through BAC end sequence analysis. Plant Mol. Biol. 2013, 83, 591–606. [Google Scholar] [CrossRef]
- Middleton, C.P.; Stein, N.; Keller, B.; Kilian, B.; Wicker, T. Comparative analysis of genome composition in Triticeae reveals strong variation in transposable element dynamics and nucleotide diversity. Plant J. 2013, 73, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Guyot, R.; Darré, T.; Dupeyron, M.; de Kochko, A.; Hamon, S.; Couturon, E.; Crouzillat, D.; Rigoreau, M.; Rakotomalala, J.-J.; Raharimalala, N.E.; et al. Partial sequencing reveals the transposable element composition of Coffea genomes and provides evidence for distinct evolutionary stories. Mol. Genet. Genomics 2016, 291, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.C.; Guyot, R.; do Valle, C.B.; Chiari, L.; Techio, V.H.; Heslop-Harrison, P.; Vanzela, A.L.L. Chromosomal distribution and evolution of abundant retrotransposons in plants: Gypsy elements in diploid and polyploid brachiaria forage grasses. Chromosome Res. 2015, 23, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Piégu, B.; Bire, S.; Arensburger, P.; Bigot, Y. A survey of transposable element classification systems—A call for a fundamental update to meet the challenge of their diversity and complexity. Mol. Phylogenet. Evol. 2015, 86, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Arkhipova, I.R. Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mob. DNA 2017, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Büchen-Osmond, C. ICTVdb-The Universal Virus Database, Version 4; Columbia University: New York, NY, USA, 2006. [Google Scholar]
- Abrusán, G.; Grundmann, N.; Demester, L.; Makalowski, W. TEclass—A tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 2009, 25, 1329–1330. [Google Scholar] [CrossRef] [PubMed]
- Pang, E.; Cao, H.; Zhang, B.; Lin, K. Crop Genome Annotation: A Case Study for the Brassica Rapa Genome; Springer: Berlin/Heidelberg, Germany, 2015; pp. 53–64. [Google Scholar] [CrossRef]
- Loureiro, T.; Camacho, R.; Vieira, J.; Fonseca, N.A. Improving the performance of transposable elements detection tools. J. Integr. Bioinform. 2013, 10, 231. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Waminal, N.E.; Perumal, S.; Liu, S.; Chalhoub, B.; Kim, H.H.; Yang, T.-J. Quantity, distribution, and evolution of major repeats in Brassica Napus. In The Brassica Napus Genome; Liu, S., Snowdon, R., Chalhoub, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 111–129. [Google Scholar] [CrossRef]
- Rawal, K.; Ramaswamy, R. Genome-wide analysis of mobile genetic element insertion sites. Nucleic Acids Res. 2011, 39, 6864–6878. [Google Scholar] [CrossRef]
- Garbus, I.; Romero, J.R.; Valarik, M.; Vanžurová, H.; Karafiátová, M.; Cáccamo, M.; Doležel, J.; Tranquilli, G.; Helguera, M.; Echenique, V. Characterization of repetitive DNA landscape in wheat Homeologous group 4 chromosomes. BMC Genomics 2015, 16, 375. [Google Scholar] [CrossRef]
- Nicolas, J.; Peterlongo, P.; Tempel, S. Finding and characterizing repeats in plant genomes. In Plant Bioinformatics; Edwards, D., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 293–337. [Google Scholar] [CrossRef]
- Chiusano, M.L.; Colantuono, C. Repeat sequences in the tomato genome. In The Tomato Genome; Causse, M., Giovannoni, J., Bouzayen, M., Zouine, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 173–199. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker. 1996. Available online: http://www.repeatmasker.org (accessed on 3 August 2019).
- An, M.M.; Guo, C.; Lin, P.P.; Zhou, M.B. Heterogeneous Evolution of Ty3-Gypsy Retroelements among Bamboo Species. Genet. Mol. Res. 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Klonowski, P.; Dagman, V.; Pelton, P. CENSOR-a program for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 1996, 20, 119–121. [Google Scholar] [CrossRef]
- Rho, M.; Choi, J.H.; Kim, S.; Lynch, M.; Tang, H. De novo identification of LTR retrotransposons in eukaryotic genomes. BMC Genomics 2007, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, A.; Lecroq, T.; Dauchel, H.; Alexandre, J. FORRepeats: Detects repeats on entire chromosomes and between genomes. Bioinformatics 2003, 19, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR-FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Vini Pereira. Insertion bias and purifying selection of retrotransposons in the Arabidopsis Thaliana genome. Genome Biol. 2004, 5, 1–10. [Google Scholar]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal-repeat retrotransposons. Plant Physiol. 2017, 176, 1410–1422. [Google Scholar] [CrossRef]
- McCarthy, E.M.; McDonald, J.F. LTR STRUC: A novel search and identification program for LTR retrotransposons. Bioinformatics 2003, 19, 362–367. [Google Scholar] [CrossRef]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 14, 18. [Google Scholar] [CrossRef]
- Steinbiss, S.; Kastens, S.; Kurtz, S. LTRsift: A graphical user interface for semi-automatic classification and postprocessing of de novo detected LTR retrotransposons. Mob. DNA 2012, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.-C.; Zhao, Y.-J.; Zhang, Q.-J.; Gao, L.-Z. LTRtype, an efficient tool to characterize structurally complex LTR retrotransposons and nested insertions on genomes. Front. Plant Sci. 2017, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Castoe, T.A.; Hedges, D.J.; Batzer, M.A.; Pollock, D.D. Identification of repeat structure in large genomes using repeat probability clouds. Anal. Biochem. 2008, 380, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Hoede, C.; Arnoux, S.; Moisset, M.; Chaumier, T.; Inizan, O.; Jamilloux, V.; Quesneville, H. PASTEC: An automatic transposable element classification tool. PLoS ONE 2014, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Campagna, D.; Romualdi, C.; Vitulo, N.; Del Favero, M.; Lexa, M.; Cannata, N.; Valle, G. RAP: A new computer program for de novo identification of repeated sequences in whole genomes. Bioinformatics 2005, 21, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V. Automated paleontology of repetitive DNA with REANNOTATE. BMC Genomics 2008, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ye, J.; Li, S.; Wang, J.; Han, Y.; Ye, C.; Wang, J.; Yang, H.; Yu, J.; Wong, G.K.S.; et al. ReAS: Recovery of ancestral sequences for transposable elements from the unassembled reads of a whole genome shotgun. PLoS Comput. Biol. 2005, 1, 0313–0321. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Feschotte, C.; Keswani, U.; Ranganathan, N.; Guibotsy, M.L.; Levine, D. Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol. Evol. 2009, 1, 205–220. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; States, D.J. The Repeat Pattern Toolkit (RPT): Analyzing the structure and evolution of the C. elegans genome. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology, Stanford, CA, USA, 14–17 August 1994; Volume 2, pp. 1–9. [Google Scholar]
- Quesneville, H.; Bergman, C.M.; Andrieu, O.; Autard, D.; Nouaud, D.; Ashburner, M.; Anxolabehere, D. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput. Biol. 2005, 1, 0166–0175. [Google Scholar] [CrossRef]
- Achaz, G.; Boyer, F.; Rocha, E.P.C.; Viari, A.; Coissac, E. Repseek, a tool to retrieve approximate repeats from large DNA sequences. Bioinformatics 2007, 23, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete henomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [PubMed]
- Ramachandran, D.; Hawkins, J.S. Methods for accurate quantification of LTR-retrotransposon copy number using short-read sequence data: A case study in Sorghum. Mol. Genet. Genomics 2016, 291, 1871–1883. [Google Scholar] [CrossRef] [PubMed]
- Choulet, F.; Caccamo, M.; Wright, J.; Alaux, M.; Šimková, H.; Šafář, J.; Leroy, P.; Doležel, J.; Rogers, J.; Eversole, K.; et al. The Wheat Black Jack: Advances towards sequencing the 21 chromosomes of bread wheat. In Genomics of Plant Genetic Resources; Springer: Dordrecht, The Netherlands, 2014; pp. 405–438. [Google Scholar] [CrossRef]
- Natali, L.; Cossu, R.; Barghini, E.; Giordani, T.; Buti, M.; Mascagni, F.; Morgante, M.; Gill, N.; Kane, N.C.; Rieseberg, L.; et al. The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genomics 2013, 14, 686. [Google Scholar] [CrossRef]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Yu, Z.; Pan, Y. A deep learning method for lincRNA detection using auto-encoder algorithm. BMC Bioinform. 2017, 18, 511. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019. [Google Scholar] [CrossRef]
- Rosen, G.L. Signal Processing for Biologically-Inspired Gradient Source Localization and DNA Sequence Analysis. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2006. [Google Scholar]
- Yu, N.; Guo, X.; Gu, F.; Pan, Y. DNA AS X: An information-coding-based model to improve the sensitivity in comparative gene analysis. In Proceedings of the International Symposium on Bioinformatics Research and Applications, Norfolk, VA, USA, 7–10 June 2015; pp. 366–377. [Google Scholar]
- Nair, A.S.; Sreenadhan, S.P. A coding measure scheme employing Electron-Ion Interaction Pseudopotential (EIIP). Bioinformation 2006, 1, 197. [Google Scholar] [PubMed]
- Akhtar, M.; Epps, J.; Ambikairajah, E. Signal processing in sequence analysis: Advances in eukaryotic gene prediction. IEEE J. Sel. Top. Signal Process. 2008, 2, 310–321. [Google Scholar] [CrossRef]
- Kauer, G.; Blöcker, H. Applying signal theory to the analysis of biomolecules. Bioinformatics 2003, 19, 2016–2021. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S.; Bach, F. Bioinformatics: The Machine Learning Approach; MIT Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Iquebal, M.A.; Jaiswal, S.; Mukhopadhyay, C.S.; Sarkar, C.; Rai, A.; Kumar, D. Applications of bioinformatics in plant and agriculture. In PlantOmics: The Omics of Plant Science; Springer: New Delhi, India, 2015; pp. 755–789. [Google Scholar] [CrossRef]
- Touati, R.; Messaoudi, I.; Oueslati, A.E.; Lachiri, Z. A combined support vector machine-FCGS classification based on the wavelet transform for helitrons recognition in C. Elegans. Multimed. Tools Appl. 2018, 78, 13047–13066. [Google Scholar] [CrossRef]
- Tsafnat, G.; Setzermann, P.; Partridge, S.R.; Grimm, D. Computational inference of difficult word boundaries in DNA languages. In Proceedings of the ACM International Conference Proceeding Series, Barcelona, Spain, 14–18 November 2011. [Google Scholar]
- Nakano, F.K.; Martiello Mastelini, S.; Barbon, S.; Cerri, R. Stacking methods for hierarchical classification. In Proceedings of the 16th IEEE International Conference on Machine Learning and Applications, ICMLA 2017, Cancun, Mexico, 18–21 December 2017; pp. 289–296. [Google Scholar]
- Nakano, F.K.; Pinto, W.J.; Pappa, G.L.; Cerri, R. Top-down strategies for hierarchical classification of transposable elements with neural networks. In Proceedings of the International Joint Conference on Neural Networks, Anchorage, AK, USA, 14–19 May 2017; pp. 2539–2546. [Google Scholar]
- Nakano, F.K.; Mastelini, S.M.; Barbon, S.; Cerri, R. Improving Hierarchical Classification of Transposable Elements Using Deep Neural Networks. In Proceedings of the International Joint Conference on Neural Networks, Rio de Janeiro, Brazil, 8–13 July 2018. [Google Scholar]
- Cheng, S.; Melkonian, M.; Smith, S.A.; Brockington, S.; Archibald, J.M.; Delaux, P.-M.; Li, F.-W.; Melkonian, B.; Mavrodiev, E.V.; Sun, W.; et al. 10KP: A phylodiverse genome sequencing plan. Gigascience 2018, 7, giy013. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.N.; Campos, V.D.A.; Barella, V.H. On the search for retrotransposons: Alternative protocols to obtain sequences to learn profile hidden markov models. J. Comput. Biol. 2018, 25, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Arias, S.; Tabares-Soto, R.; Ceballos, D.; Guyot, R. Parallel programming in biological sciences, taking advantage of supercomputing in genomics. In Advances in Computing; Solano, A., Ordoñez, H., Eds.; Springer: Zurich, Switzerland, 2017; Volume 735, pp. 627–643. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complete Gene Name | Short Name | Function |
|---|---|---|
| Reverse transcriptase | RT | Responsible for DNA synthesis using RNA as a template |
| RNase H | RNAseH | Responsible for the degradation of the RNA template in the DNA-RNA hybrid |
| Intregrase | INT | Responsible for catalyzing the insertion of the retrotransposon cDNA into the genome of a host cell |
| Aspartic protease | AP | Responsible for processing large transposon transcripts into smaller protein products |
| Envelope | ENV | Responsible for cell-to-cell transfer of retroviruses. |
| Group specific antigen | GAG | Structural protein for virus-like particles |
| Chromodomain | Chrod | Responsible for targeting the insertion of new LTR retrotransposon copies into heterochromatic regions by recognizing specific heterochromatic histone marks and/or other factors |
| Retrotransposon | Stresses by External Conditions | Species | Reference |
|---|---|---|---|
| Tnt1 | Protoplast and tissue culture, pathogens, pathogen elicitors, compounds related to plant defense, wounding, freezing, in vitro regeneration, mechanical damage, and microbial factors. | Tobacco | [92] |
| Tto1 | Wounding, methyl jasmonate, tissue culture, fungal elicitors, chilling, cytosine demethylation, resistance to bacterial blight, and plant development. | Tobacco | [93] |
| Tos17 | Tissue culture and viral infection. | Rice | [94] |
| OARE-1 | Wounding, jasmonic and salicylic acid, UV light, infection with an incompatible race of the crown rust fungus. | Oat | [95] |
| Reme1 | UV light. | Melon | [96] |
| ONSEN | Heat stress. | A. thaliana and other members of the Brassicaceae family | [97] |
| GBRE-1 | Heat stress. | Gossypium | [98] |
| FaRE1 | Hormonal treatments. | Strawberry | [99] |
| BARE-1 | Water-induced stress. | Barley, Hordeum spontaneum | [100] |
| Tlc1 | Phytohormones, wounding, protoplast preparation, high salt concentration and stress-associated signaling molecules. | Solanum chilense | [101] |
| Erika | Fungal infection. | Wild wheat | [57] |
| Bs1 | Barley stripe mosaic virus infection. | Maize | [102] |
| ZmMI1 | Cold. | Maize | [103] |
| CLCoi1 | Wounding and salt stress. | Lemon | [90] |
| MAGGY | Heat shock. | Rice | [104] |
| Wis2-1A | Interspecific hybridization. | Wheat | [57] |
| LORE1 | Tissue culture. | Lotus japonicus | [105] |
| Superfamilies | |||
|---|---|---|---|
| REXdb a | Wicker and Keller b | GyDB c | ICTV d |
| Copia | Copia | Ty1/Copia | Pseudoviridae |
| Gypsy | Gypsy | Ty3/Gypsy | Metaviridae |
| Bel-pao | Bel-pao | Bel-pao | Semotiviruses |
| Lineages (Copia) | |||
| Ale | Ale | Sirevirus/Retrofit | pseudovirus |
| Alesia | Ale | - | - |
| Angela | Angela | - | pseudovirus |
| Bianca | Bianca | - | - |
| Bryco | - | - | - |
| Lyco | - | - | - |
| Gymco-I, II, III, IV | - | - | - |
| Ikeros | Angela | Tork | pseudovirus |
| Ivana | Ivana | Sirevirus/Oryco | - |
| Osser | - | Osser | hemivirus |
| SIRE | Maximus | Sirevirus/SIRE | Sirevirus |
| TAR | TAR | Tork | - |
| Tork | - | Tork | pseudovirus |
| Lineages (Gypsy) | |||
| chromovirus|CRM | - | chromoviruses|CRM | - |
| chromovirus|Chlamyvir | - | - | - |
| chromovirus|Galadriel | - | chromoviruses|Galadriel | - |
| chromovirus|Reina | - | chromoviruses|Reina | - |
| chromovirus|Tekay | - | chromoviruses|Del | Metavirus (Del1) |
| non-chromovirus|OTA|Athila | - | Athila/Tat|Athila | Metavirus (Athila) |
| non-chromovirus|OTA|Tat|TatI | - | - | - |
| non-chromovirus|OTA|Tat|TatII | - | - | - |
| non-chromovirus|OTA|Tat|TatIII | - | - | - |
| non-chromovirus|OTA|Tat|Ogre | - | Athila/Tat|Tat (Ogre) | - |
| non-chromovirus|OTA|Tat|Retand | - | Athila/Tat|Tat | Metavirus (Tat4) |
| non-chromovirus|Phygy | - | - | - |
| non-chromovirus|Selgy | - | - | - |
| Software | Approach | TE Class or Order | Applies ML | Input Format Files | Tasks | Reference |
|---|---|---|---|---|---|---|
| Censor | Homology-based | Any | NO | Any | I | [178] |
| Find_ltr | Structure-based, Homology-based | Complete LTR RTs, and solo LTRs | NO | Assembled sequences | I | [179] |
| FORRepeats | Homology-based | Any | NO | Any | I | [180] |
| Inpactor | Structure-based, Homology-based | LTR RTs | NO | Assembled sequences, LTR_Struc output or REPET output | C, O | [23] |
| LTR-FINDER | Structure-based | LTR RTs | NO | Assembled sequences | I | [181] |
| LTR_MINER | Structure-based | LTR RTs | NO | RepeatMasker output | I | [182] |
| LTR_retriever | Structure-based | LTR RTs | NO | Assembled sequences | I | [183] |
| LTR_STRUC | Structure-based | LTR RTs | NO | Assembled sequences | I | [184] |
| LTRClassifier | Homology-based | LTR RTs | NO | Assembled sequences | C | [22] |
| LTRdigest | Structure-based, Homology-based | LTR RTs | NO | LTRharvest output | C | [185] |
| LTRHarvest | Structure-based | LTR RTs | NO | Assembled sequences | I | [186] |
| LTRSift | Structure-based | LTR RTs | NO | LTRdigest output | C | [187] |
| LTRType | Homology-based | LTR RTs | NO | Assembled sequences | I | [188] |
| P-Clouds | De novo | Any | NO | Assembled sequences | I | [189] |
| PASTEC | Structure-based, Homology-based | Any | NO | Assembled sequences | C | [190] |
| PILER | Structure-based, De novo | Any | NO | Assembled sequences | I | [191] |
| RAP | De novo | Any | NO | Assembled sequences | I | [192] |
| REannotate | Other | Any | NO | RepeatMasker output | O | [193] |
| ReAS | De novo | Any | NO | Unassembled sequence reads | I | [194] |
| RECON | De novo | Any | NO | Unassembled and assembled sequences | I | [195] |
| Red | De novo (HMM) | Any | YES | Unassembled and assembled sequences | I | [18] |
| REDdenovo | De novo | Any | NO | Unassembled sequence reads | I | [21] |
| REPCLASS | Structure-based, Homology-based | Any | NO | Assembled sequences | I | [196] |
| RepeatExplorer | De novo | Any | NO | Unassembled sequence reads | I | [197] |
| RepeatMasker | Homology-based | Any | NO | Assembled sequences | O | http://www.repeatmasker.org/ |
| RepeatModeler | De novo | Any | NO | Assembled sequences | I | http://www.repeatmasker.org/RepeatModeler/ |
| RepeatScout | De novo | Any | NO | Assembled sequences | I | [198] |
| Repeat Pattern | De novo | Any | NO | Assembled sequences | I | [199] |
| REPET | De novo, Structure-based, | Any | NO | Assembled sequences | I, C | [200] |
| Repseek | De novo | Any | NO | Assembled sequences | I | [201] |
| REPuter | De novo | Any | NO | Assembled sequences | I | [202] |
| TEClass | De novo (SVM) | Any | YES | Assembled sequences | C | [17] |
| TEdna | De novo | Any | NO | Unassembled sequence reads | I | [19] |
| transposome | De novo | Any | NO | Unassembled sequence reads | I | [20] |
| Database | Genomes | Data Composition | URL |
|---|---|---|---|
| Gypsy database | Several plant genomes | Domains from LTR Retrotransposons | http://gydb.org/index.php/Main_Page |
| MASiVEdb | Several plant genomes | Sire Retrotransposons | http://databases.bat.ina.certh.gr/masivedb/ |
| Repbase | Several plant genomes | All TEs | https://www.girinst.org/repbase/ |
| RepPop | Populus trichocarpa | All TEs | http://csbl.bmb.uga.edu/~ffzhou/RepPop/ |
| RetrOryza | Rice | LTR Retrotransposons | http://retroryza.fr |
| REXdb | Several plant genomes | Domains form LTR Retrotransposons | http://repeatexplorer.org/?page_id=918 |
| SINEBase | Several plant genomes | SINEs | http://sines.eimb.ru/ |
| SoyTEdb | Soybean | All TEs | https://soybase.org/soytedb/ |
| TIGR Maize repeat database | Maize | All TEs | http://maize.jcvi.org/repeat_db.shtml |
| TRansposable Elements Platform (TREP) database | Several cereal genomes | All TEs | http://botserv2.uzh.ch/kelldata/trep-db/ |
| Plant Genome and System Biology (PGSB) Repeat Database | Several plant genomes | All TEs | http://pgsb.helmholtz-muenchen.de/plant/recat/ |
| RepetDB | Several plant genomes | All TE consensus | http://urgi.versailles.inra.fr/repetdb/begin.do |
| Coding Schemes | Codebook | Reference |
|---|---|---|
| DAX | {‘C’:0, ‘T’:1, ‘A’:2, ‘G’:3} | [210] |
| EIIP | {‘C’:0.1340, ‘T’:0.1335, ‘A’:0.1260, ‘G’:0.0806} | [211] |
| Complementary | {‘C’:-1, ‘T’:-2, ‘A’:2, ‘G’:1} | [212] |
| Enthalpy | {‘CC’:0.11, ‘TT’:0.091, ‘AA’:0.091, ‘GG’:0.11, ‘CT’:0.078, ‘TA’:0.06, ‘AG’:0.078, ‘CA’:0.058, ‘TG’:0.058, ‘CG’: 0.119, ‘TC’:0.056, ‘AT’:0.086, ‘GA’:0.056, ‘AC’:0.065, ‘GT’:0.065, ‘GC’:0.1111} | [213] |
| Galois (4) | {‘CC’:0.0, ‘CT’:1.0, ‘CA’:2.0, ‘CG’:3.0, ‘TC’:4.0, ‘TT’:5.0, ‘TA’:6.0, ‘TG’:7.0, ‘AC:8.0, ‘AT: 9.0, ‘AA’:1.0, ‘AG:11.0, ‘GC’:12.0, ‘GT’:13.0, ‘GA’:14.0, ‘GG’:15.0 } | [209] |
| Orthogonal Encoding | {‘A’: [1, 0, 0, 0], ‘C’: [0, 1, 0, 0], ‘T’: [0, 0, 1, 0], ‘G’: [0, 0, 0, 1]} | [214] |
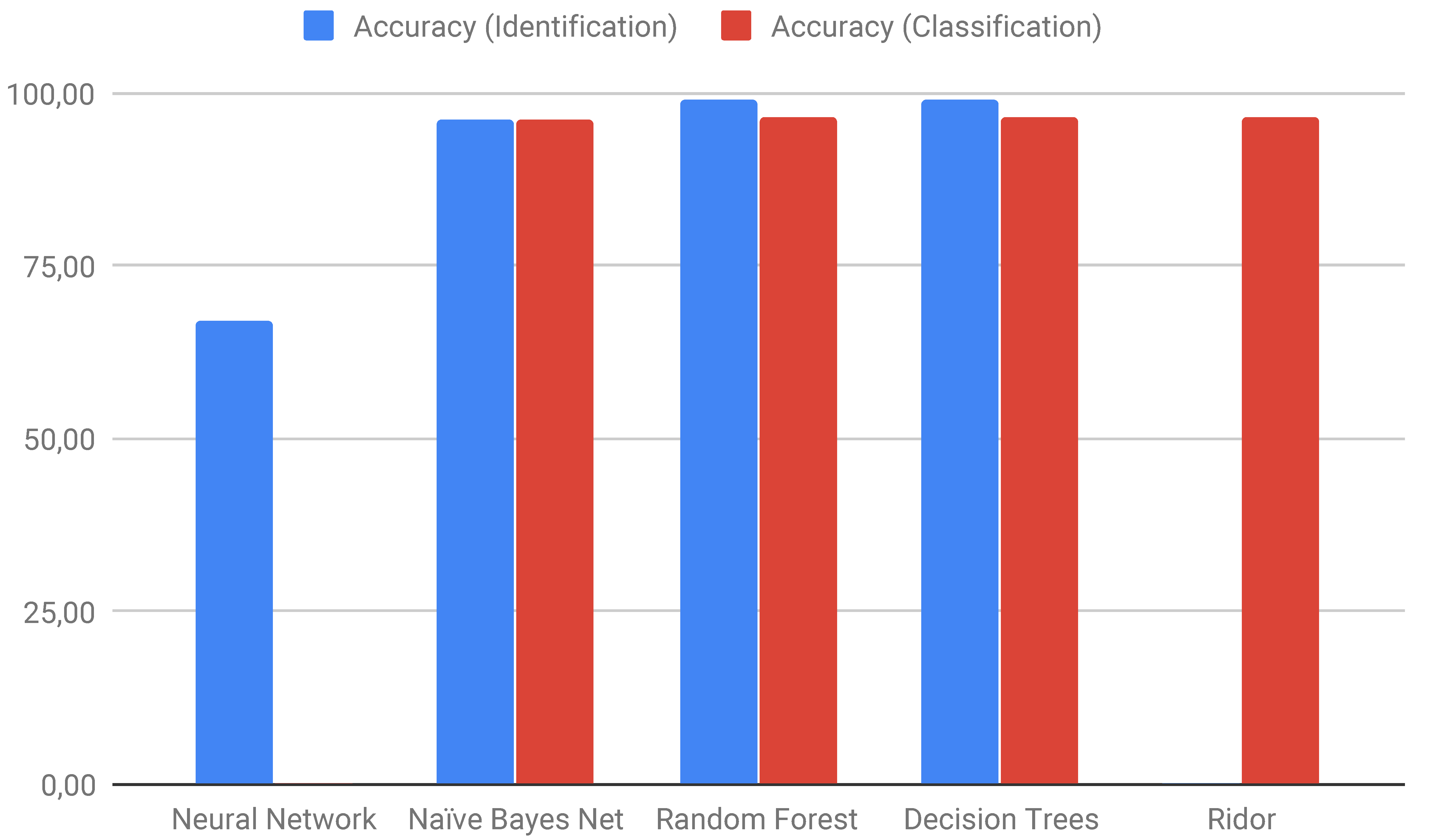
| Identification | Classification | ||
|---|---|---|---|
| Algorithm | Accuracy | Algorithm | Accuracy |
| Neural Network | 67.01 | Ridor | 96.43 |
| Naïve Bayes Net | 96.30 | Naïve Bayes Net | 96.37 |
| Random Forest | 98.90 | Random Forest | 96.56 |
| Decision Trees | 98.92 | Decision Trees | 96.56 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orozco-Arias, S.; Isaza, G.; Guyot, R. Retrotransposons in Plant Genomes: Structure, Identification, and Classification through Bioinformatics and Machine Learning. Int. J. Mol. Sci. 2019, 20, 3837. https://doi.org/10.3390/ijms20153837
Orozco-Arias S, Isaza G, Guyot R. Retrotransposons in Plant Genomes: Structure, Identification, and Classification through Bioinformatics and Machine Learning. International Journal of Molecular Sciences. 2019; 20(15):3837. https://doi.org/10.3390/ijms20153837
Chicago/Turabian StyleOrozco-Arias, Simon, Gustavo Isaza, and Romain Guyot. 2019. "Retrotransposons in Plant Genomes: Structure, Identification, and Classification through Bioinformatics and Machine Learning" International Journal of Molecular Sciences 20, no. 15: 3837. https://doi.org/10.3390/ijms20153837
APA StyleOrozco-Arias, S., Isaza, G., & Guyot, R. (2019). Retrotransposons in Plant Genomes: Structure, Identification, and Classification through Bioinformatics and Machine Learning. International Journal of Molecular Sciences, 20(15), 3837. https://doi.org/10.3390/ijms20153837