Hurdles to Cardioprotection in the Critically Ill


Abstract
1. Introduction
2. Critically Ill Cardiac Patients
3. Mechanical Circulatory Support (MCS)
3.1. Short-Term MCS
3.1.1. Extracorporeal Membrane Oxygenation (ECMO)
3.1.2. Percutaneous VADs
3.1.3. Durable VADs
3.2. MCS-Associated Complications
3.2.1. ECMO Complications
3.2.2. PVAD Complications
3.2.3. Durable VAD Complications
3.3. Cardioprotective Strategies for MCS
3.3.1. Acute Mechanical Unloading
3.3.2. Pulsatile Flow
3.3.3. Heparin
3.3.4. Mitochondrial Transplant
3.3.5. Mesenchymal Stem Cells
4. Heart Transplantation
4.1. Immunosuppression
4.2. Immunosuppression Can Impair Cardiac Function
4.2.1. Induction Therapy
4.2.2. Maintenance Therapy
4.3. Cardioprotection and HTx
4.3.1. Intrinsic Immune Responses Aid Cardiac Recovery
4.3.2. Cardioprotection Despite Immunosuppression
4.3.3. Cardioprotective Cellular Therapies and Immunosuppression
5. Cardioprotection With Inotropic Support
5.1. Adrenergic Stimulants
5.1.1. Beta-Adrenoceptors
5.1.2. Alpha-Adrenoceptors
5.1.3. Dopamine Receptors
5.2. Complications with Adrenergic Inotrope Support
5.2.1. Adrenergic Desensitization
5.2.2. Energetic Imbalance
5.3. Myofilament Calcium Sensitisers
5.4. Enhancers of Cytosolic Ca2+ Handling
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HF | Heart Failure |
| HTx | Heart Transplant |
| BD | Brain dead |
| PGD | Primary graft dysfunction |
| IRI | Ischemia-reperfusion injury |
| IR | Ischemia-reperfusion |
| MCS | Mechanical circulatory support |
| VAD | Ventricular assist device |
| ECMO | Extracorporeal membrane oxygenation |
| V-A | Veno-arterial |
| PVAD | Percutaneous ventricular assist device |
| IABP | Intra-aortic balloon pump |
| LV | Left ventricle |
| RV | Right ventricle |
| RVF | Right ventricular failure |
| AMI | Acute myocardial infarction |
| PCI | Percutaneous coronary intervention |
| ICT | Intracardiac thrombosis |
| VA | Ventricular arrhythmia |
| AI | Aortic insufficiency |
| MSC | Mesenchymal stromal cells |
| MPC | Mesenchymal precursor cells |
| TAC | Tacrolimus (TAC) |
| CsA | Cyclosporine A |
| MMF | Mycophenolate mofetil |
| AZA | Azathioprine |
| Treg | Regulatory T cells |
| CRS | Cytokine release syndrome |
| GPCR | G protein-coupled receptors |
| ATG | Anti-thymocyte globulin |
| LMWH | Low molecular weight derivatives |
| HLA-G | Human leukocyte antigen-G |
| PGD | Prostaglandin D |
| PGD2 | Prostaglandin D2 |
| MP | Methylprednisolone |
| β-AR | β-adrenoceptors |
| α-AR | α-adrenoceptor |
| GC | Glucocorticoids |
References
- Ritchie, H.; Roser, M. Causes of Death. Available online: OurWorldInData.org (accessed on 15 April 2019).
- Ponikowski, P.; Anker, S.D.; AlHabib, K.F.; Cowie, M.R.; Force, T.L.; Hu, S.; Jaarsma, T.; Krum, H.; Rastogi, V.; Rohde, L.E.; et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail. 2014, 1, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States A Policy Statement from the American Heart Association. Circ. Heart Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. Shattuck lecture—Cardiovascular medicine at the turn of the millennium: Triumphs, concerns, and opportunities. N. Engl. J. Med. 1997, 337, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.; Pivetta, A.; Pinamonti, B.; Stolfo, D.; Zecchin, M.; Barbati, G.; Di Lenarda, A.; Sinagra, G. Long-term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: Changing mortality over the last 30 years. Eur. J. Heart Fail. 2014, 16, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Dunlay, S.M.; Redfield, M.M.; Weston, S.A.; Therneau, T.M.; Hall Long, K.; Shah, N.D.; Roger, V.L. Hospitalizations after heart failure diagnosis a community perspective. J. Am. Coll. Cardiol. 2009, 54, 1695–1702. [Google Scholar] [CrossRef]
- Gheorghiade, M.; Vaduganathan, M.; Fonarow, G.C.; Bonow, R.O. Rehospitalization for heart failure: Problems and perspectives. J. Am. Coll. Cardiol. 2013, 61, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef]
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Bolli, R.; Becker, L.; Gross, G.; Mentzer Jr, R.; Balshaw, D.; Lathrop, D.A. Myocardial protection at a crossroads: The need for translation into clinical therapy. Circ. Res. 2004, 95, 125–134. [Google Scholar] [CrossRef]
- Heusch, G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 2013, 381, 166–175. [Google Scholar] [CrossRef]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef]
- Rosenberg, J.H.; Werner, J.H.; Moulton, M.J.; Agrawal, D.K. Current Modalities and Mechanisms Underlying Cardioprotection by Ischemic Conditioning. J. Cardiovasc. Transl. Res. 2018, 11, 292–307. [Google Scholar] [CrossRef]
- Peart, J.N.; Headrick, J.P. Clinical cardioprotection and the value of conditioning responses. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1705–H1720. [Google Scholar] [CrossRef]
- See Hoe, L.; Patel, H.H.; Peart, J.N. Delta Opioid Receptors and Cardioprotection. Handb. Exp. Pharmacol. 2018, 247, 301–334. [Google Scholar]
- Gregory, S.D.; Stevens, M.C.; Pauls, J.P.; Schummy, E.; Diab, S.; Thomson, B.; Anderson, B.; Tansley, G.; Salamonsen, R.; Fraser, J.F.; et al. In Vivo Evaluation of Active and Passive Physiological Control Systems for Rotary Left and Right Ventricular Assist Devices. Artif. Organs 2016, 40, 894–903. [Google Scholar] [CrossRef]
- Passmore, M.R.; Fung, Y.L.; Simonova, G.; Foley, S.R.; Dunster, K.R.; Diab, S.D.; Tung, J.P.; Minchinton, R.M.; McDonald, C.I.; Anstey, C.M.; et al. Inflammation and lung injury in an ovine model of extracorporeal membrane oxygenation support. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1202–L1212. [Google Scholar] [CrossRef]
- See Hoe, L.E.; Engkilde-Pedersen, S.; Obonyo, N.G.; Wells, M.A.; Boon, C.; Bartnikowski, N.; Passmore, M.; McDonald, C.; Black, D.; Molenaar, P.; et al. Development of an Ovine Model of Heart Transplantation Following 24-Hour Brain Stem Death. Heart Lung Circ. 2018, 27, S92. [Google Scholar] [CrossRef]
- Habal, M.V.; Garan, A.R. Long-term management of end-stage heart failure. Best Pract. Res. Clin. Anaesthesiol. 2017, 31, 153–166. [Google Scholar] [CrossRef]
- Chaudhry, S.P.; Stewart, G.C. Advanced Heart Failure: Prevalence, Natural History, and Prognosis. Heart Fail. Clin. 2016, 12, 323–333. [Google Scholar] [CrossRef]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef]
- Atherton, J.J.; Sindone, A.; De Pasquale, C.G.; Driscoll, A.; MacDonald, P.S.; Hopper, I.; Kistler, P.; Briffa, T.G.; Wong, J.; Abhayaratna, W.P.; et al. National Heart Foundation of Australia and Cardiac Society of Australia and New Zealand: Australian clinical guidelines for the management of heart failure 2018. Med. J. Aust. 2018, 209, 363–369. [Google Scholar] [CrossRef]
- Kong, C.H.; Lin, X.Y.; Woo, C.C.; Wong, H.C.; Lee, C.N.; Richards, A.M.; Sorokin, V.A. Characteristics of aortic wall extracellular matrix in patients with acute myocardial infarction: Tissue microarray detection of collagen I, collagen III and elastin levels. Interact. Cardiovasc. Thorac. Surg. 2013, 16, 11–15. [Google Scholar] [CrossRef]
- See Hoe, L.E.; May, L.T.; Headrick, J.P.; Peart, J.N. Sarcolemmal dependence of cardiac protection and stress-resistance: Roles in aged or diseased hearts. Br. J. Pharmacol. 2016, 173, 2966–2991. [Google Scholar] [CrossRef]
- See Hoe, L.E.; Schilling, J.M.; Tarbit, E.; Kiessling, C.J.; Busija, A.R.; Niesman, I.R.; Du Toit, E.; Ashton, K.J.; Roth, D.M.; Headrick, J.P.; et al. Sarcolemmal cholesterol and caveolin-3 dependence of cardiac function, ischemic tolerance, and opioidergic cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H895–H903. [Google Scholar] [CrossRef]
- De Carvalho, L.P.; Tan, S.H.; Ow, G.S.; Tang, Z.; Ching, J.; Kovalik, J.P.; Poh, S.C.; Chin, C.T.; Richards, A.M.; Martinez, E.C.; et al. Plasma Ceramides as Prognostic Biomarkers and Their Arterial and Myocardial Tissue Correlates in Acute Myocardial Infarction. JACC Basic Transl. Sci. 2018, 3, 163–175. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Distinctive molecular signature and activated signaling pathways in aortic smooth muscle cells of patients with myocardial infarction. Atherosclerosis 2018, 271, 237–244. [Google Scholar] [CrossRef]
- Derda, A.A.; Woo, C.C.; Wongsurawat, T.; Richards, M.; Lee, C.N.; Kofidis, T.; Kuznetsov, V.A.; Sorokin, V.A. Gene expression profile analysis of aortic vascular smooth muscle cells reveals upregulation of cadherin genes in myocardial infarction patients. Physiol. Genom. 2018, 50, 648–657. [Google Scholar] [CrossRef]
- Feldman, D.; Pamboukian, S.V.; Teuteberg, J.J. The 2013 International Society for Heart and Lung Transplantation Guidelines for mechanical circulatory support: Executive summary. J. Heart Lung Transplant. 2013, 32, 157–187. [Google Scholar] [CrossRef]
- Kirklin James, K.; Naftel David, C. Mechanical Circulatory Support. Circ. Heart Fail. 2008, 1, 200–205. [Google Scholar] [CrossRef]
- Shekar, K.; Gregory, S.D.; Fraser, J.F. Mechanical circulatory support in the new era: An overview. Crit. Care 2016, 20, 66. [Google Scholar] [CrossRef]
- Paliwal, S.; Kakkar, A.; Sharma, R.; Airan, B.; Mohanty, S. Differential reduction of reactive oxygen species by human tissuespecific mesenchymal stem cells from different donors under oxidative stress. J. Biosci. 2017, 42, 373–382. [Google Scholar] [CrossRef]
- Reynolds, H.R.; Hochman, J.S. Cardiogenic shock: Current concepts and improving outcomes. Circulation 2008, 117, 686–697. [Google Scholar] [CrossRef]
- Thiele, H.; Ohman, E.M.; Desch, S.; Eitel, I.; de Waha, S. Management of cardiogenic shock. Eur. Heart J. 2015, 36, 1223–1230. [Google Scholar] [CrossRef]
- Thiele, H.; Zeymer, U.; Neumann, F.J. Intraaortic balloon support for myocardial infarction with cardiogenic shock. N. Engl. J. Med. 2012, 367, 1287–1296. [Google Scholar] [CrossRef]
- Lauten, E.A.; Engström, W.A.; Jung, F.C.; Empen, G.K.; Erne, P.S.P.; Cook, R.S.; Windecker, R.S.; Bergmann, R.M.; Klingenberg, R.R.; Lüscher, R.T.; et al. Percutaneous Left-Ventricular Support with the Impella-2.5–Assist Device in Acute Cardiogenic Shock: Results of the Impella–EUROSHOCK-Registry. Circ. Heart Fail. 2013, 6, 23–30. [Google Scholar] [CrossRef]
- Myat, A.; Patel, N.; Tehrani, S.; Banning, A.P.; Redwood, S.R.; Bhatt, D.L. Percutaneous Circulatory Assist Devices for High-Risk Coronary Intervention. JACC Cardiovasc. Interv. 2015, 8, 229–244. [Google Scholar] [CrossRef]
- O’Neill William, W.; Kleiman Neal, S.; Moses, J.; Henriques Jose, P.S.; Dixon, S.; Massaro, J.; Palacios, I.; Maini, B.; Mulukutla, S.; Džavík, V.; et al. A Prospective, Randomized Clinical Trial of Hemodynamic Support with Impella 2.5 Versus Intra-Aortic Balloon Pump in Patients Undergoing High-Risk Percutaneous Coronary Intervention. Circulation 2012, 126, 1717–1727. [Google Scholar]
- Takayama, H.; Truby, L.; Takeda, K.; Naka, Y. Short-Term Ventricular Assist Devices (Implantable and Percutaneous). Curr. Surg. Rep. 2014, 2, 58. [Google Scholar] [CrossRef][Green Version]
- Sorokin, V.; MacLaren, G.; Vidanapathirana, P.C.; Delnoij, T.; Lorusso, R. Choosing the appropriate configuration and cannulation strategies for extracorporeal membrane oxygenation: The potential dynamic process of organ support and importance of hybrid modes. Eur. J. Heart Fail. 2017, 19 (Suppl. 2), 75–83. [Google Scholar] [CrossRef]
- Peart, J.N.; Pepe, S.; Reichelt, M.E.; Beckett, N.; See Hoe, L.; Ozberk, V.; Niesman, I.R.; Patel, H.H.; Headrick, J.P. Dysfunctional survival-signaling and stress-intolerance in aged murine and human myocardium. Exp. Gerontol. 2014, 50, 72–81. [Google Scholar] [CrossRef]
- Vakayil, V.; Thompson, D.A.; Sundin, A.; Chandrashekar, M.; Harmon, J.V., Jr.; Brunsvold, M.E. Indicators of Mortality for Patients on Veno-Arterial Extracorporeal Membrane Oxygenation (ECMO): A Single Center Experience. J. Am. Coll. Surg. 2017, 225, e74–e75. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Yang, F.; Wang, X.; Xie, H.; Fan, E.; Ogino, M.; Brodie, D.; Wang, H.; Hou, X. Predicting mortality in patients undergoing VA-ECMO after coronary artery bypass grafting: The REMEMBER score. Crit. Care 2019, 23, 11. [Google Scholar] [CrossRef]
- Lawson, W.E.; Koo, M. Percutaneous Ventricular Assist Devices and ECMO in the Management of Acute Decompensated Heart Failure. Clin. Med. Insights Cardiol. 2015, 9 (Suppl. 1), 41–48. [Google Scholar] [CrossRef]
- Gilotra, N.A.; Stevens, G.R. Temporary mechanical circulatory support: A review of the options, indications, and outcomes. Clin. Med. Insights Cardiol. 2015, 8 (Suppl. 1), 75–85. [Google Scholar] [CrossRef]
- Burkhoff, D.; Cohen, H.; Brunckhorst, C.; O’Neill, W.W. A randomized multicenter clinical study to evaluate the safety and efficacy of the TandemHeart percutaneous ventricular assist device versus conventional therapy with intraaortic balloon pumping for treatment of cardiogenic shock. Am. Heart J. 2006, 152, 469.e1–469.e8. [Google Scholar] [CrossRef]
- Thiele, H.; Sick, P.; Boudriot, E.; Diederich, K.W.; Hambrecht, R.; Niebauer, J.; Schuler, G. Randomized comparison of intra-aortic balloon support with a percutaneous left ventricular assist device in patients with revascularized acute myocardial infarction complicated by cardiogenic shock. Eur. Heart J. 2005, 26, 1276–1283. [Google Scholar] [CrossRef]
- Miller, M.A.; Dukkipati, S.R.; Mittnacht, A.J.; Chinitz, J.S.; Belliveau, L.; Koruth, J.S.; Gomes, J.A.; d’Avila, A.; Reddy, V.Y. Activation and Entrainment Mapping of Hemodynamically Unstable Ventricular Tachycardia Using a Percutaneous Left Ventricular Assist Device. J. Am. Coll. Cardiol. 2011, 58, 1363–1371. [Google Scholar] [CrossRef]
- Seyfarth, M.; Sibbing, D.; Bauer, I.; Fröhlich, G.; Bott-Flügel, L.; Byrne, R.; Dirschinger, J.; Kastrati, A.; Schömig, A. A Randomized Clinical Trial to Evaluate the Safety and Efficacy of a Percutaneous Left Ventricular Assist Device Versus Intra-Aortic Balloon Pumping for Treatment of Cardiogenic Shock Caused by Myocardial Infarction. J. Am. Coll. Cardiol. 2008, 52, 1584–1588. [Google Scholar] [CrossRef]
- Schrage, B.; Ibrahim, K.; Loehn, T.; Werner, N.; Sinning, J.M.; Pappalardo, F.; Pieri, M.; Skurk, C.; Lauten, A.; Landmesser, U.; et al. Impella Support for Acute Myocardial Infarction Complicated by Cardiogenic Shock. Circulation 2019, 139, 1249–1258. [Google Scholar] [CrossRef]
- Kapur, N.K.; Qiao, X.; Paruchuri, V.; Morine, K.J.; Syed, W.; Dow, S.; Shah, N.; Pandian, N.; Karas, R.H. Mechanical Pre-Conditioning with Acute Circulatory Support Before Reperfusion Limits Infarct Size in Acute Myocardial Infarction. JACC Heart Fail. 2015, 3, 873–882. [Google Scholar] [CrossRef]
- Myers, T. Temporary Ventricular Assist Devices in the Intensive Care Unit as a Bridge to Decision. AACN Adv. Crit. Care 2012, 23, 55–68. [Google Scholar] [CrossRef]
- Nicolini, F.; Gherli, T. Alternatives to transplantation in the surgical therapy for heart failure. Eur. J. Cardio-Thoracic Surg. 2009, 35, 214–228. [Google Scholar] [CrossRef]
- Slaughter, M.S.; Rogers, J.G.; Milano, C.A.; Russell, S.D.; Conte, J.V.; Feldman, D.; Sun, B.; Tatooles, A.J.; Delgado, R.M.; Long, J.W.; et al. Advanced Heart Failure Treated with Continuous-Flow Left Ventricular Assist Device. N. Engl. J. Med. 2009, 361, 2241–2251. [Google Scholar] [CrossRef]
- Dandel, M.; Hetzer, R. Recovery of failing hearts by mechanical unloading: Pathophysiologic insights and clinical relevance. Am. Heart J. 2018, 206, 30–50. [Google Scholar] [CrossRef]
- Kirklin, J.K.; Pagani, F.D.; Kormos, R.L.; Stevenson, L.W.; Blume, E.D.; Myers, S.L.; Miller, M.A.; Baldwin, J.T.; Young, J.B.; Naftel, D.C. Eighth annual INTERMACS report: Special focus on framing the impact of adverse events. J. Hear. Lung Transplant. 2017, 36, 1080–1086. [Google Scholar] [CrossRef]
- Dandel, M.; Hetzer, R. Myocardial recovery during mechanical circulatory support: Long-term outcome and elective ventricular assist device implantation to promote recovery as a treatment goal. Heart Lung Vessel. 2015, 7, 289. [Google Scholar]
- Kirklin, J.K.; Naftel, D.C.; Pagani, F.D. Sixth INTERMACS annual report: A 10,000-patient database. J. Heart Lung Transplant. 2014, 33, 555–564. [Google Scholar] [CrossRef]
- Drakos, S.G.; Kfoury, A.G.; Stehlik, J.; Selzman, C.H.; Reid, B.B.; Terrovitis, J.V.; Nanas, J.N.; Li, D.Y. Bridge to recovery: Understanding the disconnect between clinical and biological outcomes. Circulation 2012, 126, 230–241. [Google Scholar] [CrossRef]
- Mehra, M.R.; Goldstein, D.J.; Uriel, N.; Cleveland, J.C.; Yuzefpolskaya, M.; Salerno, C.; Walsh, M.N.; Milano, C.A.; Patel, C.B.; Ewald, G.A.; et al. Two-Year Outcomes with a Magnetically Levitated Cardiac Pump in Heart Failure. N. Engl. J. Med. 2018, 378, 1386–1395. [Google Scholar] [CrossRef]
- Thomas, J.; Kostousov, V.; Teruya, J. Bleeding and Thrombotic Complications in the Use of Extracorporeal Membrane Oxygenation. Semin. Thromb. Hemost. 2018, 44, 20–29. [Google Scholar] [CrossRef]
- Dalton, H.J.; Reeder, R.; Garcia-Filion, P.; Holubkov, R.; Berg, R.A.; Zuppa, A.; Moler, F.W.; Shanley, T.; Pollack, M.M.; Newth, C.; et al. Factors Associated with Bleeding and Thrombosis in Children Receiving Extracorporeal Membrane Oxygenation. Am. J. Respir. Crit. Care Med. 2017, 196, 762–771. [Google Scholar] [CrossRef]
- Mehta, H.; Eisen, H.; Cleveland, J.C. Indications and Complications for VA-ECMO for Cardiac Failure; American College of Cardiology Perspectives and Analysis: Washington, DC, USA, 2015. [Google Scholar]
- Alhussein, M.; Moayedi, Y.; Posada, J.D.; Ross, H.; Hickey, E.; Rao, V.; Billia, F. Ventricular Thrombosis Post-Venoarterial Extracorporeal Membrane Oxygenation. Circ. Heart Fail. 2017, 10, e003757. [Google Scholar] [CrossRef]
- Makdisi, G.; Hashmi, Z.A.; Wozniak, T.C.; Wang, I.W. Left ventricular thrombus associated with arteriovenous extra corporeal membrane oxygenation. J. Thorac. Dis. 2015, 7, E552–E554. [Google Scholar]
- Williams, B.; Bernstein, W. Review of Venoarterial Extracorporeal Membrane Oxygenation and Development of Intracardiac Thrombosis in Adult Cardiothoracic Patients. J. Extra Corpor. Technol. 2016, 48, 162–167. [Google Scholar]
- Weber, C.; Deppe, A.C.; Sabashnikov, A.; Slottosch, I.; Kuhn, E.; Eghbalzadeh, K.; Scherner, M.; Choi, Y.H.; Madershahian, N.; Wahlers, T. Left ventricular thrombus formation in patients undergoing femoral veno-arterial extracorporeal membrane oxygenation. Perfusion 2018, 33, 283–288. [Google Scholar] [CrossRef]
- Bhat, A.G.; Golchin, A.; Pasupula, D.K.; Hernandez-Montfort, J.A. Right Sided Intracardiac Thrombosis during Veno-Arterial Extracorporeal Membrane Oxygenation: A Case Report and Literature Review. Case Rep. Crit. Care 2019, 2019, 8594681. [Google Scholar] [CrossRef]
- Riahi, M.; Baruteau, A.E. Left ventricular distention under venoarterial extracorporeal membrane oxygenation support: When should we consider percutaneous left heart decompression? J. Thorac. Dis. 2017, 9, 4919–4921. [Google Scholar] [CrossRef]
- Meani, P.; Pappalardo, F. The step forward for VA ECMO: Left ventricular unloading! J. Thorac. Dis. 2017, 9, 4149–4151. [Google Scholar] [CrossRef]
- Donker, D.W.; Brodie, D.; Henriques, J.P.S.; Broome, M. Left ventricular unloading during veno-arterial ECMO: A review of percutaneous and surgical unloading interventions. Perfusion 2019, 34, 98–105. [Google Scholar] [CrossRef]
- Conrad, S.A. Persistent hypoxemia on ECMO. Qatar Med. J. 2017, 1, 18. [Google Scholar] [CrossRef]
- Cove, M.E. Disrupting differential hypoxia in peripheral veno-arterial extracorporeal membrane oxygenation. Crit. Care 2015, 19, 280. [Google Scholar] [CrossRef]
- Hou, X.; Yang, X.; Du, Z.; Xing, J.; Li, H.; Jiang, C.; Wang, J.; Xing, Z.; Li, S.; Li, X.; et al. Superior vena cava drainage improves upper body oxygenation during veno-arterial extracorporeal membrane oxygenation in sheep. Crit. Care 2015, 19, 68. [Google Scholar] [CrossRef]
- Sidebotham, D. Troubleshooting adult ECMO. J. Extra Corpor. Technol. 2011, 43, P27–P32. [Google Scholar]
- Millar, J.E.; Fanning, J.P.; McDonald, C.I.; McAuley, D.F.; Fraser, J.F. The inflammatory response to extracorporeal membrane oxygenation (ECMO): A review of the pathophysiology. Crit. Care 2016, 20, 387. [Google Scholar] [CrossRef]
- Hayes, R.A.; Shekar, K.; Fraser, J.F. Is hyperoxaemia helping or hurting patients during extracorporeal membrane oxygenation? Review of a complex problem. Perfusion 2013, 28, 184–193. [Google Scholar] [CrossRef]
- McDonald, C.I.; Fraser, J.F.; Coombes, J.S.; Fung, Y.L. Oxidative stress during extracorporeal circulation. Eur. J. Cardio Thorac. Surg. 2014, 46, 937–943. [Google Scholar] [CrossRef]
- Gregoric, I.D.; Bruckner, B.A.; Jacob, L.; Loyalka, P.; Kar, B.; La Francesca, S.; Myers, T.; Frazier, O.H. Techniques and Complications of TandemHeart Ventricular Assist Device Insertion During Cardiac Procedures. ASAIO J. 2009, 55, 251–254. [Google Scholar] [CrossRef]
- De Souza, C.F.; De Souza Brito, F.; De Lima, V.C.; De Camargo Carvalho, A.C. Percutaneous Mechanical Assistance for the Failing Heart. J. Interv. Cardiol. 2010, 23, 195–202. [Google Scholar] [CrossRef]
- Kirklin, J.K.; Naftel, D.C.; Pagani, F.D.; Kormos, R.L.; Stevenson, L.W.; Blume, E.D.; Myers, S.L.; Miller, M.A.; Baldwin, J.T.; Young, J.B. Seventh INTERMACS annual report: 15,000 patients and counting. J. Hear. Lung Transplant. 2015, 34, 1495–1504. [Google Scholar] [CrossRef]
- Stewart, C.G.; Givertz, M.M. Mechanical Circulatory Support for Advanced Heart Failure: Patients and Technology in Evolution. Circulation 2012, 125, 1304–1315. [Google Scholar] [CrossRef]
- Ziv, O.; Dizon, J.; Thosani, A.; Naka, Y.; Magnano, A.R.; Garan, H. Effects of Left Ventricular Assist Device Therapy on Ventricular Arrhythmias. J. Am. Coll. Cardiol. 2005, 45, 1428–1434. [Google Scholar] [CrossRef]
- Refaat, M.; Chemaly, E.; Lebeche, D.; Gwathmey, J.K.; Hajjar, R.J. Ventricular Arrhythmias after Left Ventricular Assist Device Implantation. Pacing Clin. Electrophysiol. 2008, 31, 1246–1252. [Google Scholar] [CrossRef]
- Grzywacz, F.W.; Piacentino, V., III; Marble, J.; Bozorgnia, B.; Gaughan, J.P.; Rothman, S.A.; Margulies, K.B. Effect of Acute Unloading Via Head-Up Tilt on QTc Prolongation in Patients with Ischemic or Non-Ischemic Cardiomyopathy. Am. J. Cardiol. 2006, 97, 412–415. [Google Scholar] [CrossRef]
- Oswald, H.; Schultz-Wildelau, C.; Gardiwal, A.; Lüsebrink, U.; König, T.; Meyer, A.; Duncker, D.; Pichlmaier, M.A.; Klein, G.; Strüber, M. Implantable defibrillator therapy for ventricular tachyarrhythmia in left ventricular assist device patients. Eur. J. Fail. 2010, 12, 593–599. [Google Scholar] [CrossRef]
- Aggarwal, A.; Raghuvir, R.; Eryazici, P.; Macaluso, G.; Sharma, P.; Blair, C.; Tatooles, A.J.; Pappas, P.S.; Bhat, G. The Development of Aortic Insufficiency in Continuous-Flow Left Ventricular Assist Device–Supported Patients. Ann. Thorac. Surg. 2013, 95, 493–498. [Google Scholar] [CrossRef]
- Deo, S.V.; Sharma, V.; Cho, Y.H.; Shah, I.K.; Park, S.J. De Novo Aortic Insufficiency During Long-Term Support on a Left Ventricular Assist Device: A Systematic Review and Meta-Analysis. ASAIO J. 2014, 60, 183–188. [Google Scholar] [CrossRef]
- May-Newman, K.; Hillen, B.; Dembitsky, W. Effect of Left Ventricular Assist Device Outflow Conduit Anastomosis Location on Flow Patterns in the Native Aorta. ASAIO J. 2006, 52, 132–139. [Google Scholar] [CrossRef]
- Cowger, J.; Pagani, F.D.; Haft, J.W.; Romano, M.A.; Aaronson, K.D.; Kolias, T.J. The development of aortic insufficiency in left ventricular assist device-supported patients. Circ. Heart Fail. 2010, 3, 668–674. [Google Scholar] [CrossRef]
- Drakos, S.; Janicki, L.; Horne, B.D.; Kfoury, A.; Reid, B.B.; Clayson, S.; Horton, K.; Haddad, F.; Li, D.Y.; Renlund, D.G.; et al. Risk Factors Predictive of Right Ventricular Failure After Left Ventricular Assist Device Implantation. Am. J. Cardiol. 2010, 105, 1030–1035. [Google Scholar] [CrossRef]
- Lampert, B.C.; Teuteberg, J.J. Right ventricular failure after left ventricular assist devices. J. Heart Lung Transplant. 2015, 34, 1123–1130. [Google Scholar] [CrossRef]
- Argiriou, M.; Kolokotron, S.M.; Sakellaridis, T.; Argiriou, O.; Charitos, C.; Zarogoulidis, P.; Katsikogiannis, N.; Kougioumtzi, I.; Machairiotis, N.; Tsiouda, T.; et al. Right heart failure post left ventricular assist device implantation. J. Thorac. Dis. 2014, 6, S52–S59. [Google Scholar]
- Hayek, S.; Sims Daniel, B.; Markham David, W.; Butler, J.; Kalogeropoulos Andreas, P. Assessment of Right Ventricular Function in Left Ventricular Assist Device Candidates. Circ. Cardiovasc. Imaging 2014, 7, 379–389. [Google Scholar] [CrossRef]
- Turner, K.R. Right Ventricular Failure After Left Ventricular Assist Device Placement—The Beginning of the End or Just Another Challenge? J. Cardiothorac. Vasc. Anesth. 2019, 33, 1105–1121. [Google Scholar] [CrossRef]
- Deschka, H.; Holthaus, A.J.; Sindermann, J.R.; Welp, H.; Schlarb, D.; Monsefi, N.; Martens, S.; Scherer, M. Can Perioperative Right Ventricular Support Prevent Postoperative Right Heart Failure in Patients with Biventricular Dysfunction Undergoing Left Ventricular Assist Device Implantation? J. Cardiothorac. Vasc. Anesth. 2016, 30, 619–626. [Google Scholar] [CrossRef]
- Dell’Italia, L.J. Anatomy and Physiology of the Right Ventricle. Cardiol. Clin. 2012, 30, 167–187. [Google Scholar] [CrossRef]
- Topilsky, Y.; Hasin, T.; Oh Jae, K.; Borgeson Daniel, D.; Boilson Barry, A.; Schirger John, A.; Clavell Alfredo, L.; Frantz Robert, P.; Tsutsui, R.; Liu, M.; et al. Echocardiographic Variables After Left Ventricular Assist Device Implantation Associated with Adverse Outcome. Circ. Cardiovasc. Imaging 2011, 4, 648–661. [Google Scholar] [CrossRef]
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship Between Infarct Size and Outcomes Following Primary PCI: Patient-Level Analysis from 10 Randomized Trials. J. Am. Coll. Cardiol. 2016, 67, 1674–1683. [Google Scholar] [CrossRef]
- Curran, J.; Burkhoff, D.; Kloner, R.A. Beyond Reperfusion: Acute Ventricular Unloading and Cardioprotection During Myocardial Infarction. J. Cardiovasc. Transl. Res. 2019, 12, 95–106. [Google Scholar] [CrossRef]
- Uriel, N.; Sayer, G.; Annamalai, S.; Kapur, N.K.; Burkhoff, D. Mechanical Unloading in Heart Failure. J. Am. Coll. Cardiol. 2018, 72, 569–580. [Google Scholar] [CrossRef]
- Meyns, B.; Stolinski, J.; Leunens, V.; Verbeken, E.; Flameng, W. Left ventricular support by Catheter-Mountedaxial flow pump reduces infarct size. J. Am. Coll. Cardiol. 2003, 41, 1087–1095. [Google Scholar] [CrossRef]
- Achour, H.; Boccalandro, F.; Felli, P.; Amirian, J.; Uthman, M.; Buja, M.; Smalling, R.W. Mechanical left ventricular unloading prior to reperfusion reduces infarct size in a canine infarction model. Catheter. Cardiovasc. Interv. 2005, 64, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Tamareille, S.; Achour, H.; Amirian, J.; Felli, P.; Bick, R.J.; Poindexter, B.; Geng, Y.J.; Barry, W.H.; Smalling, R.W. Left ventricular unloading before reperfusion reduces endothelin-1 release and calcium overload in porcine myocardial infarction. J. Thorac. Cardiovasc. Surg. 2008, 136, 343–351. [Google Scholar] [CrossRef]
- Kapur Navin, K.; Paruchuri, V.; Urbano-Morales Jose, A.; Mackey Emily, E.; Daly Gerard, H.; Qiao, X.; Pandian, N.; Perides, G.; Karas Richard, H. Mechanically Unloading the Left Ventricle Before Coronary Reperfusion Reduces Left Ventricular Wall Stress and Myocardial Infarct Size. Circulation 2013, 128, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.L.; Zhang, Y.; Qiao, X.; Reyelt, L.; Paruchuri, V.; Schnitzler, G.R.; Morine, K.J.; Annamalai, S.K.; Bogins, C.; Natov, P.S.; et al. Left Ventricular Unloading Before Reperfusion Promotes Functional Recovery After Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 72, 501–514. [Google Scholar] [CrossRef]
- O’Neill, W.W.; Schreiber, T.; Wohns, D.H.W.; Rihal, C.; Naidu, S.S.; Civitello, A.B.; Dixon, S.R.; Massaro, J.M.; Maini, B.; Ohman, E.M. The Current Use of Impella 2.5 in Acute Myocardial Infarction Complicated by Cardiogenic Shock: Results from the USpella Registry. J. Interv. Cardiol. 2014, 27, 1–11. [Google Scholar] [CrossRef]
- Basir, M.B.; Schreiber, T.L.; Grines, C.L.; Dixon, S.R.; Moses, J.W.; Maini, B.S.; Khandelwal, A.K.; Ohman, E.M.; O’Neill, W.W.; Dixon, S.F. Effect of Early Initiation of Mechanical Circulatory Support on Survival in Cardiogenic Shock. Am. J. Cardiol. 2017, 119, 845–851. [Google Scholar] [CrossRef]
- Russo, J.J.; Aleksova, N.; Pitcher, I.; Couture, E.; Parlow, S.; Faraz, M.; Visintini, S.; Simard, T.; Di Santo, P.; Mathew, R.; et al. Left Ventricular Unloading During Extracorporeal Membrane Oxygenation in Patients with Cardiogenic Shock. J. Am. Coll. Cardiol. 2019, 73, 654–662. [Google Scholar] [CrossRef]
- Gass, A.; Palaniswamy, C.; Aronow, W.S.; Kolte, D.; Khera, S.; Ahmad, H.; Cuomo, L.J.; Timmermans, R.; Cohen, M.; Tang, G.H.; et al. Peripheral venoarterial extracorporeal membrane oxygenation in combination with intra-aortic balloon counterpulsation in patients with cardiovascular compromise. Cardiology 2014, 129, 137–143. [Google Scholar] [CrossRef]
- Cheng, A.; Swartz, M.F.; Massey, H.T. Impella to unload the left ventricle during peripheral extracorporeal membrane oxygenation. ASAIO J. 2013, 59, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.M.; Lipinski, J.; Al-Kindi, S.G.; Patel, T.; Saric, P.; Li, J.; Nadeem, F.; Ladas, T.; Alaiti, A.; Phillips, A.; et al. Simultaneous Venoarterial Extracorporeal Membrane Oxygenation and Percutaneous Left Ventricular Decompression Therapy with Impella Is Associated with Improved Outcomes in Refractory Cardiogenic Shock. ASAIO J. 2019, 65, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, F.; Ruggeri, L. LV distention on VA-ECMO, what to do? Qatar Med. J. 2017, 2017, 23. [Google Scholar] [CrossRef]
- Undar, A.; Rosenberg, G.; Myers, J.L. Major factors in the controversy of pulsatile versus nonpulsatile flow during acute and chronic cardiac support. ASAIO J. 2005, 51, 173–175. [Google Scholar] [PubMed]
- Guan, Y.; Karkhanis, T.; Wang, S.; Rider, A.; Koenig, S.C.; Slaughter, M.S.; El Banayosy, A.; Undar, A. Physiologic benefits of pulsatile perfusion during mechanical circulatory support for the treatment of acute and chronic heart failure in adults. Artif. Organs 2010, 34, 529–536. [Google Scholar] [CrossRef]
- Kato Tomoko, S.; Chokshi, A.; Singh, P.; Khawaja, T.; Cheema, F.; Akashi, H.; Shahzad, K.; Iwata, S.; Homma, S.; Takayama, H.; et al. Effects of Continuous-Flow Versus Pulsatile-Flow Left Ventricular Assist Devices on Myocardial Unloading and Remodeling. Circ. Heart Fail. 2011, 4, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, C.R.; Giridharan, G.A.; Litwak, K.N.; Sobieski, M.; Prabhu, S.D.; Slaughter, M.S.; Koenig, S.C. Hemodynamic Responses to Continuous versus Pulsatile Mechanical Unloading of the Failing Left Ventricle. ASAIO J. 2010, 56, 410–416. [Google Scholar] [CrossRef]
- Alvarez, J.; Rao, V. HeartMate 3-a “Step” in the right direction. J. Thorac. Dis. 2017, 9, E457–E460. [Google Scholar] [CrossRef]
- Mehra, M.R.; Naka, Y.; Uriel, N.; Goldstein, D.J.; Cleveland, J.C.; Colombo, P.C.; Walsh, M.N.; Milano, C.A.; Patel, C.B.; Jorde, U.P.; et al. A Fully Magnetically Levitated Circulatory Pump for Advanced Heart Failure. N. Engl. J. Med. 2016, 376, 440–450. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Lee, C.T.; Wang, C.H.; Tu, Y.K.; Lai, C.H.; Wang, Y.C.; Chao, A.; Huang, C.H.; Cheng, Y.J.; Chen, Y.S. Investigation of microcirculation in patients with venoarterial extracorporeal membrane oxygenation life support. Crit. Care 2018, 22, 200. [Google Scholar] [CrossRef]
- Wang, S.; Izer, J.M.; Clark, J.B.; Patel, S.; Pauliks, L.; Kunselman, A.R.; Leach, D.; Cooper, T.K.; Wilson, R.P.; Undar, A. In Vivo Hemodynamic Performance Evaluation of Novel Electrocardiogram-Synchronized Pulsatile and Nonpulsatile Extracorporeal Life Support Systems in an Adult Swine Model. Artif. Organs 2015, 39, E90–E101. [Google Scholar] [CrossRef] [PubMed]
- Sezai, A.; Shiono, M.; Nakata, K.; Hata, M.; Iida, M.; Saito, A.; Hattori, T.; Wakui, S.; Soeda, M.; Taoka, M.; et al. Effects of pulsatile CPB on interleukin-8 and endothelin-1 levels. Artif. Organs 2005, 29, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Orime, Y.; Shiono, M.; Nakata, K.; Hata, M.; Sezai, A.; Yamada, H.; Iida, M.; Kashiwazaki, S.; Nemoto, M.; Kinoshita, J.; et al. The role of pulsatility in end-organ microcirculation after cardiogenic shock. ASAIO J. 1996, 42, M724–M729. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.J.; Hunt, B.J. Current Understanding of How Extracorporeal Membrane Oxygenators Activate Haemostasis and Other Blood Components. Front. Med. 2018, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Camporese, G.; Bernardi, E.; Noventa, F. Update on the clinical use of the low-molecular-weight heparin, parnaparin. Vasc. Health Risk Manag. 2009, 5, 819–831. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Friedrichs, G.S.; Kilgore, K.S.; Manley, P.J.; Gralinski, M.R.; Lucchesi, B.R. Effects of heparin and N-acetyl heparin on ischemia/reperfusion-induced alterations in myocardial function in the rabbit isolated heart. Circ. Res. 1994, 75, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Park, J.L.; Kilgore, K.S.; Naylor, K.B.; Booth, E.A.; Murphy, K.L.; Lucchesi, B.R. N-Acetylheparin pretreatment reduces infarct size in the rabbit. Pharmacology 1999, 58, 120–131. [Google Scholar] [CrossRef]
- Black, S.C.; Gralinski, M.R.; Friedrichs, G.S.; Kilgore, K.S.; Driscoll, E.M.; Lucchesi, B.R. Cardioprotective effects of heparin or N-acetylheparin in an in vivo model of myocardial ischaemic and reperfusion injury. Cardiovasc. Res. 1995, 29, 629–636. [Google Scholar] [CrossRef]
- Gralinski, M.R.; Driscoll, E.M.; Friedrichs, G.S.; DeNardis, M.R.; Lucchesi, B.R. Reduction of Myocardial Necrosis After Glycosaminoglycan Administration: Effects of a Single Intravenous Administration of Heparin or N-Acetylheparin 2 Hours Before Regional Ischemia and Reperfusion. J. Cardiovasc. Pharmacol. Ther. 1996, 1, 219–228. [Google Scholar]
- Ziarek, J.J.; Veldkamp, C.T.; Zhang, F.; Murray, N.J.; Kartz, G.A.; Liang, X.; Su, J.; Baker, J.E.; Linhardt, R.J.; Volkman, B.F. Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 2013, 288, 737–746. [Google Scholar] [CrossRef]
- Krueger, K.; Schmutz, A.; Zieger, B.; Kalbhenn, J. Venovenous Extracorporeal Membrane Oxygenation with Prophylactic Subcutaneous Anticoagulation Only: An Observational Study in More Than 60 Patients. Artif. Organs 2017, 41, 186–192. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS ONE 2016, 11, e0160889. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; McCully, J.D. Mitochondrial transplantation: Applications for pediatric patients with congenital heart disease. Transl. Pediatr. 2018, 7, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef]
- Moskowitzova, K.; Shin, B.; Liu, K.; Ramirez-Barbieri, G.; Guariento, A.; Blitzer, D.; Thedsanamoorthy, J.K.; Yao, R.; Snay, E.R.; Inkster, J.A.H.; et al. Mitochondrial transplantation prolongs cold ischemia time in murine heart transplantation. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2019, 38, 92–99. [Google Scholar] [CrossRef]
- Pacak, C.A.; Preble, J.M.; Kondo, H.; Seibel, P.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B.; McCully, J.D. Actin-dependent mitochondrial internalization in cardiomyocytes: Evidence for rescue of mitochondrial function. Biol. Open 2015, 4, 622–626. [Google Scholar] [CrossRef]
- Preble, J.M.; Pacak, C.A.; Kondo, H.; MacKay, A.A.; Cowan, D.B.; McCully, J.D. Rapid isolation and purification of mitochondria for transplantation by tissue dissociation and differential filtration. J. Vis. Exp. 2014, 91, e51682. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Barbieri, G.; Moskowitzova, K.; Shin, B.; Blitzer, D.; Orfany, A.; Guariento, A.; Iken, K.; Friehs, I.; Zurakowski, D.; del Nido, P.J.; et al. Alloreactivity and allorecognition of syngeneic and allogeneic mitochondria. Mitochondrion 2019, 46, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Cowan, D.B.; Emani, S.M.; del Nido, P.J.; McCully, J.D. Mitochondrial Transplantation in Myocardial Ischemia and Reperfusion Injury. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 595–619. [Google Scholar]
- Bertero, E.; Maack, C.; O’Rourke, B. Mitochondrial transplantation in humans: “magical” cure or cause for concern? J. Clin. Investig. 2018, 128, 5191–5194. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Davies, L.C. Mesenchymal stromal cells and the innate immune response. Immunol. Lett. 2015, 168, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143. [Google Scholar] [CrossRef]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J. Biomed. Sci. 2018, 25, 31. [Google Scholar] [CrossRef] [PubMed]
- Singer, N.G.; Caplan, A.I. Mesenchymal Stem Cells: Mechanisms of Inflammation. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 457–478. [Google Scholar] [CrossRef]
- Liu, H.; Li, D.; Zhang, Y.; Li, M. Inflammation, mesenchymal stem cells and bone regeneration. Histochem. Cell Biol. 2018, 149, 393–404. [Google Scholar] [CrossRef]
- Millar, J.E.; von Bahr, V.; Malfertheiner, M.V.; Ki, K.K.; Redd, M.A.; Bartnikowski, N.; Suen, J.Y.; McAuley, D.F.; Fraser, J.F. Administration of mesenchymal stem cells during ECMO results in a rapid decline in oxygenator performance. Thorax 2019, 74, 194. [Google Scholar] [CrossRef]
- Von Bahr, V.; Millar, J.E.; Malfertheiner, M.V.; Ki, K.K.; Passmore, M.R.; Bartnikowski, N.; Redd, M.A.; Cavaye, M.; Suen, J.Y.; McAuley, D.F.; et al. Mesenchymal stem cells may ameliorate inflammation in an ex vivo model of extracorporeal membrane oxygenation. Perfusion 2019, 34, 15–21. [Google Scholar] [CrossRef]
- Dandel, M.; Knosalla, C.; Hetzer, R. Contribution of ventricular assist devices to the recovery of failing hearts: A review and the Berlin Heart Center Experience. Eur. J. Fail. 2014, 16, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sampaio, L.C.; Li, K.; Silva, G.V.; Cabreira-Hansen, M.; Vela, D.; Segura, A.M.; Bovè, C.; Perin, E.C. Safety and feasibility of mapping and stem cell delivery in the presence of an implanted left ventricular assist device: A preclinical investigation in sheep. Texas Heart Inst. J. 2013, 40, 229–234. [Google Scholar]
- Ascheim, D.D.; Gelijns, C.A.; Goldstein, A.D.; Moye, T.L.; Smedira, K.N.; Lee, O.S.; Klodell, A.C.; Szady, E.A.; Parides, G.M.; Jeffries, T.N.; et al. Mesenchymal Precursor Cells as Adjunctive Therapy in Recipients of Contemporary Left Ventricular Assist Devices. Circulation 2014, 129, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.M.; Pagani, F.D.; Mancini, D.M.; Chang, H.L.; Lala, A.; Woo, Y.J.; Acker, M.A.; Selzman, C.H.; Soltesz, E.G.; Kern, J.A.; et al. Intramyocardial Injection of Mesenchymal Precursor Cells and Successful Temporary Weaning from Left Ventricular Assist Device Support in Patients with Advanced Heart Failure: A Randomized Clinical TrialIntramyocardial Injection of Mesenchymal Precursor Cells and Weaning from Left Ventricular Assist Device (LVAD) SupportIntramyocardial Injection of Mesenchymal Precursor Cells and Weaning from Left Ventricular Assist Device (LVAD) Support. JAMA 2019, 321, 1176–1186. [Google Scholar] [PubMed]
- ANZOD. ANZOD Registry, 2017 Annual Report, Section 7: Deceased Donor Heart Donation; Australia and New Zealand Dialysis and Transplant Registry: Adelaide, Australia, 2017. [Google Scholar]
- ANZOD. ANZOD Registry, 2017 Annual Report, Section 12: Organ Waiting Lists; Australia and New Zealand Dialysis and Transplant Registry: Adelaide, Australia, 2017. [Google Scholar]
- The Transplantation Society of Australia and New Zealand (TSANZ). Clinical Guidelines for Organ Transplantation from Deceased Donors; Version 1.3; The Transplantation Society of Australia and New Zealand (TSANZ): Sydney, Australia, 2019. [Google Scholar]
- Watts, R.P.; Bilska, I.; Diab, S.; Dunster, K.R.; Bulmer, A.C.; Barnett, A.G.; Fraser, J.F. Novel 24-h ovine model of brain death to study the profile of the endothelin axis during cardiopulmonary injury. Intensive Care Med. Exp. 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.P.; Thom, O.; Fraser, J.F. Inflammatory signalling associated with brain dead organ donation: From brain injury to brain stem death and posttransplant ischaemia reperfusion injury. J. Transplant. 2013, 2013, 521369. [Google Scholar] [CrossRef]
- Kobashigawa, J.; Zuckermann, A.; Macdonald, P.; Leprince, P.; Esmailian, F.; Luu, M.; Mancini, D.; Patel, J.; Razi, R.; Reichenspurner, H.; et al. Report from a consensus conference on primary graft dysfunction after cardiac transplantation. J. Heart Lung Transplant. 2014, 33, 327–340. [Google Scholar] [CrossRef]
- Wilhelm, M.J.; Pratschke, J.; Laskowski, I.A.; Paz, D.M.; Tilney, N.L. Brain death and its impact on the donor heart-lessons from animal models. J. Heart Lung Transplant. 2000, 19, 414–418. [Google Scholar] [CrossRef]
- See Hoe, L.E.; McGiffin, D.; Fraser, J.F. Untapped potential in Australian Hospitals for organ donation after circulatory death. Med. J. Aust. 2018, 208, 276. [Google Scholar] [CrossRef]
- Khush, K.K.; Cherikh, W.S.; Chambers, D.C.; Goldfarb, S.; Hayes, D., Jr.; Kucheryavaya, A.Y.; Levvey, B.J.; Meiser, B.; Rossano, J.W.; Stehlik, J.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-fifth Adult Heart Transplantation Report-2018; Focus Theme: Multiorgan Transplantation. J. Heart Lung Transplant. 2018, 37, 1155–1168. [Google Scholar] [CrossRef]
- Furiasse, N.; Kobashigawa, J.A. Immunosuppression and adult heart transplantation: Emerging therapies and opportunities. Expert Rev. Cardiovasc. Ther. 2017, 15, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.; von Scheidt, W. Cardiac allograft vasculopathy: A review. Circulation 1997, 96, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Lindenfeld, J.; Miller, G.G.; Shakar, S.F.; Zolty, R.; Lowes, B.D.; Wolfel, E.E.; Mestroni, L.; Page, R.L.; Kobashigawa, J. Drug therapy in the heart transplant recipient: Part II: Immunosuppressive drugs. Circulation 2004, 110, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Pihusch, R.; Holler, E.; Muhlbayer, D.; Gohring, P.; Stotzer, O.; Pihusch, M.; Hiller, E.; Kolb, H.J. The impact of antithymocyte globulin on short-term toxicity after allogeneic stem cell transplantation. Bone Marrow Transpl. 2002, 30, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Wing, M.G.; Moreau, T.; Greenwood, J.; Smith, R.M.; Hale, G.; Isaacs, J.; Waldmann, H.; Lachmann, P.J.; Compston, A. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: Involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J. Clin. Investig. 1996, 98, 2819–2826. [Google Scholar] [CrossRef]
- Winkler, U.; Jensen, M.; Manzke, O.; Schulz, H.; Diehl, V.; Engert, A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 1999, 94, 2217–2224. [Google Scholar]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Keogh, A. Calcineurin inhibitors in heart transplantation. J. Heart Lung Transplant. 2004, 23, S202–S206. [Google Scholar] [CrossRef]
- Jarzembowski, T.M.; John, E.; Panaro, F.; Manzelli, A.; Cabrera, A.; Greco, A.; Varga, P.; Sankary, H.; Testa, G.; Benedetti, E. Reversal of tacrolimus-related hypertrophic obstructive cardiomyopathy 5 years after kidney transplant in a 6-year-old recipient. Pediatr. Transplant. 2005, 9, 117–121. [Google Scholar] [CrossRef]
- Pappas, P.A.; Weppler, D.; Pinna, A.D.; Rusconi, P.; Thompson, J.F.; Jaffe, J.S.; Tzakis, A.G. Sirolimus in pediatric gastrointestinal transplantation: The use of sirolimus for pediatric transplant patients with tacrolimus-related cardiomyopathy. Pediatr. Transplant. 2000, 4, 45–49. [Google Scholar] [CrossRef]
- Turska-Kmiec, A.; Jankowska, I.; Pawlowska, J.; Kalicinski, P.; Kawalec, W.; Tomyn, M.; Markiewicz, M.; Teisseyre, J.; Czubkowski, P.; Rekawek, J.; et al. Reversal of tacrolimus-related hypertrophic cardiomyopathy after conversion to rapamycin in a pediatric liver transplant recipient. Pediatr. Transplant. 2007, 11, 319–323. [Google Scholar] [CrossRef]
- Cox, T.H.; Baillie, G.M.; Baliga, P. Bradycardia associated with intravenous administration of tacrolimus in a liver transplant recipient. Pharmacotherapy 1997, 17, 1328–1330. [Google Scholar]
- Hodak, S.P.; Moubarak, J.B.; Rodriguez, I.; Gelfand, M.C.; Alijani, M.R.; Tracy, C.M. QT prolongation and near fatal cardiac arrhythmia after intravenous tacrolimus administration: A case report. Transplantation 1998, 66, 535–537. [Google Scholar] [CrossRef]
- McLeod, J.; Wu, S.; Grazette, L.; Sarcon, A. Tacrolimus-Associated Dilated Cardiomyopathy in Adult Patient After Orthotopic Liver Transplant. J. Investig. Med. High Impact Case Rep. 2017, 5, 2324709617706087. [Google Scholar] [CrossRef]
- Naesens, M.; Kuypers, D.R.; Sarwal, M. Calcineurin inhibitor nephrotoxicity. Clin. J. Am. Soc. Nephrol. 2009, 4, 481–508. [Google Scholar] [CrossRef]
- Lyson, T.; Ermel, L.D.; Belshaw, P.J.; Alberg, D.G.; Schreiber, S.L.; Victor, R.G. Cyclosporine- and FK506-induced sympathetic activation correlates with calcineurin-mediated inhibition of T-cell signaling. Circ. Res. 1993, 73, 596–602. [Google Scholar] [CrossRef]
- Pham, S.M.; Kormos, R.L.; Hattler, B.G.; Kawai, A.; Tsamandas, A.C.; Demetris, A.J.; Murali, S.; Fricker, F.J.; Chang, H.C.; Jain, A.B.; et al. A prospective trial of tacrolimus (FK 506) in clinical heart transplantation: Intermediate-term results. J. Thorac. Cardiovasc. Surg. 1996, 111, 764–772. [Google Scholar] [CrossRef][Green Version]
- Meiser, B.M.; Billingham, M.E.; Morris, R.E. Effects of cyclosporin, FK506, and rapamycin on graft-vessel disease. Lancet 1991, 338, 1297–1298. [Google Scholar] [CrossRef]
- Arai, S.; Teramoto, S.; Senoo, Y. The impact of FK506 on graft coronary disease of rat cardiac allograft—A comparison with cyclosporine. J. Heart Lung Transplant. 1992, 11, 757–762. [Google Scholar]
- Chi, J.; Zhu, Y.; Fu, Y.; Liu, Y.; Zhang, X.; Han, L.; Yin, X.; Zhao, D. Cyclosporin A induces apoptosis in H9c2 cardiomyoblast cells through calcium-sensing receptor-mediated activation of the ERK MAPK and p38 MAPK pathways. Mol. Cell. Biochem. 2012, 367, 227–236. [Google Scholar] [CrossRef]
- Buetler, T.M.; Cottet-Maire, F.; Krauskopf, A.; Ruegg, U.T. Does cyclosporin A generate free radicals? Trends Pharmacol. Sci. 2000, 21, 288–290. [Google Scholar] [CrossRef]
- Selcoki, Y.; Uz, E.; Bayrak, R.; Sahin, S.; Kaya, A.; Uz, B.; Karanfil, A.; Ozkara, A.; Akcay, A. The protective effect of erdosteine against cyclosporine A-induced cardiotoxicity in rats. Toxicology 2007, 239, 53–59. [Google Scholar] [CrossRef]
- Tang, J.; Wang, G.; Liu, Y.; Fu, Y.; Chi, J.; Zhu, Y.; Zhao, Y.; Yin, X. Cyclosporin A induces cardiomyocyte injury through calcium-sensing receptor-mediated calcium overload. Pharmazie 2011, 66, 52–57. [Google Scholar]
- Serkova, N.J.; Christians, U.; Benet, L.Z. Biochemical mechanisms of cyclosporine neurotoxicity. Mol. Interv. 2004, 4, 97–107. [Google Scholar] [CrossRef]
- Omar, A.G.; El-Mas, M.M. Time-domain evaluation of cyclosporine interaction with hemodynamic variability in rats. Cardiovasc. Drugs Ther. 2004, 18, 461–468. [Google Scholar] [CrossRef]
- Moien-Afshari, F.; McManus, B.M.; Laher, I. Immunosuppression and transplant vascular disease: Benefits and adverse effects. Pharmacol. Ther. 2003, 100, 141–156. [Google Scholar] [CrossRef]
- Reichart, B.; Meiser, B.; Vigano, M.; Rinaldi, M.; Martinelli, L.; Yacoub, M.; Banner, N.R.; Gandjbakhch, I.; Dorent, R.; Hetzer, R.; et al. European Multicenter Tacrolimus (FK506) Heart Pilot Study: One-year results—European Tacrolimus Multicenter Heart Study Group. J. Heart Lung Transplant. 1998, 17, 775–781. [Google Scholar]
- Meiser, B.M.; Uberfuhr, P.; Fuchs, A.; Schmidt, D.; Pfeiffer, M.; Paulus, D.; Schulze, C.; Wildhirt, S.; Scheidt, W.V.; Angermann, C.; et al. Single-center randomized trial comparing tacrolimus (FK506) and cyclosporine in the prevention of acute myocardial rejection. J. Heart Lung Transplant. 1998, 17, 782–788. [Google Scholar]
- Teebken, O.E.; Struber, M.; Harringer, W.; Pichlmaier, M.A.; Haverich, A. Primary immunosuppression with tacrolimus and mycophenolate mofetil versus cyclosporine and azathioprine in heart transplant recipients. Transplant. Proc. 2002, 34, 1265–1268. [Google Scholar] [CrossRef]
- Rinaldi, M.; Pellegrini, C.; Martinelli, L.; Goggi, C.; Gavazzi, A.; Campana, C.; Arbustini, E.; Grossi, P.; Regazzi, M.; Ippoliti, G.; et al. FK506 effectiveness in reducing acute rejection after heart transplantation: A prospective randomized study. J. Heart Lung Transplant. 1997, 16, 1001–1010. [Google Scholar]
- Czerny, M.; Sahin, V.; Fasching, P.; Zuckermann, A.; Zimpfer, D.; Kilo, J.; Wolner, E.; Grimm, M. The impact of diabetes mellitus at the time of heart transplantation on long-term survival. Diabetologia 2002, 45, 1498–1508. [Google Scholar]
- Taylor, D.O.; Barr, M.L.; Radovancevic, B.; Renlund, D.G.; Mentzer, R.M., Jr.; Smart, F.W.; Tolman, D.E.; Frazier, O.H.; Young, J.B.; VanVeldhuisen, P. A randomized, multicenter comparison of tacrolimus and cyclosporine immunosuppressive regimens in cardiac transplantation: Decreased hyperlipidemia and hypertension with tacrolimus. J. Heart Lung Transplant. 1999, 18, 336–345. [Google Scholar] [CrossRef]
- Kristeller, J.L.; Jankowski, A.; Reinaker, T. Role of corticosteroids during cardiopulmonary bypass. Hosp. Pharm. 2014, 49, 232–236. [Google Scholar] [CrossRef]
- Williams, C.M.; Coleman, J.W. Induced expression of mRNA for IL-5, IL-6, TNF-alpha, MIP-2 and IFN-gamma in immunologically activated rat peritoneal mast cells: Inhibition by dexamethasone and cyclosporin A. Immunology 1995, 86, 244–249. [Google Scholar]
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef]
- Silverman, H.S.; Pfeifer, M.P. Relation between use of anti-inflammatory agents and left ventricular free wall rupture during acute myocardial infarction. Am. J. Cardiol. 1987, 59, 363–364. [Google Scholar] [CrossRef]
- Hammerman, H.; Schoen, F.J.; Braunwald, E.; Kloner, R.A. Drug-induced expansion of infarct: Morphologic and functional correlations. Circulation 1984, 69, 611–617. [Google Scholar] [CrossRef]
- Mannisi, J.A.; Weisman, H.F.; Bush, D.E.; Dudeck, P.; Healy, B. Steroid administration after myocardial infarction promotes early infarct expansion. A study in the rat. J. Clin. Investig. 1987, 79, 1431–1439. [Google Scholar] [CrossRef]
- Garcia, R.A.; Go, K.V.; Villarreal, F.J. Effects of timed administration of doxycycline or methylprednisolone on post-myocardial infarction inflammation and left ventricular remodeling in the rat heart. Mol. Cell. Biochem. 2007, 300, 159–169. [Google Scholar] [CrossRef]
- Costanzo, M.R.; Dipchand, A.; Starling, R.; Anderson, A.; Chan, M.; Desai, S.; Fedson, S.; Fisher, P.; Gonzales-Stawinski, G.; Martinelli, L.; et al. The International Society of Heart and Lung Transplantation Guidelines for the care of heart transplant recipients. J. Heart Lung Transplant. 2010, 29, 914–956. [Google Scholar] [CrossRef] [PubMed]
- Teuteberg, J.J.; Shullo, M.; Zomak, R.; McNamara, D.; McCurry, K.; Kormos, R.L. Aggressive steroid weaning after cardiac transplantation is possible without the additional risk of significant rejection. Clin. Transplant. 2008, 22, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.O.; Bristow, M.R.; O’Connell, J.B.; Price, G.D.; Hammond, E.H.; Doty, D.B.; Karwande, S.V.; Gay, W.A., Jr.; Jones, K.W.; Lappe, D.; et al. Improved long-term survival after heart transplantation predicted by successful early withdrawal from maintenance corticosteroid therapy. J. Heart Lung Transplant. 1996, 15, 1039–1046. [Google Scholar] [PubMed]
- Ballantyne, C.M.; Radovancevic, B.; Farmer, J.A.; Frazier, O.H.; Chandler, L.; Payton-Ross, C.; Cocanougher, B.; Jones, P.H.; Young, J.B.; Gotto, A.M., Jr. Hyperlipidemia after heart transplantation: Report of a 6-year experience, with treatment recommendations. J. Am. Coll. Cardiol. 1992, 19, 1315–1321. [Google Scholar] [CrossRef]
- Bamgbola, O. Metabolic consequences of modern immunosuppressive agents in solid organ transplantation. Ther. Adv. Endocrinol. Metab. 2016, 7, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Nussinovitch, U.; de Carvalho, J.F.; Pereira, R.M.; Shoenfeld, Y. Glucocorticoids and the cardiovascular system: State of the art. Curr. Pharm. Des. 2010, 16, 3574–3585. [Google Scholar] [CrossRef] [PubMed]
- Sartori, T.M.; Maurizio, P.G.; Sara, P.; Ugolino, L.; Annalisa, A.; Panagiotis, T.; Massimo, F.; Antonio, G. Relation between long-term steroid treatment after heart transplantation, hypofibrinolysis and myocardial microthrombi generation. J. Heart Lung Transplant. 1999, 18, 693–700. [Google Scholar] [CrossRef]
- Sack, M.N.; Murphy, E. The role of comorbidities in cardioprotection. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 267–272. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Habertheuer, A.; Kocher, A.; Laufer, G.; Andreas, M.; Szeto, W.Y.; Petzelbauer, P.; Ehrlich, M.; Wiedemann, D. Cardioprotection: A review of current practice in global ischemia and future translational perspective. BioMed Res. Int. 2014, 2014, 325725. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yamamoto, F. Myocardial protection in cardiac surgery: A historical review from the beginning to the current topics. Gen. Thorac. Cardiovasc. Surg. 2013, 61, 485–496. [Google Scholar] [CrossRef]
- Steen, S.; Paskevicius, A.; Liao, Q.; Sjoberg, T. Safe orthotopic transplantation of hearts harvested 24 hours after brain death and preserved for 24 hours. Scand. Cardiovasc. J. 2016, 50, 193–200. [Google Scholar] [CrossRef]
- Nilsson, J.; Jernryd, V.; Qin, G.; Paskevicius, A.; Sjoberg, T.; Hoglund, P.; Steen, S. Non Ischemic Heart Preservation—Results from the Safety Study. J. Heart Lung Transplant. 2019, 38, S26. [Google Scholar] [CrossRef]
- Michel, S.G.; LaMuraglia Ii, G.M.; Madariaga, M.L.; Anderson, L.M. Innovative cold storage of donor organs using the Paragonix Sherpa Pak devices. Heart Lung Vessel 2015, 7, 246–255. [Google Scholar]
- Naito, N.; Funamoto, M.; Pierson, R.; Villavicencio, M.; Riley, W.; Lewis, G.; D’Alessandro, D. The First Clinical Use of a Novel Cold Storage System of Donor Hearts. J. Heart Lung Transplant. 2019, 38, S44. [Google Scholar] [CrossRef]
- Ardehali, A.; Esmailian, F.; Deng, M.; Soltesz, E.; Hsich, E.; Naka, Y.; Mancini, D.; Camacho, M.; Zucker, M.; Leprince, P.; et al. Ex-vivo perfusion of donor hearts for human heart transplantation (PROCEED II): A prospective, open-label, multicentre, randomised non-inferiority trial. Lancet 2015, 385, 2577–2584. [Google Scholar] [CrossRef]
- Wood, K.J.; Bushell, A.; Hester, J. Regulatory immune cells in transplantation. Nat. Rev. Immunol. 2012, 12, 417–430. [Google Scholar] [CrossRef]
- Hester, J.; Schiopu, A.; Nadig, S.N.; Wood, K.J. Low-dose rapamycin treatment increases the ability of human regulatory T cells to inhibit transplant arteriosclerosis in vivo. Am. J. Transplant. 2012, 12, 2008–2016. [Google Scholar] [CrossRef]
- Kingsley, C.I.; Karim, M.; Bushell, A.R.; Wood, K.J. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J. Immunol. 2002, 168, 1080–1086. [Google Scholar] [CrossRef]
- Xia, N.; Jiao, J.; Tang, T.T.; Lv, B.J.; Lu, Y.Z.; Wang, K.J.; Zhu, Z.F.; Mao, X.B.; Nie, S.F.; Wang, Q.; et al. Activated regulatory T-cells attenuate myocardial ischaemia/reperfusion injury through a CD39-dependent mechanism. Clin. Sci. 2015, 128, 679–693. [Google Scholar] [CrossRef]
- Hoerbelt, R.; Benjamin, C.L.; Shoji, T.; Houser, S.L.; Muniappan, A.; Hasse, R.S.; Ledgerwood, L.G.; Allan, J.S.; Sachs, D.H.; Madsen, J.C. The effects of tolerance on allograft damage caused by the innate immune system. Transplantation 2008, 85, 314–322. [Google Scholar] [CrossRef]
- Lila, N.; Amrein, C.; Guillemain, R.; Chevalier, P.; Latremouille, C.; Fabiani, J.N.; Dausset, J.; Carosella, E.D.; Carpentier, A. Human leukocyte antigen-G expression after heart transplantation is associated with a reduced incidence of rejection. Circulation 2002, 105, 1949–1954. [Google Scholar] [CrossRef]
- Lila, N.; Amrein, C.; Guillemain, R.; Chevalier, P.; Fabiani, J.N.; Carpentier, A. Soluble human leukocyte antigen-G: A new strategy for monitoring acute and chronic rejections after heart transplantation. J. Heart Lung Transplant. 2007, 26, 421–422. [Google Scholar] [CrossRef]
- Sheshgiri, R.; Gustafsson, F.; Sheedy, J.; Rao, V.; Ross, H.J.; Delgado, D.H. Everolimus but not mycophenolate mofetil therapy is associated with soluble HLA-G expression in heart transplant patients. J. Heart Lung Transplant. 2009, 28, 1193–1197. [Google Scholar] [CrossRef]
- Ferjani, H.; Timoumi, R.; Amara, I.; Abid, S.; Achour, A.; Bacha, H.; Boussema-Ayed, I. Beneficial effects of mycophenolate mofetil on cardiotoxicity induced by tacrolimus in wistar rats. Exp. Biol. Med. (Maywood) 2017, 242, 448–455. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Imam, F.; Nadeem, A.; Al-Harbi, M.M.; Iqbal, M.; Rahman, S.; Al-Hosaini, K.A.; Bahashwan, S. Protection against tacrolimus-induced cardiotoxicity in rats by olmesartan and aliskiren. Toxicol. Mech. Methods 2014, 24, 697–702. [Google Scholar] [CrossRef]
- Behbod, F.; Erwin-Cohen, R.A.; Wang, M.E.; Trawick, B.W.; Qu, X.; Verani, R.; Kahan, B.D.; Stepkowski, S.M.; Kirken, R.A. Concomitant inhibition of Janus kinase 3 and calcineurin-dependent signaling pathways synergistically prolongs the survival of rat heart allografts. J. Immunol. 2001, 166, 3724–3732. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Mewton, N.; Croisille, P.; Gahide, G.; Rioufol, G.; Bonnefoy, E.; Sanchez, I.; Cung, T.T.; Sportouch, C.; Angoulvant, D.; Finet, G.; et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J. Am. Coll. Cardiol. 2010, 55, 1200–1205. [Google Scholar] [CrossRef]
- Ikeda, G.; Matoba, T.; Nakano, Y.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Funamoto, D.; Sunagawa, K.; Egashira, K. Nanoparticle-Mediated Targeting of Cyclosporine a Enhances Cardioprotection Against Ischemia-Reperfusion Injury Through Inhibition of Mitochondrial Permeability Transition Pore Opening. Sci. Rep. 2016, 6, 20467. [Google Scholar] [CrossRef]
- Xue, Q.; Patterson, A.J.; Xiao, D.; Zhang, L. Glucocorticoid modulates angiotensin II receptor expression patterns and protects the heart from ischemia and reperfusion injury. PLoS ONE 2014, 9, e106827. [Google Scholar] [CrossRef]
- Libby, P.; Maroko, P.R.; Bloor, C.M.; Sobel, B.E.; Braunwald, E. Reduction of experimental myocardial infarct size by corticosteroid administration. J. Clin. Investig. 1973, 52, 599–607. [Google Scholar] [CrossRef]
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.C.; Rebsamen, M.C.; Hsieh, C.M.; Chui, D.S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479. [Google Scholar] [CrossRef]
- Tuuminen, R.; Syrjala, S.; Krebs, R.; Arnaudova, R.; Rouvinen, E.; Nykanen, A.I.; Lemstrom, K.B. Combined donor simvastatin and methylprednisolone treatment prevents ischemia-reperfusion injury in rat cardiac allografts through vasculoprotection and immunomodulation. Transplantation 2013, 95, 1084–1091. [Google Scholar] [CrossRef]
- Enc, Y.; Karaca, P.; Ayoglu, U.; Camur, G.; Kurc, E.; Cicek, S. The acute cardioprotective effect of glucocorticoid in myocardial ischemia-reperfusion injury occurring during cardiopulmonary bypass. Heart Vessel. 2006, 21, 152–156. [Google Scholar] [CrossRef]
- Schmidbauer, G.; Homeyer, A.; Bohle, R.M.; Grimm, H.; Binder, J.; Kupiec-Weglinski, J.W. Donor pretreatment with methylprednisolone synergistically prolongs survival of cardiac allografts in sensitized rat recipients conditioned with rapamycin. Transplant. Proc. 1997, 29, 607–608. [Google Scholar] [CrossRef]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Thiemermann, C. Corticosteroids and cardioprotection. Nat. Med. 2002, 8, 453–455. [Google Scholar] [CrossRef]
- Tokudome, S.; Sano, M.; Shinmura, K.; Matsuhashi, T.; Morizane, S.; Moriyama, H.; Tamaki, K.; Hayashida, K.; Nakanishi, H.; Yoshikawa, N.; et al. Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin-type prostaglandin D synthase-derived PGD2 biosynthesis. J. Clin. Investig. 2009, 119, 1477–1488. [Google Scholar] [CrossRef]
- Eguchi, Y.; Eguchi, N.; Oda, H.; Seiki, K.; Kijima, Y.; Matsu-ura, Y.; Urade, Y.; Hayaishi, O. Expression of lipocalin-type prostaglandin D synthase (beta-trace) in human heart and its accumulation in the coronary circulation of angina patients. Proc. Natl. Acad. Sci. USA 1997, 94, 14689–14694. [Google Scholar] [CrossRef]
- Katsumata, Y.; Shinmura, K.; Sugiura, Y.; Tohyama, S.; Matsuhashi, T.; Ito, H.; Yan, X.; Ito, K.; Yuasa, S.; Ieda, M.; et al. Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia-reperfusion injury by activating Nrf2. Hypertension 2014, 63, 80–87. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Belhaj, A.; Dewachter, L.; Rorive, S.; Remmelink, M.; Weynand, B.; Melot, C.; Galanti, L.; Hupkens, E.; Sprockeels, T.; Dewachter, C.; et al. Roles of inflammation and apoptosis in experimental brain death-induced right ventricular failure. J. Heart Lung Transplant. 2016, 35, 1505–1518. [Google Scholar] [CrossRef]
- McLean, K.M.; Duffy, J.Y.; Pandalai, P.K.; Lyons, J.M.; Bulcao, C.F.; Wagner, C.J.; Akhter, S.A.; Pearl, J.M. Glucocorticoids alter the balance between pro- and anti-inflammatory mediators in the myocardium in a porcine model of brain death. J. Heart Lung Transplant. 2007, 26, 78–84. [Google Scholar] [CrossRef]
- Wan, S.; DeSmet, J.M.; Antoine, M.; Goldman, M.; Vincent, J.L.; LeClerc, J.L. Steroid administration in heart and heart-lung transplantation: Is the timing adequate? Ann. Thorac. Surg. 1996, 61, 674–678. [Google Scholar] [CrossRef]
- Sandha, J.K.; White, C.W.; Muller, A.; Avery, E.; Thliveris, J.; Dixon, I.M.C.; Arora, R.C.; Tian, G.; Hryshko, L.V.; Nagendran, J.; et al. Steroids Limit Myocardial Edema During Ex Vivo Perfusion of Hearts Donated After Circulatory Death. Ann. Thorac. Surg. 2018, 105, 1763–1770. [Google Scholar] [CrossRef]
- Pesonen, E.; Keski-Nisula, J.; Passov, A.; Vahatalo, R.; Puntila, J.; Andersson, S.; Suominen, P.K. Heart-Type Fatty Acid Binding Protein and High-Dose Methylprednisolone in Pediatric Cardiac Surgery. J. Cardiothorac. Vasc. Anesth. 2017, 31, 1952–1956. [Google Scholar] [CrossRef]
- Dhar, R.; Cotton, C.; Coleman, J.; Brockmeier, D.; Kappel, D.; Marklin, G.; Wright, R. Comparison of high- and low-dose corticosteroid regimens for organ donor management. J. Crit. Care 2013, 28, 111-e1. [Google Scholar] [CrossRef]
- Segel, L.D.; Follette, D.M.; Castellanos, L.M.; Hayes, R.; Baker, J.M.; Smolens, I.V. Steroid pretreatment improves graft recovery in a sheep orthotopic heart transplantation model. J. Heart Lung Transplant. 1997, 16, 371–380. [Google Scholar]
- Huber, B.C.; Ransohoff, J.D.; Ransohoff, K.J.; Riegler, J.; Ebert, A.; Kodo, K.; Gong, Y.; Sanchez-Freire, V.; Dey, D.; Kooreman, N.G.; et al. Costimulation-adhesion blockade is superior to cyclosporine A and prednisone immunosuppressive therapy for preventing rejection of differentiated human embryonic stem cells following transplantation. Stem Cells 2013, 31, 2354–2363. [Google Scholar] [CrossRef]
- Foldes, G.; Mioulane, M.; Kodagoda, T.; Lendvai, Z.; Iqbal, A.; Ali, N.N.; Schneider, M.D.; Harding, S.E. Immunosuppressive agents modulate function, growth, and survival of cardiomyocytes and endothelial cells derived from human embryonic stem cells. Stem Cells Dev. 2014, 23, 467–476. [Google Scholar] [CrossRef]
- Crockford, D.; Turjman, N.; Allan, C.; Angel, J. Thymosin beta4: Structure, function, and biological properties supporting current and future clinical applications. Ann. N. Y. Acad. Sci. 2010, 1194, 179–189. [Google Scholar] [CrossRef]
- Hinkel, R.; El-Aouni, C.; Olson, T.; Horstkotte, J.; Mayer, S.; Muller, S.; Willhauck, M.; Spitzweg, C.; Gildehaus, F.J.; Munzing, W.; et al. Thymosin beta4 is an essential paracrine factor of embryonic endothelial progenitor cell-mediated cardioprotection. Circulation 2008, 117, 2232–2240. [Google Scholar] [CrossRef]
- Postrach, J.; Schmidt, M.; Thormann, M.; Thein, E.; Burdorf, L.; Reichart, B.; Sotlar, K.; Walz, C.; Faber, C.; Bauer, A.; et al. Adeno-associated viral vector 2.9 thymosin ss4 application attenuates rejection after heart transplantation: Results of a preclinical study in the pig. Transplantation 2014, 98, 835–843. [Google Scholar] [CrossRef]
- Schuleri, K.H.; Amado, L.C.; Boyle, A.J.; Centola, M.; Saliaris, A.P.; Gutman, M.R.; Hatzistergos, K.E.; Oskouei, B.N.; Zimmet, J.M.; Young, R.G.; et al. Early improvement in cardiac tissue perfusion due to mesenchymal stem cells. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2002–H2011. [Google Scholar] [CrossRef]
- Hatzistergos, K.E.; Quevedo, H.; Oskouei, B.N.; Hu, Q.; Feigenbaum, G.S.; Margitich, I.S.; Mazhari, R.; Boyle, A.J.; Zambrano, J.P.; Rodriguez, J.E.; et al. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ. Res. 2010, 107, 913–922. [Google Scholar] [CrossRef]
- Karantalis, V.; Hare, J.M. Use of mesenchymal stem cells for therapy of cardiac disease. Circ. Res. 2015, 116, 1413–1430. [Google Scholar] [CrossRef]
- Sanina, C.; Hare, J.M. Mesenchymal Stem Cells as a Biological Drug for Heart Disease: Where Are We with Cardiac Cell-Based Therapy? Circ. Res. 2015, 117, 229–233. [Google Scholar] [CrossRef]
- Popp, F.C.; Eggenhofer, E.; Renner, P.; Slowik, P.; Lang, S.A.; Kaspar, H.; Geissler, E.K.; Piso, P.; Schlitt, H.J.; Dahlke, M.H. Mesenchymal stem cells can induce long-term acceptance of solid organ allografts in synergy with low-dose mycophenolate. Transpl. Immunol. 2008, 20, 55–60. [Google Scholar] [CrossRef]
- Inoue, S.; Popp, F.C.; Koehl, G.E.; Piso, P.; Schlitt, H.J.; Geissler, E.K.; Dahlke, M.H. Immunomodulatory effects of mesenchymal stem cells in a rat organ transplant model. Transplantation 2006, 81, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Jelbart, M.E.; Dorsch, S.E. Suppressor T cells in rats with prolonged cardiac allograft survival after treatment with cyclosporine. Transplantation 1984, 37, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Pearce, N.W.; Gurley, K.E.; Dorsch, S.E. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. III. Further characterization of the CD4+ suppressor cell and its mechanisms of action. J. Exp. Med. 1990, 171, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Bushell, A.; Wood, K. GITR ligation blocks allograft protection by induced CD25+CD4+ regulatory T cells without enhancing effector T-cell function. Am. J. Transplant. 2007, 7, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Tanriver, Y.; Jiang, S.; Xue, S.A.; Ratnasothy, K.; Chen, D.; Stauss, H.J.; Bucy, R.P.; Lombardi, G.; Lechler, R. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J. Clin. Investig. 2008, 118, 3619–3628. [Google Scholar] [CrossRef]
- McMurchy, A.N.; Bushell, A.; Levings, M.K.; Wood, K.J. Moving to tolerance: Clinical application of T regulatory cells. Semin. Immunol. 2011, 23, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Fueyo, A.; Sandner, S.; Habicht, A.; Mariat, C.; Kenny, J.; Degauque, N.; Zheng, X.X.; Strom, T.B.; Turka, L.A.; Sayegh, M.H. Specificity of CD4+CD25+ regulatory T cell function in alloimmunity. J. Immunol. 2006, 176, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Tang, Q.; Bluestone, J.A. CD4+CD25+ regulatory T cells in transplantation: Progress, challenges and prospects. Am. J. Transplant. 2007, 7, 1457–1463. [Google Scholar] [CrossRef]
- Raimondi, G.; Sumpter, T.L.; Matta, B.M.; Pillai, M.; Corbitt, N.; Vodovotz, Y.; Wang, Z.; Thomson, A.W. Mammalian target of rapamycin inhibition and alloantigen-specific regulatory T cells synergize to promote long-term graft survival in immunocompetent recipients. J. Immunol. 2010, 184, 624–636. [Google Scholar] [CrossRef]
- Pilat, N.; Farkas, A.M.; Mahr, B.; Schwarz, C.; Unger, L.; Hock, K.; Oberhuber, R.; Aumayr, K.; Wrba, F.; Wekerle, T. T-regulatory cell treatment prevents chronic rejection of heart allografts in a murine mixed chimerism model. J. Heart Lung Transplant. 2014, 33, 429–437. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Migliavacca, B.; Horejs-Hoeck, J.; Kaupper, T.; Roncarolo, M.G. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 2006, 177, 8338–8347. [Google Scholar] [CrossRef] [PubMed]
- Scotta, C.; Esposito, M.; Fazekasova, H.; Fanelli, G.; Edozie, F.C.; Ali, N.; Xiao, F.; Peakman, M.; Afzali, B.; Sagoo, P.; et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4(+)CD25(+)FOXP3(+) T regulatory cell subpopulations. Haematologica 2013, 98, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Segundo, D.S.; Ruiz, J.C.; Izquierdo, M.; Fernandez-Fresnedo, G.; Gomez-Alamillo, C.; Merino, R.; Benito, M.J.; Cacho, E.; Rodrigo, E.; Palomar, R.; et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4+CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation 2006, 82, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Pearl, J.P.; Parris, J.; Hale, D.A.; Hoffmann, S.C.; Bernstein, W.B.; McCoy, K.L.; Swanson, S.J.; Mannon, R.B.; Roederer, M.; Kirk, A.D. Immunocompetent T-cells with a memory-like phenotype are the dominant cell type following antibody-mediated T-cell depletion. Am. J. Transplant. 2005, 5, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Wever-Pinzon, O.; Edwards, L.B.; Taylor, D.O.; Kfoury, A.G.; Drakos, S.G.; Selzman, C.H.; Fang, J.C.; Lund, L.H.; Stehlik, J. Association of recipient age and causes of heart transplant mortality: Implications for personalization of post-transplant management-An analysis of the International Society for Heart and Lung Transplantation Registry. J. Heart Lung Transplant. 2017, 36, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Bangash, M.N.; Kong, M.L.; Pearse, R.M. Use of inotropes and vasopressor agents in critically ill patients. Br. J. Pharmacol. 2012, 165, 2015–2033. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, F.; Pieri, M.; Arnaez Corada, B.; Ajello, S.; Melisurgo, G.; De Bonis, M.; Zangrillo, A. Timing and Strategy for Weaning from Venoarterial ECMO are Complex Issues. J. Cardiothorac. Vasc. Anesth. 2015, 29, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; Eschenhagen, T.; Hamdani, N.; Heinzel, F.R.; Lyon, A.R.; Manstein, D.J.; Metzger, J.; Papp, Z.; Tocchetti, C.G.; Yilmaz, M.B.; et al. Treatments targeting inotropy. Eur. Heart J. 2018, 39. [Google Scholar] [CrossRef] [PubMed]
- Fellahi, J.L.; Fischer, M.O.; Daccache, G.; Gerard, J.L.; Hanouz, J.L. Positive inotropic agents in myocardial ischemia-reperfusion injury: A benefit/risk analysis. Anesthesiology 2013, 118, 1460–1465. [Google Scholar] [CrossRef]
- Da Luz, V.F.; Otsuki, D.A.; Gonzalez, M.M.; Negri, E.M.; Caldini, E.G.; Damaceno-Rodrigues, N.R.; Malbouisson, L.M.; Viana, B.G.; Vane, M.F.; Carmona, M.J. Myocardial protection induced by fentanyl in pigs exposed to high-dose adrenaline. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1098–1107. [Google Scholar] [CrossRef]
- Najafi, A.; Sequeira, V.; Kuster, D.W.; van der Velden, J. beta-adrenergic receptor signalling and its functional consequences in the diseased heart. Eur. J. Clin. Investig. 2016, 46, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Rajadurai, M.; Stanely Mainzen Prince, P. Preventive effect of naringin on cardiac markers, electrocardiographic patterns and lysosomal hydrolases in normal and isoproterenol-induced myocardial infarction in Wistar rats. Toxicology 2007, 230, 178–188. [Google Scholar] [CrossRef]
- Rajadurai, M.; Stanely Mainzen Prince, P. Preventive effect of naringin on lipid peroxides and antioxidants in isoproterenol-induced cardiotoxicity in Wistar rats: Biochemical and histopathological evidences. Toxicology 2006, 228, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Rajadurai, M.; Stanely Mainzen Prince, P. Preventive effect of naringin on lipids, lipoproteins and lipid metabolic enzymes in isoproterenol-induced myocardial infarction in Wistar rats. J. Biochem. Mol. Toxicol. 2006, 20, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Nichtova, Z.; Novotova, M.; Kralova, E.; Stankovicova, T. Morphological and functional characteristics of models of experimental myocardial injury induced by isoproterenol. Gen. Physiol. Biophys. 2012, 31, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Berman, M.; Ali, A.; Ashley, E.; Freed, D.; Clarke, K.; Tsui, S.; Parameshwar, J.; Large, S. Is stress cardiomyopathy the underlying cause of ventricular dysfunction associated with brain death? J. Heart Lung Transplant. 2010, 29, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Couch, L.S.; Harding, S.E. Takotsubo Syndrome: Stress or NO Stress? JACC Basic Transl. Sci. 2018, 3, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Ansari, U.; El-Battrawy, I.; Fastner, C.; Behnes, M.; Sattler, K.; Huseynov, A.; Baumann, S.; Tulumen, E.; Borggrefe, M.; Akin, I. Clinical outcomes associated with catecholamine use in patients diagnosed with Takotsubo cardiomyopathy. BMC Cardiovasc. Disord. 2018, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Nunez Gil, I.J.; Santoro, F.; Madias, J.E.; Pelliccia, F.; Brunetti, N.D.; Salmoirago-Blotcher, E.; Sharkey, S.W.; Eitel, I.; Akashi, Y.J.; et al. Takotsubo syndrome: State-of-the-art review by an expert panel—Part 1. Cardiovasc. Revasc. Med. 2019, 20, 70–79. [Google Scholar] [CrossRef]
- Paur, H.; Wright, P.T.; Sikkel, M.B.; Tranter, M.H.; Mansfield, C.; O’Gara, P.; Stuckey, D.J.; Nikolaev, V.O.; Diakonov, I.; Pannell, L.; et al. High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: A new model of Takotsubo cardiomyopathy. Circulation 2012, 126, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Rees, P.S.; Prasad, S.; Poole-Wilson, P.A.; Harding, S.E. Stress (Takotsubo) cardiomyopathy—A novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.M.; Eiswirth, C.C.; Mealey, P.C.; Larrabee, P.; Herrick, C.M.; Bristow, M.R. Beta-adrenergic supersensitivity of the transplanted human heart is presynaptic in origin. Circulation 1989, 79, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Theodoropoulos, S.; Mathias, C.J.; Dhalla, N.; Wittes, J.; Mitchell, A.; Yacoub, M. Increased sensitivity of the denervated transplanted human heart to isoprenaline both before and after beta-adrenergic blockade. Circulation 1987, 75, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.; Nim, S.; de Montigny, C.; Rousseau, G. Alterations of beta-adrenoceptor responsiveness in postischemic myocardium after 72 h of reperfusion. Eur. J. Pharmacol. 2004, 495, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Anthonio, R.L.; Brodde, O.E.; van Veldhuisen, D.J.; Scholtens, E.; Crijns, H.J.; van Gilst, W.H. Beta-adrenoceptor density in chronic infarcted myocardium: A subtype specific decrease of beta1-adrenoceptor density. Int. J. Cardiol. 2000, 72, 137–141. [Google Scholar] [CrossRef]
- Brinks, H.; Boucher, M.; Gao, E.; Chuprun, J.K.; Pesant, S.; Raake, P.W.; Huang, Z.M.; Wang, X.; Qiu, G.; Gumpert, A.; et al. Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ. Res. 2010, 107, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- See Hoe, L.E.; Schilling, J.M.; Busija, A.R.; Haushalter, K.J.; Ozberk, V.; Keshwani, M.M.; Roth, D.M.; Toit, E.D.; Headrick, J.P.; Patel, H.H.; et al. Chronic beta1-adrenoceptor blockade impairs ischaemic tolerance and preconditioning in murine myocardium. Eur. J. Pharmacol. 2016, 789, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Otani, H.; Oishi, C.; Fujikawa, M.; Yamashita, K.; Okazaki, T.; Sato, D.; Ueyama, T.; Iwasaka, J.; Yamamoto, Y.; et al. Efficacy of intracoronary administration of a short-acting beta-blocker landiolol during reperfusion in pigs. Int. J. Cardiol. 2011, 146, 347–353. [Google Scholar] [CrossRef]
- Spear, J.F.; Prabu, S.K.; Galati, D.; Raza, H.; Anandatheerthavarada, H.K.; Avadhani, N.G. beta1-Adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2459–H2466. [Google Scholar] [CrossRef]
- Salie, R.; Moolman, J.A.; Lochner, A. The role of beta-adrenergic receptors in the cardioprotective effects of beta-preconditioning (betaPC). Cardiovasc. Drugs Ther. 2011, 25, 31–46. [Google Scholar] [CrossRef]
- Lochner, A.; Genade, S.; Tromp, E.; Podzuweit, T.; Moolman, J.A. Ischemic preconditioning and the beta-adrenergic signal transduction pathway. Circulation 1999, 100, 958–966. [Google Scholar] [CrossRef] [PubMed]
- See Hoe, L.E.; Foster, S.R.; Wendt, L.; Patel, H.H.; Headrick, J.P.; Peart, J.N. Regulation of the beta-Adrenergic Receptor Signaling Pathway in Sustained Ligand-Activated Preconditioning. J. Pharmacol. Exp. Ther. 2019, 369, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Bernstein, D.; Murphy, E.; Steenbergen, C. The role of beta-adrenergic receptor signaling in cardioprotection. FASEB J. 2005, 19, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.P. Cell logic for dual coupling of a single class of receptors to G(s) and G(i) proteins. Circ. Res. 2000, 87, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Peart, J.N.; Gross, G.J. Chronic exposure to morphine produces a marked cardioprotective phenotype in aged mouse hearts. Exp. Gerontol. 2004, 39, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- See Hoe, L.; Zemljic-Harpf, A.; Schilling, J.M.; Finley, C.; Kellerhals, S.E.; Roth, D.M.; Peart, J.N.; Patel, H.H.; Headrick, J.P. Opioid-mediated sustained ligand-activated preconditioning induces cardiac protection in type II diabetes mellitus. FASEB J. 2013, 27, 1169. [Google Scholar]
- O’Connell, T.D.; Jensen, B.C.; Baker, A.J.; Simpson, P.C. Cardiac alpha1-adrenergic receptors: Novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol. Rev. 2014, 66, 308–333. [Google Scholar] [CrossRef]
- Laks, M.M.; Morady, F.; Swan, H.J. Myocardial hypertrophy produced by chronic infusion of subhypertensive doses of norepinephrine in the dog. Chest 1973, 64, 75–78. [Google Scholar] [CrossRef]
- Vecchione, C.; Fratta, L.; Rizzoni, D.; Notte, A.; Poulet, R.; Porteri, E.; Frati, G.; Guelfi, D.; Trimarco, V.; Mulvany, M.J.; et al. Cardiovascular influences of alpha1b-adrenergic receptor defect in mice. Circulation 2002, 105, 1700–1707. [Google Scholar] [CrossRef]
- Tejero-Taldo, M.I.; Gursoy, E.; Zhao, T.C.; Kukreja, R.C. Alpha-adrenergic receptor stimulation produces late preconditioning through inducible nitric oxide synthase in mouse heart. J. Mol. Cell. Cardiol. 2002, 34, 185–195. [Google Scholar] [CrossRef]
- Meng, X.; Brown, J.M.; Ao, L.; Banerjee, A.; Harken, A.H. Norepinephrine induces cardiac heat shock protein 70 and delayed cardioprotection in the rat through alpha 1 adrenoceptors. Cardiovasc. Res. 1996, 32, 374–383. [Google Scholar] [CrossRef]
- Baghelai, K.; Graham, L.J.; Wechsler, A.S.; Jakoi, E.R. Delayed myocardial preconditioning by alpha1-adrenoceptors involves inhibition of apoptosis. J. Thorac. Cardiovasc. Surg. 1999, 117, 980–986. [Google Scholar] [CrossRef]
- Zhu, H.; McElwee-Witmer, S.; Perrone, M.; Clark, K.L.; Zilberstein, A. Phenylephrine protects neonatal rat cardiomyocytes from hypoxia and serum deprivation-induced apoptosis. Cell Death Differ. 2000, 7, 773–784. [Google Scholar] [CrossRef][Green Version]
- O’Connell, T.D.; Swigart, P.M.; Rodrigo, M.C.; Ishizaka, S.; Joho, S.; Turnbull, L.; Tecott, L.H.; Baker, A.J.; Foster, E.; Grossman, W.; et al. Alpha1-adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J. Clin. Investig. 2006, 116, 1005–1015. [Google Scholar] [CrossRef]
- Tonnarini, G.; Parlapiano, C.; Cavallotti, D.; Tego, A.; Curione, M.; Giancaspro, G.; Vincentelli, G.M.; Leone, S.; Cavallotti, C. Dopamine receptor subtypes in the human coronary vessels of healthy subjects. J. Recept. Signal Transduct. Res. 2011, 31, 33–38. [Google Scholar] [CrossRef]
- Cavallotti, C.; Mancone, M.; Bruzzone, P.; Sabbatini, M.; Mignini, F. Dopamine receptor subtypes in the native human heart. Heart Vessel. 2010, 25, 432–437. [Google Scholar] [CrossRef]
- Li, H.; Shi, S.; Sun, Y.H.; Zhao, Y.J.; Li, Q.F.; Li, H.Z.; Wang, R.; Xu, C.Q. Dopamine D2 receptor stimulation inhibits angiotensin II-induced hypertrophy in cultured neonatal rat ventricular myocytes. Clin. Exp. Pharmacol. Physiol. 2009, 36, 312–318. [Google Scholar] [CrossRef]
- Richmond, M.E.; Easterwood, R.; Singh, R.K.; Gilmore, L.; Beddows, K.; Zuckerman, W.A.; McFeely, E.D.; Chen, J.M.; Addonizio, L.J. Low-Dose Donor Dopamine Is Associated with a Decreased Risk of Right Heart Failure in Pediatric Heart Transplant Recipients. Transplantation 2016, 100, 2729–2734. [Google Scholar] [CrossRef]
- Benck, U.; Hoeger, S.; Brinkkoetter, P.T.; Gottmann, U.; Doenmez, D.; Boesebeck, D.; Lauchart, W.; Gummert, J.; Karck, M.; Lehmkuhl, H.B.; et al. Effects of donor pre-treatment with dopamine on survival after heart transplantation: A cohort study of heart transplant recipients nested in a randomized controlled multicenter trial. J. Am. Coll. Cardiol. 2011, 58, 1768–1777. [Google Scholar] [CrossRef]
- Roy, N.; Friehs, I.; Cowan, D.B.; Zurakowski, D.; McGowan, F.X.; del Nido, P.J. Dopamine induces postischemic cardiomyocyte apoptosis in vivo: An effect ameliorated by propofol. Ann. Thorac. Surg. 2006, 82, 2192–2199. [Google Scholar] [CrossRef]
- Port, J.D.; Gilbert, E.M.; Larrabee, P.; Mealey, P.; Volkman, K.; Ginsburg, R.; Hershberger, R.E.; Murray, J.; Bristow, M.R. Neurotransmitter depletion compromises the ability of indirect-acting amines to provide inotropic support in the failing human heart. Circulation 1990, 81, 929–938. [Google Scholar] [CrossRef]
- Lazou, A.; Markou, T.; Zioga, M.; Vasara, E.; Efstathiou, A.; Gaitanaki, C. Dopamine mimics the cardioprotective effect of ischemic preconditioning via activation of alpha1-adrenoceptors in the isolated rat heart. Physiol. Res. 2006, 55, 1–8. [Google Scholar]
- Gupta, V.; Goyal, R.; Sharma, P.L. Preconditioning offers cardioprotection in hyperlipidemic rat hearts: Possible role of Dopamine (D2) signaling. BMC Cardiovasc. Disord. 2015, 15, 77. [Google Scholar] [CrossRef]
- Wei, C.; Gao, J.; Li, M.; Li, H.; Wang, Y.; Li, H.; Xu, C. Dopamine D2 receptors contribute to cardioprotection of ischemic post-conditioning via activating autophagy in isolated rat hearts. Int. J. Cardiol. 2016, 203, 837–839. [Google Scholar] [CrossRef]
- Gao, J.; Guo, J.; Li, H.; Bai, S.; Li, H.; Wu, B.; Wang, L.; Xi, Y.; Tian, Y.; Yang, G.; et al. Involvement of dopamine D2 receptors activation in ischemic post-conditioning-induced cardioprotection through promoting PKC-epsilon particulate translocation in isolated rat hearts. Mol. Cell. Biochem. 2013, 379, 267–276. [Google Scholar] [CrossRef]
- Li, H.; Wei, C.; Gao, J.; Bai, S.; Li, H.; Zhao, Y.; Li, H.; Han, L.; Tian, Y.; Yang, G.; et al. Mediation of dopamine D2 receptors activation in post-conditioning-attenuated cardiomyocyte apoptosis. Exp. Cell Res. 2014, 323, 118–130. [Google Scholar] [CrossRef]
- Vettel, C.; Hottenrott, M.C.; Spindler, R.; Benck, U.; Schnuelle, P.; Tsagogiorgas, C.; Kramer, B.K.; Hoeger, S.; El-Armouche, A.; Wieland, T.; et al. Dopamine and lipophilic derivates protect cardiomyocytes against cold preservation injury. J. Pharmacol. Exp. Ther. 2014, 348, 77–85. [Google Scholar] [CrossRef]
- Spindler, R.S.; Schnuelle, P.; Nickels, L.; Jarczyk, J.; Waldherr, R.; Theisinger, S.; Theisinger, B.; Klotz, S.; Tsagogiorgas, C.; Gottmann, U.; et al. N-Octanoyl Dopamine for Donor Treatment in a Brain-death Model of Kidney and Heart Transplantation. Transplantation 2015, 99, 935–941. [Google Scholar] [CrossRef]
- Li, S.; Korkmaz-Icoz, S.; Radovits, T.; Ruppert, M.; Spindler, R.; Loganathan, S.; Hegedus, P.; Brlecic, P.; Theisinger, B.; Theisinger, S.; et al. Donor Preconditioning After the Onset of Brain Death with Dopamine Derivate n-Octanoyl Dopamine Improves Early Posttransplant Graft Function in the Rat. Am. J. Transplant. 2017, 17, 1802–1812. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Danner, S.; Bohm, M. Mechanisms of beta-adrenergic receptor desensitization: From molecular biology to heart failure. Basic Res. Cardiol. 1996, 91, 29–34. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Floerchinger, B.; Oberhuber, R.; Tullius, S.G. Effects of brain death on organ quality and transplant outcome. Transplant. Rev. (Orlando) 2012, 26, 54–59. [Google Scholar] [CrossRef]
- Gordon, J.K.; McKinlay, J. Physiological changes after brain stem death and management of the heart-beating donor. BJA Educ. 2012, 12, 225–229. [Google Scholar] [CrossRef]
- Braulio, R.; Sanches, M.D.; Teixeira Junior, A.L.; Costa, P.H.; Moreira Mda, C.; Rocha, M.A.; Andrade, S.A.; Gelape, C.L. Associated Clinical and Laboratory Markers of Donor on Allograft Function After Heart Transplant. Braz. J. Cardiovasc. Surg. 2016, 31, 89–97. [Google Scholar] [CrossRef][Green Version]
- Stoica, S.C.; Satchithananda, D.K.; White, P.A.; Parameshwar, J.; Redington, A.N.; Large, S.R. Noradrenaline use in the human donor and relationship with load-independent right ventricular contractility. Transplantation 2004, 78, 1193–1197. [Google Scholar] [CrossRef]
- Angleitner, P.; Kaider, A.; Gokler, J.; Moayedifar, R.; Osorio-Jaramillo, E.; Zuckermann, A.; Laufer, G.; Aliabadi-Zuckermann, A. High-dose catecholamine donor support and outcomes after heart transplantation. J. Heart Lung Transplant. 2018, 37, 596–603. [Google Scholar] [CrossRef]
- Costanzo, M.R. Don’t worry, be happy with intravenous norepinephrine. J. Heart Lung Transplant. 2018, 37, 572–574. [Google Scholar] [CrossRef]
- Nixon, J.L.; Kfoury, A.G.; Brunisholz, K.; Horne, B.D.; Myrick, C.; Miller, D.V.; Budge, D.; Bader, F.; Everitt, M.; Saidi, A.; et al. Impact of high-dose inotropic donor support on early myocardial necrosis and outcomes in cardiac transplantation. Clin. Transplant. 2012, 26, 322–327. [Google Scholar] [CrossRef]
- Milting, H.; Scholz, C.; Arusoglu, L.; Freitag, M.; Cebulla, R.; Jaquet, K.; Korfer, R.; Lewinski, D.V.; Kassner, A.; Brodde, O.E.; et al. Selective upregulation of beta1-adrenergic receptors and dephosphorylation of troponin I in end-stage heart failure patients supported by ventricular assist devices. J. Mol. Cell. Cardiol. 2006, 41, 441–450. [Google Scholar] [CrossRef]
- Nakahara, K.; Horimoto, H.; Nakai, Y.; Mieno, S.; Nomura, Y.; Sasaki, S. Left ventricular mechanical unloading restores Beta-2-adrenergic receptor mRNA expression and decreases susceptibility to ischemia and reperfusion in the failing heart. Eur. Surg. Res. 2003, 35, 108–114. [Google Scholar] [CrossRef]
- Pandalai, P.K.; Bulcao, C.F.; Merrill, W.H.; Akhter, S.A. Restoration of myocardial beta-adrenergic receptor signaling after left ventricular assist device support. J. Thorac. Cardiovasc. Surg. 2006, 131, 975–980. [Google Scholar] [CrossRef][Green Version]
- Akhter, S.A.; D’Souza, K.M.; Malhotra, R.; Staron, M.L.; Valeroso, T.B.; Fedson, S.E.; Anderson, A.S.; Raman, J.; Jeevanandam, V. Reversal of impaired myocardial beta-adrenergic receptor signaling by continuous-flow left ventricular assist device support. J. Heart Lung Transplant. 2010, 29, 603–609. [Google Scholar] [CrossRef][Green Version]
- Ogletree-Hughes, M.L.; Stull, L.B.; Sweet, W.E.; Smedira, N.G.; McCarthy, P.M.; Moravec, C.S. Mechanical unloading restores beta-adrenergic responsiveness and reverses receptor downregulation in the failing human heart. Circulation 2001, 104, 881–886. [Google Scholar] [CrossRef]
- Ji, X.F.; Shuo, W.; Yang, L.; Li, C.S. Impaired beta-adrenergic receptor signalling in post-resuscitation myocardial dysfunction. Resuscitation 2012, 83, 640–644. [Google Scholar] [CrossRef]
- Shin, E.; Ko, K.S.; Rhee, B.D.; Han, J.; Kim, N. Different effects of prolonged beta-adrenergic stimulation on heart and cerebral artery. Integr. Med. Res. 2014, 3, 204–210. [Google Scholar] [CrossRef]
- Chen, M.; Sato, P.Y.; Chuprun, J.K.; Peroutka, R.J.; Otis, N.J.; Ibetti, J.; Pan, S.; Sheu, S.S.; Gao, E.; Koch, W.J. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ. Res. 2013, 112, 1121–1134. [Google Scholar] [CrossRef]
- Sato, P.Y.; Chuprun, J.K.; Grisanti, L.A.; Woodall, M.C.; Brown, B.R.; Roy, R.; Traynham, C.J.; Ibetti, J.; Lucchese, A.M.; Yuan, A.; et al. Restricting mitochondrial GRK2 post-ischemia confers cardioprotection by reducing myocyte death and maintaining glucose oxidation. Sci. Signal. 2018, 11, eaau0144. [Google Scholar] [CrossRef]
- Woodall, M.C.; Ciccarelli, M.; Woodall, B.P.; Koch, W.J. G protein-coupled receptor kinase 2: A link between myocardial contractile function and cardiac metabolism. Circ. Res. 2014, 114, 1661–1670. [Google Scholar] [CrossRef]
- Bittner, H.B.; Chen, E.P.; Milano, C.A.; Kendall, S.W.; Jennings, R.B.; Sabiston, D.C., Jr.; Van Trigt, P. Myocardial beta-adrenergic receptor function and high-energy phosphates in brain death—Related cardiac dysfunction. Circulation 1995, 92, II472-8. [Google Scholar] [CrossRef]
- Pandalai, P.K.; Lyons, J.M.; Duffy, J.Y.; McLean, K.M.; Wagner, C.J.; Merrill, W.H.; Pearl, J.M.; Akhter, S.A. Role of the beta-adrenergic receptor kinase in myocardial dysfunction after brain death. J. Thorac. Cardiovasc. Surg. 2005, 130, 1183–1189. [Google Scholar] [CrossRef][Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Woo, A.Y.; Xiao, R.P. beta-Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef]
- Wells, M.A.; See Hoe, L.E.; Bouquet, M.; Molenaar, P.; Obonyo, N.G.; McDonald, C.; James, L.; Engkilde-Pedersen, S.; Hyslop, K.; Passmore, M.; et al. Donor Brain Stem Death and Cardiac Transplantation Mediate Mitochondrial Dyscoupling and Oxidative Stress; Humana Press: New York, NY, USA, 2019. [Google Scholar]
- Hartmann, C.; Radermacher, P.; Wepler, M.; Nussbaum, B. Non-Hemodynamic Effects of Catecholamines. Shock 2017, 48, 390–400. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Liaudet, L.; Calderari, B.; Pacher, P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail. Rev. 2014, 19, 815–824. [Google Scholar] [CrossRef]
- Erb, J.; Beutlhauser, T.; Feldheiser, A.; Schuster, B.; Treskatsch, S.; Grubitzsch, H.; Spies, C. Influence of levosimendan on organ dysfunction in patients with severely reduced left ventricular function undergoing cardiac surgery. J. Int. Med. Res. 2014, 42, 750–764. [Google Scholar] [CrossRef]
- Soeding, P.F.; Crack, P.J.; Wright, C.E.; Angus, J.A.; Royse, C.F. Levosimendan preserves the contractile responsiveness of hypoxic human myocardium via mitochondrial K(ATP) channel and potential pERK 1/2 activation. Eur. J. Pharmacol. 2011, 655, 59–66. [Google Scholar] [CrossRef]
- Parissis, J.T.; Adamopoulos, S.; Antoniades, C.; Kostakis, G.; Rigas, A.; Kyrzopoulos, S.; Iliodromitis, E.; Kremastinos, D. Effects of levosimendan on circulating pro-inflammatory cytokines and soluble apoptosis mediators in patients with decompensated advanced heart failure. Am. J. Cardiol. 2004, 93, 1309–1312. [Google Scholar] [CrossRef]
- Beiras-Fernandez, A.; Weis, F.C.; Kur, F.; Kaczmarek, I.; Schmoeckel, M.; Weis, M.; Reichart, B. Primary graft failure and Ca2+ sensitizers after heart transplantation. Transplant. Proc. 2008, 40, 951–952. [Google Scholar] [CrossRef]
- Affronti, A.; di Bella, I.; Carino, D.; Ragni, T. Levosimendan may improve weaning outcomes in venoarterial ECMO patients. ASAIO J. 2013, 59, 554–557. [Google Scholar] [CrossRef]
- Sponga, S.; Ivanitskaia, E.; Potapov, E.; Krabatsch, T.; Hetzer, R.; Lehmkuhl, H. Preoperative treatment with levosimendan in candidates for mechanical circulatory support. ASAIO J. 2012, 58, 6–11. [Google Scholar] [CrossRef]
- Gordon, A.C.; Santhakumaran, S.; Al-Beidh, F.; Orme, R.M.L.; Perkins, G.D.; Singer, M.; McAuley, D.F.; Mason, A.J.; Ward, J.K.; O’Dea, K.P.; et al. Levosimendan to Prevent Acute Organ Dysfunction in Sepsis: The LeoPARDS RCT; NIHR Journals Library: Southampton, UK, 2018. [Google Scholar]
- Landoni, G.; Lomivorotov, V.V.; Alvaro, G.; Lobreglio, R.; Pisano, A.; Guarracino, F.; Calabro, M.G.; Grigoryev, E.V.; Likhvantsev, V.V.; Salgado-Filho, M.F.; et al. Levosimendan for Hemodynamic Support after Cardiac Surgery. N. Engl. J. Med. 2017, 376, 2021–2031. [Google Scholar] [CrossRef]
- Mebazaa, A.; Nieminen, M.S.; Packer, M.; Cohen-Solal, A.; Kleber, F.X.; Pocock, S.J.; Thakkar, R.; Padley, R.J.; Poder, P.; Kivikko, M.; et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: The SURVIVE Randomized Trial. JAMA 2007, 297, 1883–1891. [Google Scholar] [CrossRef]
- Mehta, R.H.; Van Diepen, S.; Meza, J.; Bokesch, P.; Leimberger, J.D.; Tourt-Uhlig, S.; Swartz, M.; Parrotta, J.; Jankowich, R.; Hay, D.; et al. Levosimendan in patients with left ventricular systolic dysfunction undergoing cardiac surgery on cardiopulmonary bypass: Rationale and study design of the Levosimendan in Patients with Left Ventricular Systolic Dysfunction Undergoing Cardiac Surgery Requiring Cardiopulmonary Bypass (LEVO-CTS) trial. Am. Heart J. 2016, 182, 62–71. [Google Scholar]
- Jessup, M.; Greenberg, B.; Mancini, D.; Cappola, T.; Pauly, D.F.; Jaski, B.; Yaroshinsky, A.; Zsebo, K.M.; Dittrich, H.; Hajjar, R.J.; et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011, 124, 304–313. [Google Scholar] [CrossRef]
- Greenberg, B.; Yaroshinsky, A.; Zsebo, K.M.; Butler, J.; Felker, G.M.; Voors, A.A.; Rudy, J.J.; Wagner, K.; Hajjar, R.J. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: The CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail. 2014, 2, 84–92. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Wang, W.; Froehlich, J.P.; Huke, S.; Aon, M.A.; Wilson, G.M.; Di Benedetto, G.; O’Rourke, B.; Gao, W.D.; Wink, D.A.; et al. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ. Res. 2007, 100, 96–104. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Tocchetti, C.G.; Wang, M.; Daya, S.; Gupta, R.C.; Tunin, R.S.; Mazhari, R.; Takimoto, E.; Paolocci, N.; Cowart, D.; et al. Nitroxyl (HNO): A novel approach for the acute treatment of heart failure. Circ. Heart Fail. 2013, 6, 1250–1258. [Google Scholar] [CrossRef]
- Shah, S.J.; Blair, J.E.; Filippatos, G.S.; Macarie, C.; Ruzyllo, W.; Korewicki, J.; Bubenek-Turconi, S.I.; Ceracchi, M.; Bianchetti, M.; Carminati, P.; et al. Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: Results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: A Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial. Am. Heart J. 2009, 157, 1035–1041. [Google Scholar]




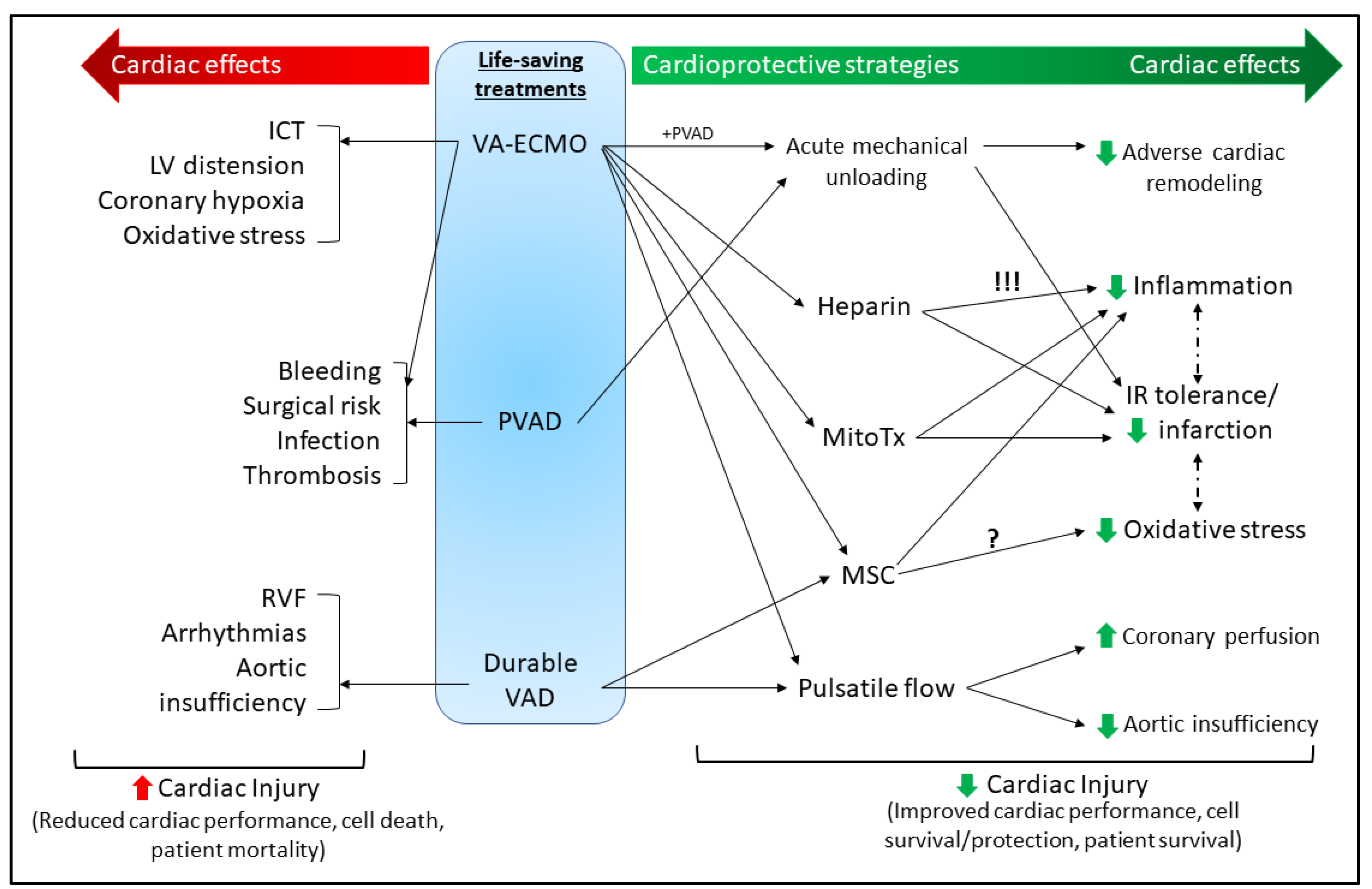{kind=link}
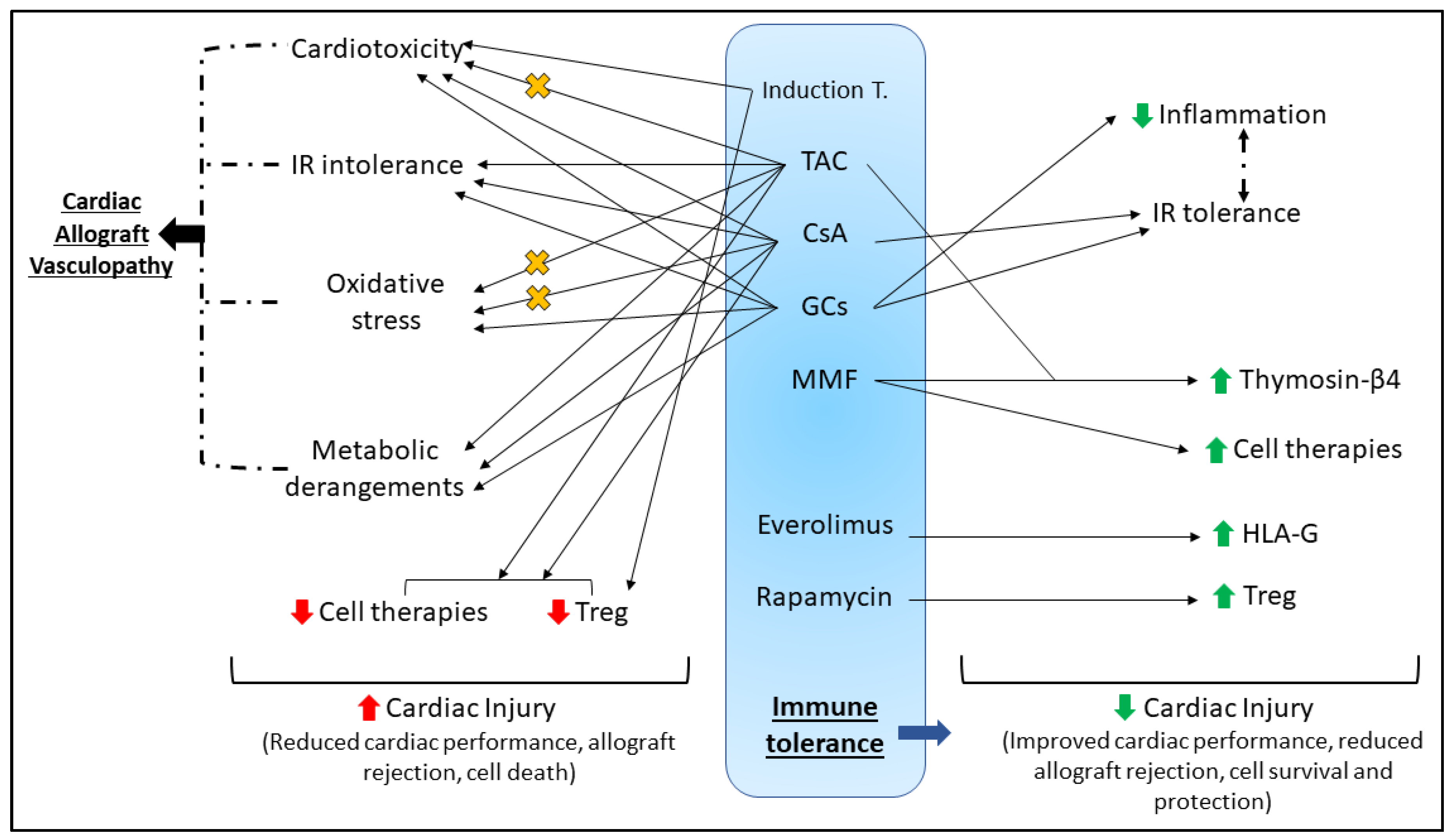{kind=link}
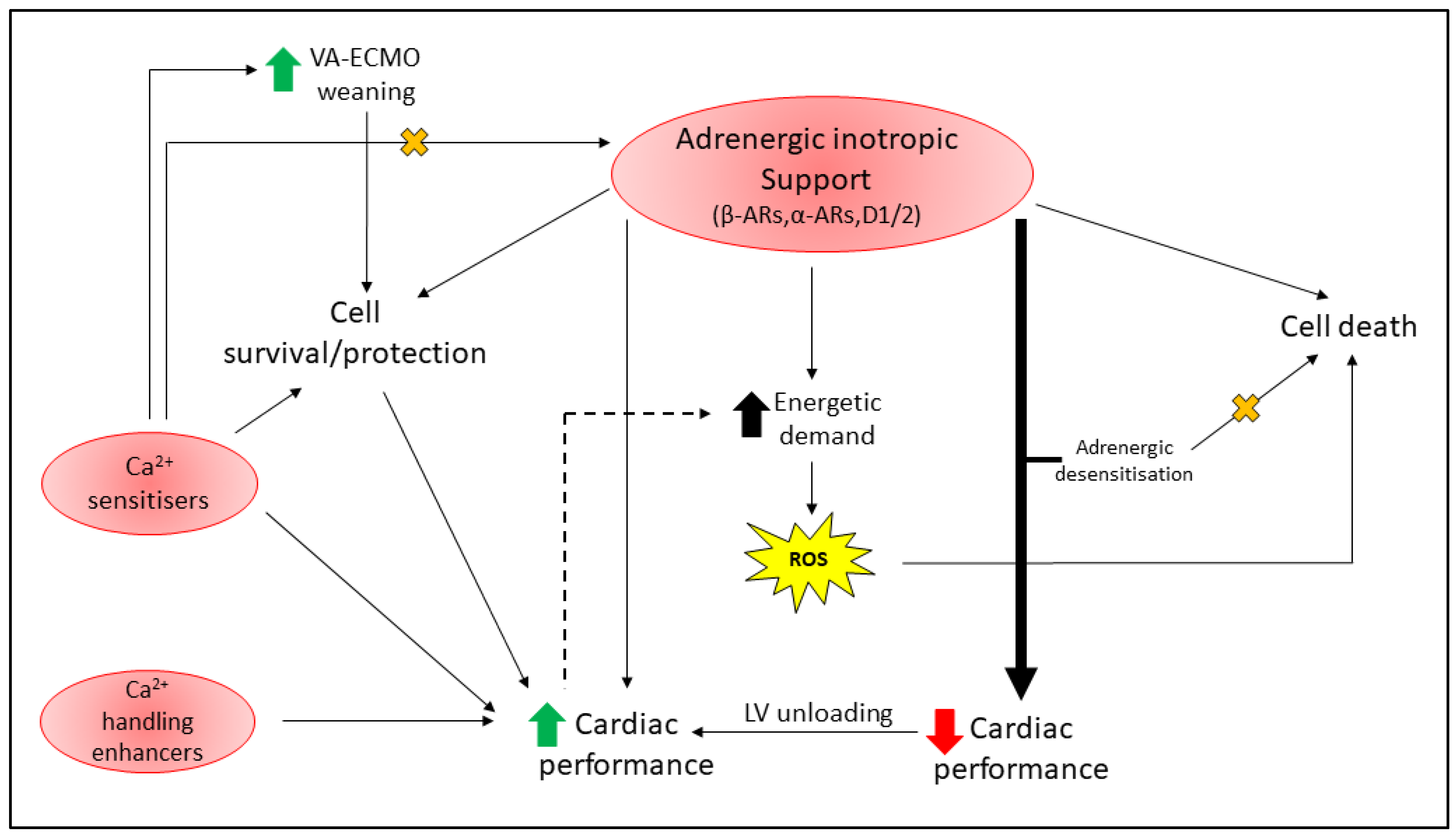{kind=link}
| Drug | Receptor Target | Therapeutic Aim | |||
|---|---|---|---|---|---|
| α1-AR | β1-AR | β2-AR | D1/2 | ||
| Adrenaline | ++++ | +++ | ++++ | 0 | Vasoconstriction Positive inotropy and chronotropy |
| Noradrenaline | ++++ | +++ | + | 0 | Positive inotropy and chronotropy Vasoconstriction |
| Dobutamine | ++ | +++ | + | 0 | Positive Inotropy and chronotropy |
| Dopamine | +++ | ++ | +++ | ++++ | Vasodilation Positive Inotropy and chronotropy |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
See Hoe, L.E.; Bartnikowski, N.; Wells, M.A.; Suen, J.Y.; Fraser, J.F. Hurdles to Cardioprotection in the Critically Ill. Int. J. Mol. Sci. 2019, 20, 3823. https://doi.org/10.3390/ijms20153823
See Hoe LE, Bartnikowski N, Wells MA, Suen JY, Fraser JF. Hurdles to Cardioprotection in the Critically Ill. International Journal of Molecular Sciences. 2019; 20(15):3823. https://doi.org/10.3390/ijms20153823
Chicago/Turabian StyleSee Hoe, Louise E, Nicole Bartnikowski, Matthew A Wells, Jacky Y Suen, and John F Fraser. 2019. "Hurdles to Cardioprotection in the Critically Ill" International Journal of Molecular Sciences 20, no. 15: 3823. https://doi.org/10.3390/ijms20153823
APA StyleSee Hoe, L. E., Bartnikowski, N., Wells, M. A., Suen, J. Y., & Fraser, J. F. (2019). Hurdles to Cardioprotection in the Critically Ill. International Journal of Molecular Sciences, 20(15), 3823. https://doi.org/10.3390/ijms20153823