Genetic Deletion or Pharmacological Inhibition of Soluble Epoxide Hydrolase Ameliorates Cardiac Ischemia/Reperfusion Injury by Attenuating NLRP3 Inflammasome Activation

 and
and
Abstract
1. Introduction
2. Results
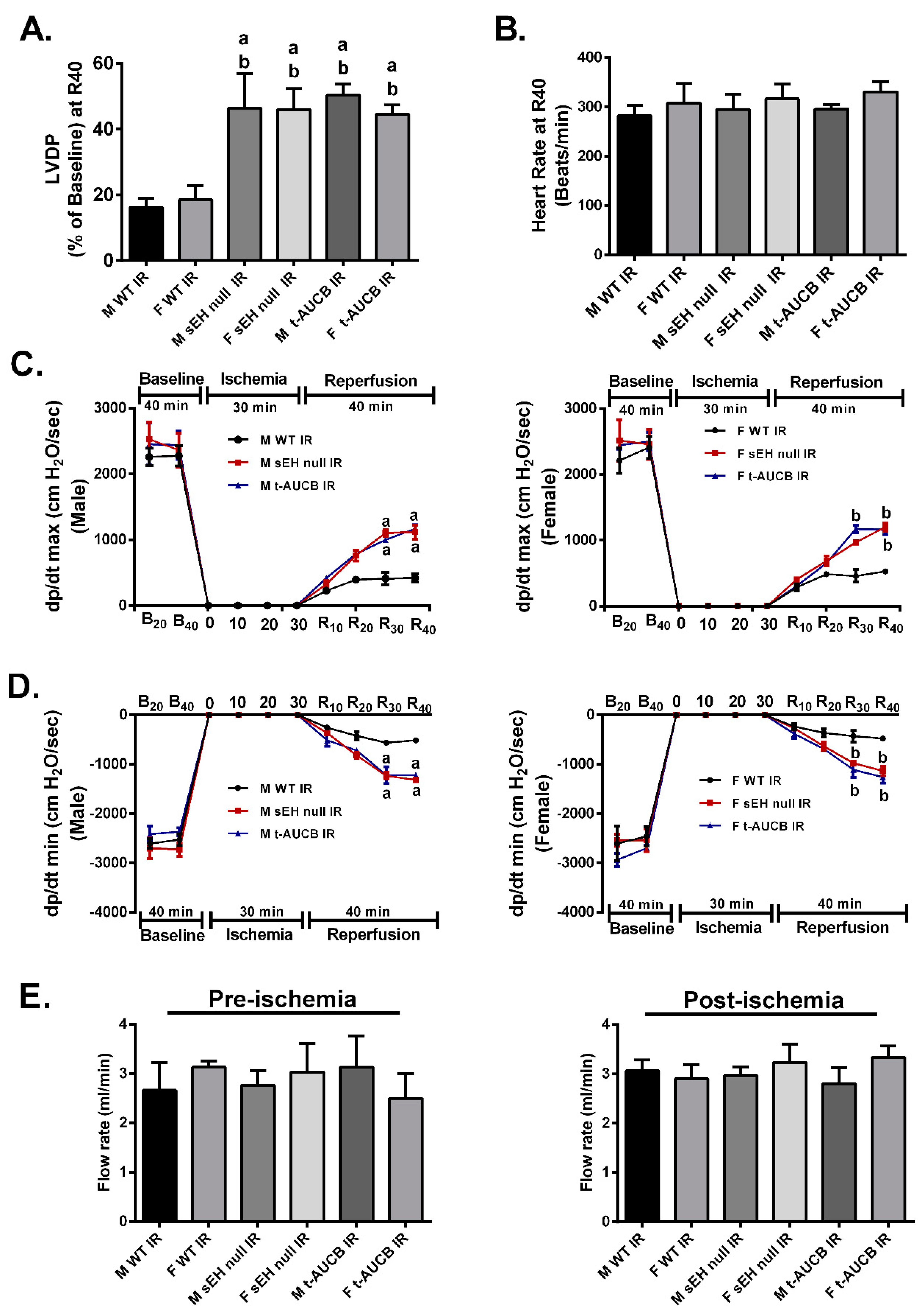
2.1. Deletion or Inhibition of sEH Improves Post-Ischemic Functional Recovery in Both Males and Females
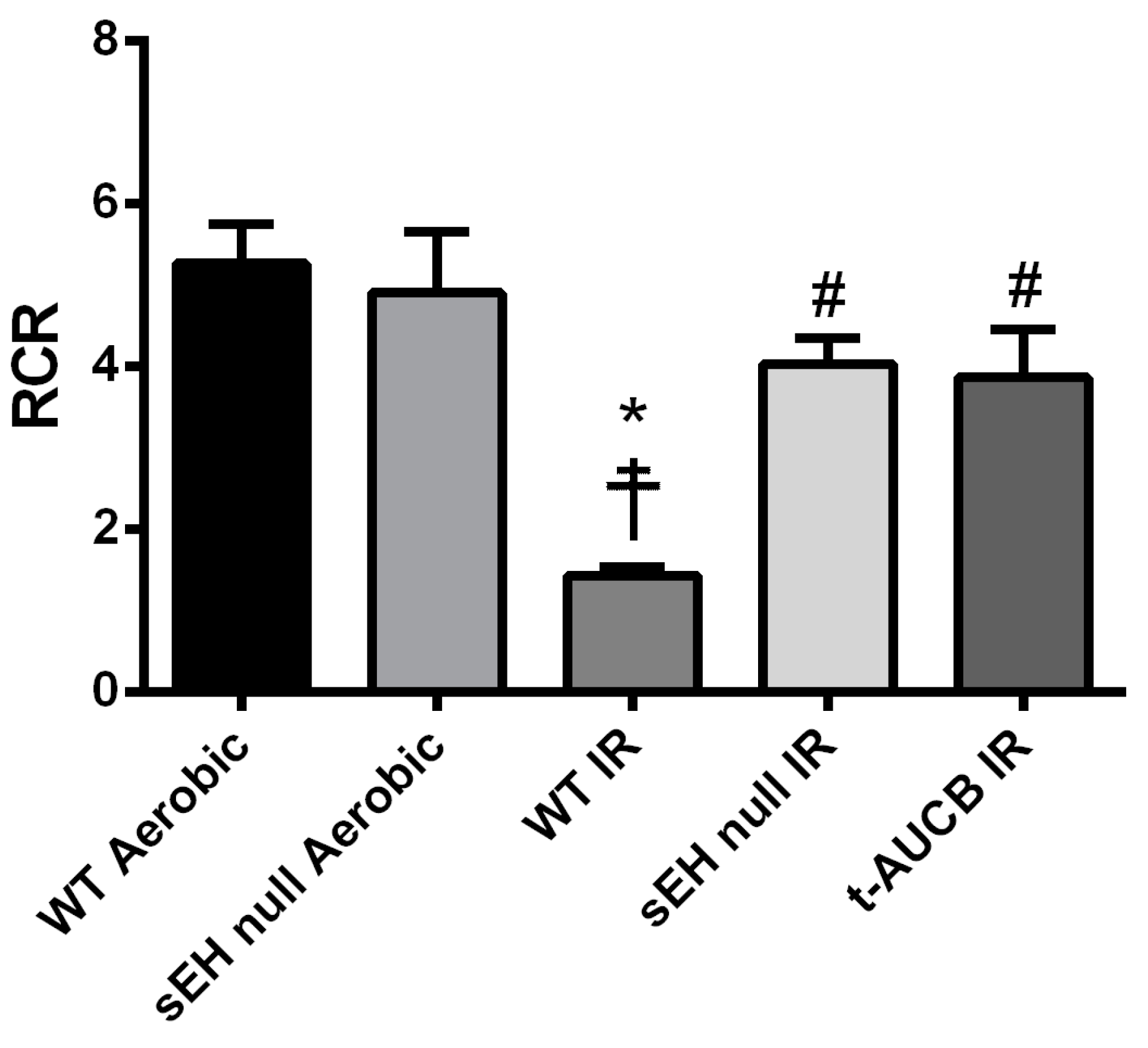
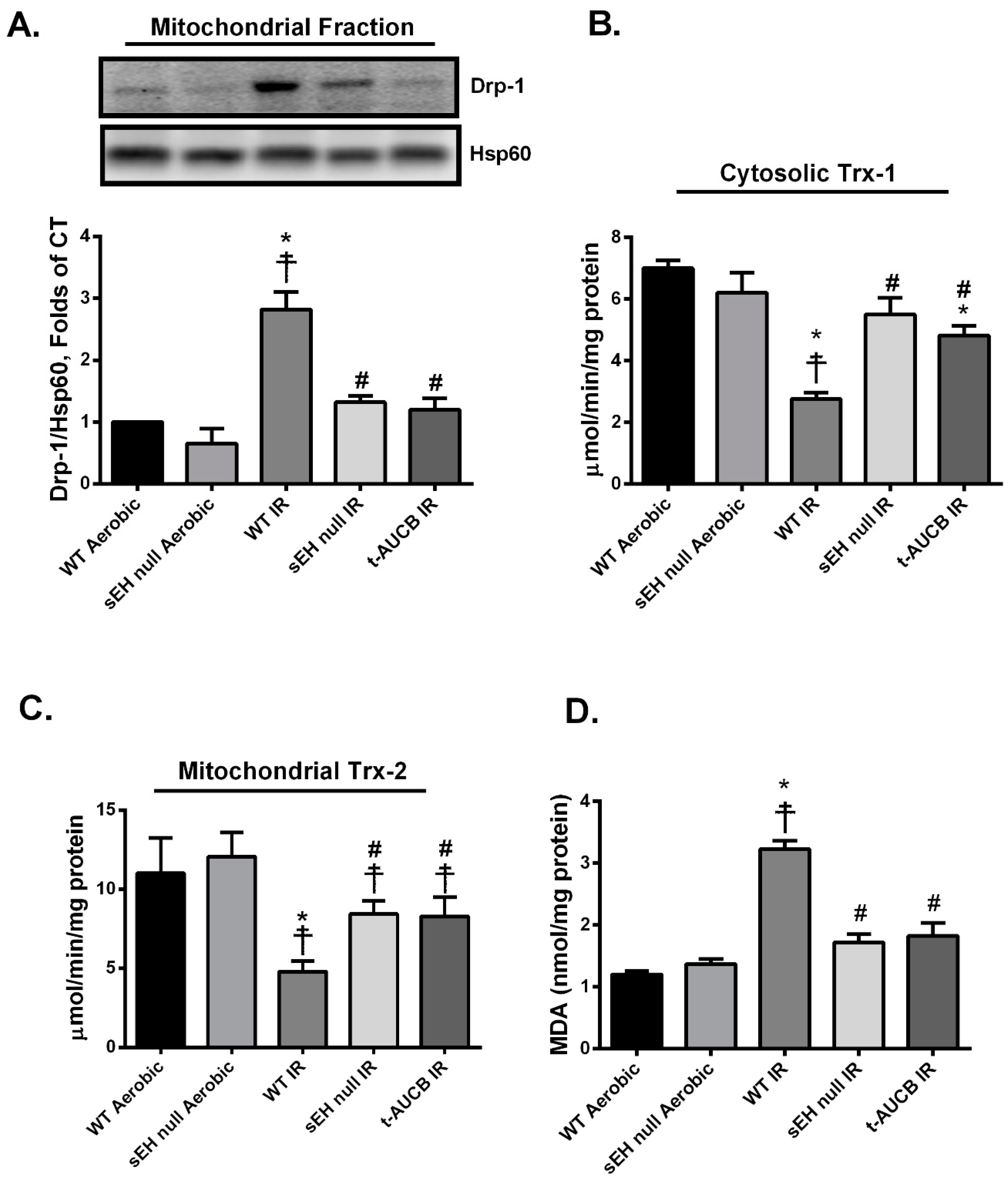
2.2. Deletion or Inhibition of sEH Limits Post-Ischemic Mitochondrial Injury
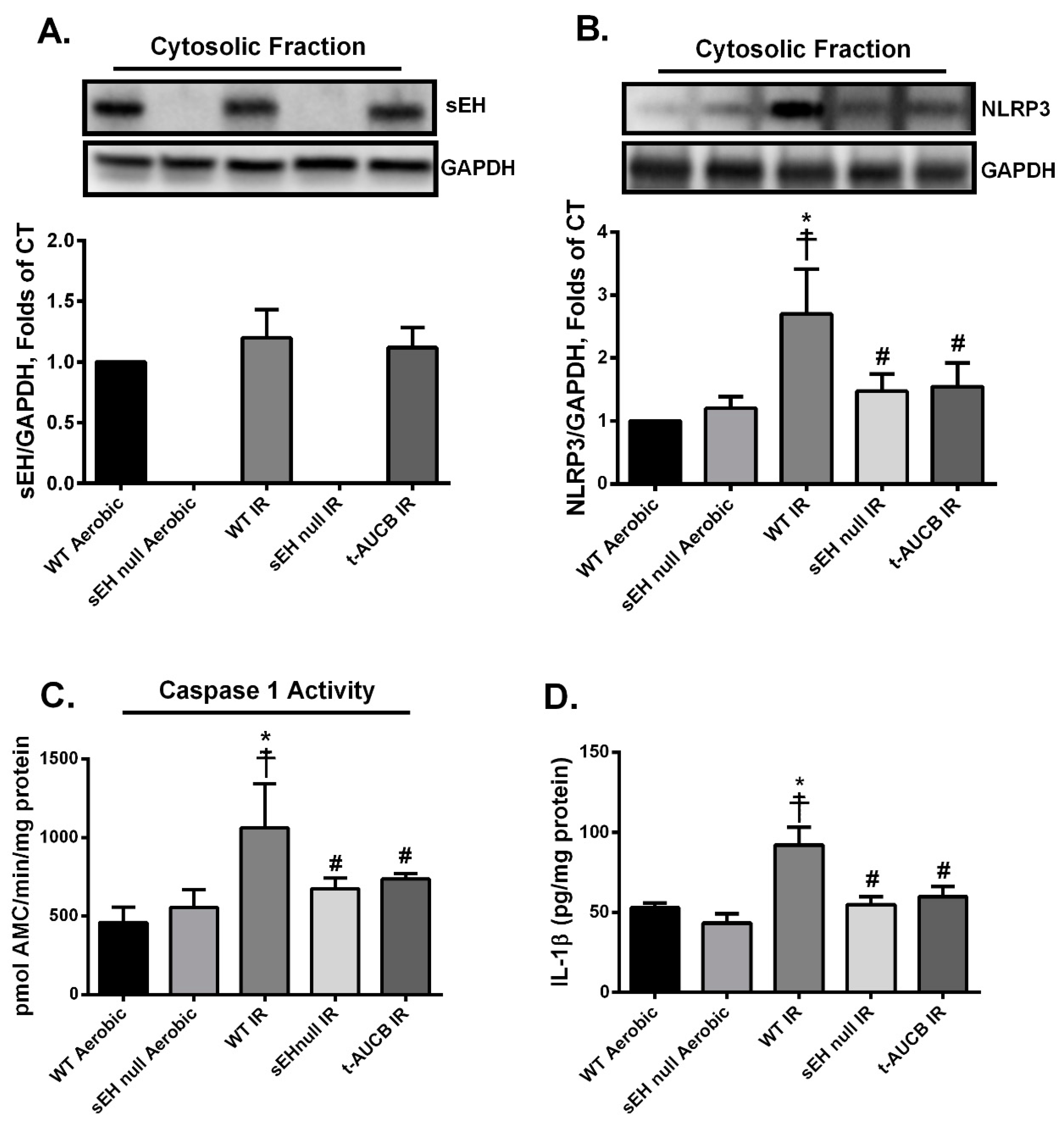
2.3. Deletion or Inhibition of sEH Abrogates the Assembly and Activation of NLRP3 Inflammasome Secondary to IR Injury
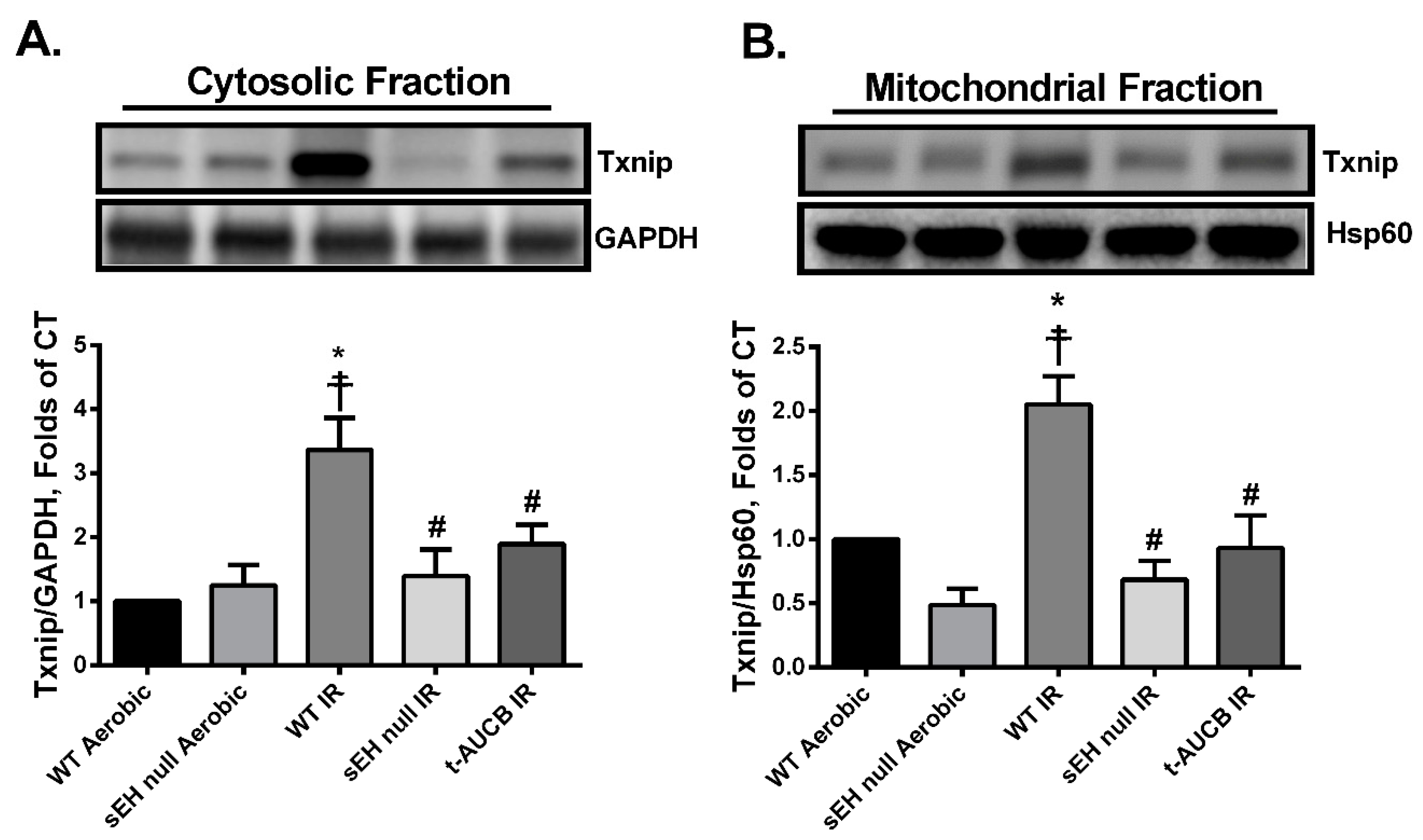
2.4. Deletion or Inhibition of sEH Blunts the Activation and Mitochondrial Translocation of Thioredoxin-Interacting Protein (Txnip) Secondary to IR Injury
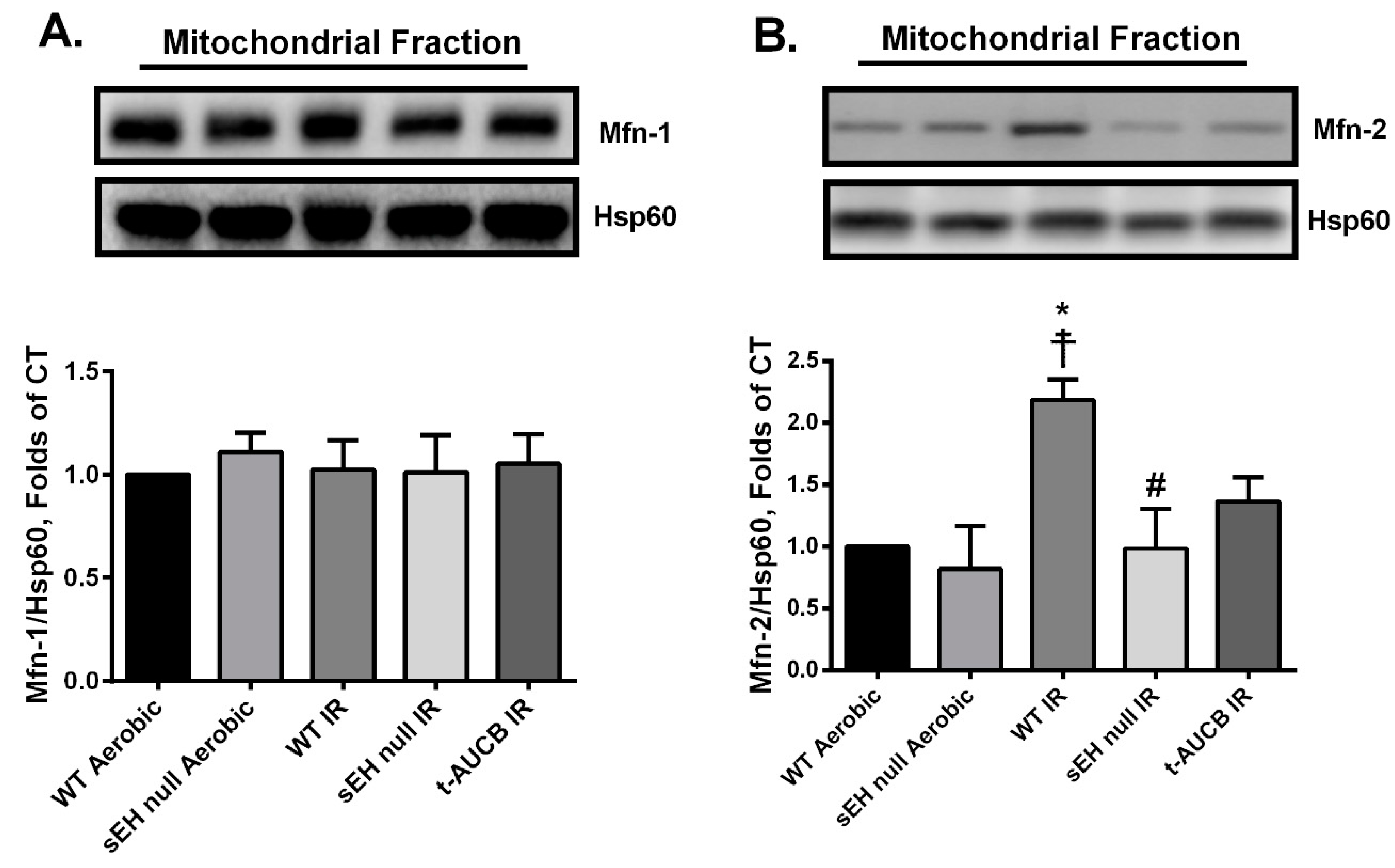
2.5. Deletion or Inhibition of sEH Abrogates the IR-Induced Upregulation of the Mitochondrial Protein Mitofusin-2
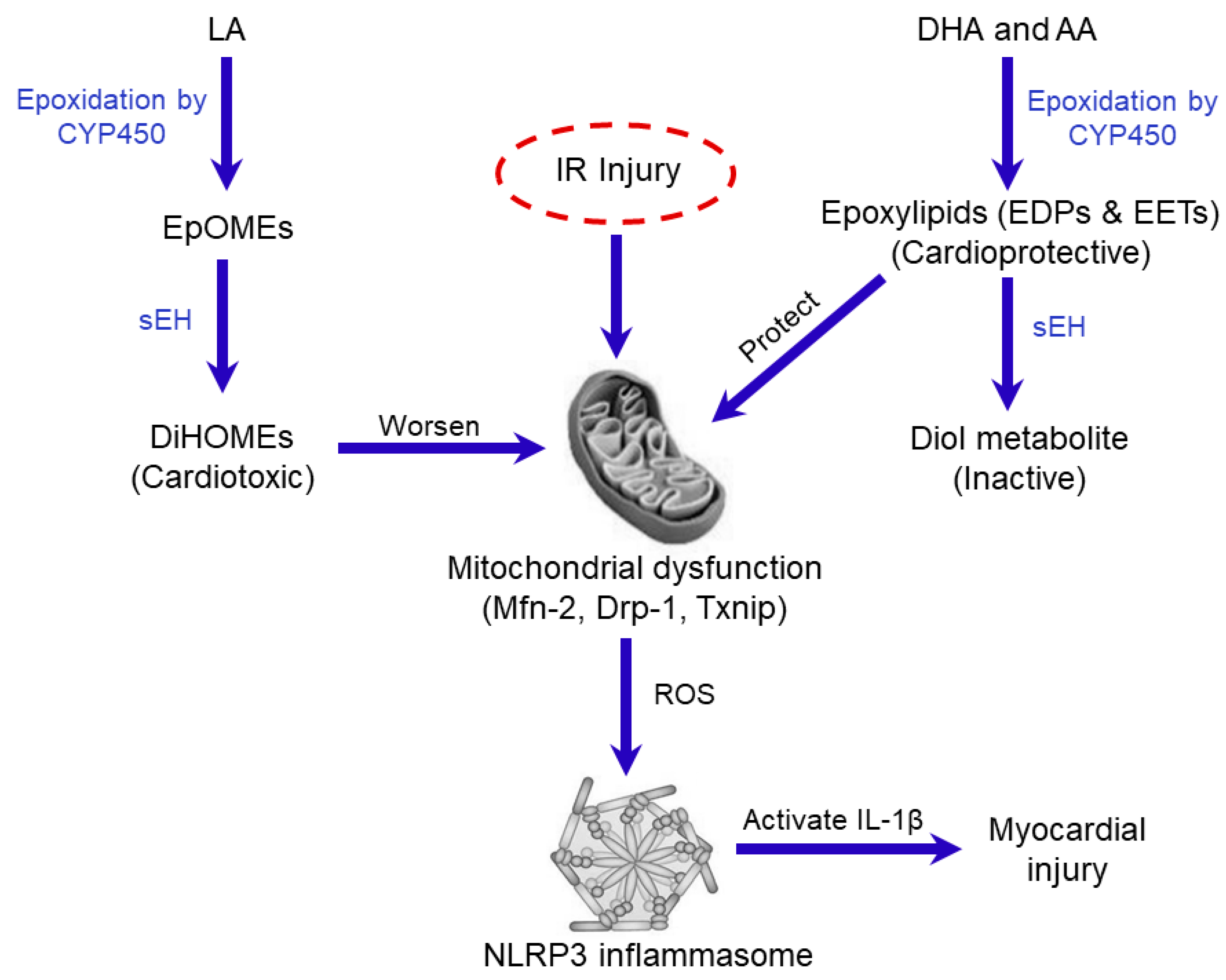
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolated Heart Perfusion
4.3. Immunoblotting
4.4. Enzyme-Linked Immunosorbent Assay
4.5. Measurement of MDA Levels
4.6. Mitochondrial Respiration
4.7. Enzymatic Assays
4.8. Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| AMC | 7-Amino-4-methylcoumarin |
| CF | Coronary flow |
| CYP | Cytochrome p450 |
| DAMPs | Damage-associated molecular patterns |
| DHA | Docosahexaenoic acid |
| DiHOME | Dihydroxyoctadecenoic acid |
| Drp-1 | Dynamin-related protein-1 |
| DTNB | 5,5’-Dithiobis-(2-nitrobenzoic acid) |
| EDPs | Epoxydocosapentaenoic acids |
| EETs | Epoxyeicosatrienoic acids |
| EpOME | Epoxyoctadecenoic acid |
| ETC | Electron transport chain |
| HR | Heart rate |
| Hsp60 | Heat shock protein 60 |
| IL-1β | Interleukin-1beta |
| IR | Ischemia-reperfusion |
| MDA | Malondialdehyde |
| Mfn | Mitofusin |
| NLRP3 | Nucleotide-binding oligomerization domain-like receptor (NLR) family pyrin domain containing 3 |
| LA | Linoleic acid |
| LPS | Lipopolysaccharide |
| LVDP | Left ventricular developed pressure |
| PUFAs | Polyunsaturated fatty acids |
| RCR | Respiratory control ratio |
| ROS | Reactive oxygen species |
| sEH | Soluble epoxide hydrolase |
| t-AUCB | trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid |
| Trx | Thioredoxin |
| Txnip | Thioredoxin-interacting protein |
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef]
- Menees, D.S.; Peterson, E.D.; Wang, Y.; Curtis, J.P.; Messenger, J.C.; Rumsfeld, J.S.; Gurm, H.S. Door-to-balloon time and mortality among patients undergoing primary PCI. N. Engl. J. Med. 2013, 369, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. NLRP3 inflammasome as a novel player in myocardial infarction. Int. Heart J. 2014, 55, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Stutz, A.; Golenbock, D.T.; Latz, E. Inflammasomes: Too big to miss. J. Clin. Investig. 2009, 119, 3502–3511. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. Role of the inflammasome in myocardial infarction. Trends Cardiovasc. Med. 2011, 21, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Lei, S.; Zhao, B.; Wu, Y.; Su, W.; Liu, M.; Meng, Q.; Zhou, B.; Leng, Y.; Xia, Z.Y. NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxidative Med. Cell. Longev. 2017, 2017, 9743280. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, O.; Ranheim, T.; Vinge, L.E.; Bliksoen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femmino, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxidative Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef] [PubMed]
- Hammock, B.D.; Ratcliff, M.; Schooley, D.A. Hydration of an 18O epoxide by a cytosolic epoxide hydrolase from mouse liver. Life Sci. 1980, 27, 1635–1641. [Google Scholar] [CrossRef]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xu, F.; Huse, L.M.; Morisseau, C.; Draper, A.J.; Newman, J.W.; Parker, C.; Graham, L.; Engler, M.M.; Hammock, B.D.; et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 2000, 87, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Weintraub, N.L.; McCaw, R.B.; Hu, S.; Harmon, S.D.; Rice, J.B.; Hammock, B.D.; Spector, A.A. Effect of soluble epoxide hydrolase inhibition on epoxyeicosatrienoic acid metabolism in human blood vessels. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2412–H2420. [Google Scholar] [CrossRef]
- Mabalirajan, U.; Rehman, R.; Ahmad, T.; Kumar, S.; Singh, S.; Leishangthem, G.D.; Aich, J.; Kumar, M.; Khanna, K.; Singh, V.P.; et al. Linoleic acid metabolite drives severe asthma by causing airway epithelial injury. Sci. Rep. 2013, 3, 1349. [Google Scholar] [CrossRef]
- Lynes, M.D.; Leiria, L.O.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, M.R.; Aoki, N.; Lefer, A.M.; Elisseou, E.M.; Zipkin, R.E. Direct cardiovascular actions of two metabolites of linoleic acid. Life Sci. 1990, 46, 427–433. [Google Scholar] [CrossRef]
- Sugiyama, S.; Hayakawa, M.; Nagai, S.; Ajioka, M.; Ozawa, T. Leukotoxin, 9, 10-epoxy-12-octadecenoate, causes cardiac failure in dogs. Life Sci. 1987, 40, 225–231. [Google Scholar] [CrossRef]
- Seubert, J.M.; Sinal, C.J.; Graves, J.; DeGraff, L.M.; Bradbury, J.A.; Lee, C.R.; Goralski, K.; Carey, M.A.; Luria, A.; Newman, J.W.; et al. Role of soluble epoxide hydrolase in post-ischemic recovery of heart contractile function. Circ. Res. 2006, 99, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Bannehr, M.; Lohr, L.; Gelep, J.; Haverkamp, W.; Schunck, W.H.; Gollasch, M.; Wutzler, A. Linoleic Acid Metabolite DiHOME Decreases Post-ischemic Cardiac Recovery in Murine Hearts. Cardiovasc. Toxicol. 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Motoki, A.; Merkel, M.J.; Packwood, W.H.; Cao, Z.; Liu, L.; Iliff, J.; Alkayed, N.J.; Van Winkle, D.M. Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2128–H2134. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef]
- Chaudhary, K.R.; Abukhashim, M.; Hwang, S.H.; Hammock, B.D.; Seubert, J.M. Inhibition of soluble epoxide hydrolase by trans-4- [4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is protective against ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 2010, 55, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Akhnokh, M.K.; Yang, F.H.; Samokhvalov, V.; Jamieson, K.L.; Cho, W.J.; Wagg, C.; Takawale, A.; Wang, X.; Lopaschuk, G.D.; Hammock, B.D.; et al. Inhibition of Soluble Epoxide Hydrolase Limits Mitochondrial Damage and Preserves Function Following Ischemic Injury. Front. Pharmacol. 2016, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.R.; Batchu, S.N.; Das, D.; Suresh, M.R.; Falck, J.R.; Graves, J.P.; Zeldin, D.C.; Seubert, J.M. Role of B-type natriuretic peptide in epoxyeicosatrienoic acid-mediated improved post-ischaemic recovery of heart contractile function. Cardiovasc. Res. 2009, 83, 362–370. [Google Scholar] [CrossRef]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and ischemia/reperfusion injury. Ann. N. Y. Acad. Sci. 2005, 1047, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Chattipakorn, S.C.; Chattipakorn, N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J. Cell. Mol. Med. 2017, 21, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Montfort, W.R. Properties and biological activities of thioredoxins. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 421–455. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Gao, E.; Bryan, N.S.; Qu, Y.; Liu, H.R.; Hu, A.; Christopher, T.A.; Lopez, B.L.; Yodoi, J.; Koch, W.J.; et al. Cardioprotective effects of thioredoxin in myocardial ischemia and reperfusion: Role of S-nitrosation [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 11471–11476. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Gao, E.; Hu, A.; Coletti, C.; Wang, Y.; Christopher, T.A.; Lopez, B.L.; Koch, W.; Ma, X.L. Thioredoxin reduces post-ischemic myocardial apoptosis by reducing oxidative/nitrative stress. Br. J. Pharmacol. 2006, 149, 311–318. [Google Scholar] [CrossRef]
- Tanaka, T.; Hosoi, F.; Yamaguchi-Iwai, Y.; Nakamura, H.; Masutani, H.; Ueda, S.; Nishiyama, A.; Takeda, S.; Wada, H.; Spyrou, G.; et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002, 21, 1695–1703. [Google Scholar] [CrossRef]
- Ozer, M.K.; Parlakpinar, H.; Cigremis, Y.; Ucar, M.; Vardi, N.; Acet, A. Ischemia-reperfusion leads to depletion of glutathione content and augmentation of malondialdehyde production in the rat heart from overproduction of oxidants: Can caffeic acid phenethyl ester (CAPE) protect the heart? Mol. Cell. Biochem. 2005, 273, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, V.; Jamieson, K.L.; Darwesh, A.M.; Keshavarz-Bahaghighat, H.; Lee, T.Y.T.; Edin, M.; Lih, F.; Zeldin, D.C.; Seubert, J.M. Deficiency of Soluble Epoxide Hydrolase Protects Cardiac Function Impaired by LPS-Induced Acute Inflammation. Front. Pharmacol. 2018, 9, 1572. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; De Keulenaer, G.W.; Lee, R.T. Vitamin D(3)-up-regulated protein-1 is a stress-responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J. Biol. Chem. 2002, 277, 26496–26500. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef]
- Shen, T.; Zheng, M.; Cao, C.; Chen, C.; Tang, J.; Zhang, W.; Cheng, H.; Chen, K.H.; Xiao, R.P. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 23354–23361. [Google Scholar] [CrossRef]
- Grant, D.F.; Storms, D.H.; Hammock, B.D. Molecular cloning and expression of murine liver soluble epoxide hydrolase. J. Biol. Chem. 1993, 268, 17628–17633. [Google Scholar] [PubMed]
- Knehr, M.; Thomas, H.; Arand, M.; Gebel, T.; Zeller, H.D.; Oesch, F. Isolation and characterization of a cDNA encoding rat liver cytosolic epoxide hydrolase and its functional expression in Escherichia coli. J. Biol. Chem. 1993, 268, 17623–17627. [Google Scholar] [PubMed]
- Beetham, J.K.; Tian, T.; Hammock, B.D. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch. Biochem. Biophys. 1993, 305, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Misawa, E.; Chan Kwo Chion, C.K.; Archer, I.V.; Woodland, M.P.; Zhou, N.Y.; Carter, S.F.; Widdowson, D.A.; Leak, D.J. Characterisation of a catabolic epoxide hydrolase from a Corynebacterium sp. Eur. J. Biochem. 1998, 253, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Rink, R.; Fennema, M.; Smids, M.; Dehmel, U.; Janssen, D.B. Primary structure and catalytic mechanism of the epoxide hydrolase from Agrobacterium radiobacter AD1. J. Biol. Chem. 1997, 272, 14650–14657. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, A.; Beetham, J.K.; Pinot, F.; Garbarino, J.E.; Rockhold, D.R.; Friedman, M.; Hammock, B.D.; Belknap, W.R. Cloning and expression of soluble epoxide hydrolase from potato. Plant J. 1994, 6, 251–258. [Google Scholar] [CrossRef]
- Kiyosue, T.; Beetham, J.K.; Pinot, F.; Hammock, B.D.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Characterization of an Arabidopsis cDNA for a soluble epoxide hydrolase gene that is inducible by auxin and water stress. Plant J. 1994, 6, 259–269. [Google Scholar] [CrossRef]
- Weintraub, N.L.; Fang, X.; Kaduce, T.L.; VanRollins, M.; Chatterjee, P.; Spector, A.A. Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporation into coronary endothelial phospholipids. Am. J. Physiol. 1999, 277, H2098–H2108. [Google Scholar] [CrossRef]
- Luria, A.; Weldon, S.M.; Kabcenell, A.K.; Ingraham, R.H.; Matera, D.; Jiang, H.; Gill, R.; Morisseau, C.; Newman, J.W.; Hammock, B.D. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J. Biol. Chem. 2007, 282, 2891–2898. [Google Scholar] [CrossRef]
- Williams, J.M.; Murphy, S.; Burke, M.; Roman, R.J. 20-hydroxyeicosatetraeonic acid: A new target for the treatment of hypertension. J. Cardiovasc. Pharmacol. 2010, 56, 336–344. [Google Scholar] [CrossRef]
- Imig, J.D. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc. Drug Rev. 2006, 24, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Marino, J.P., Jr. Soluble epoxide hydrolase, a target with multiple opportunities for cardiovascular drug discovery. Curr. Top. Med. Chem. 2009, 9, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.J.; Nithipatikom, K. Soluble epoxide hydrolase: A new target for cardioprotection. Curr. Opin. Investig. Drugs 2009, 10, 253–258. [Google Scholar] [PubMed]
- Batchu, S.N.; Lee, S.B.; Samokhvalov, V.; Chaudhary, K.R.; El-Sikhry, H.; Weldon, S.M.; Seubert, J.M. Novel soluble epoxide hydrolase inhibitor protects mitochondrial function following stress. Can. J. Physiol. Pharmacol. 2012, 90, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Seubert, J.; Yang, B.; Bradbury, J.A.; Graves, J.; Degraff, L.M.; Gabel, S.; Gooch, R.; Foley, J.; Newman, J.; Mao, L.; et al. Enhanced post-ischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ. Res. 2004, 95, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Seubert, J.M.; Zeldin, D.C.; Nithipatikom, K.; Gross, G.J. Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat. 2007, 82, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef]
- Schunck, W.H.; Konkel, A.; Fischer, R.; Weylandt, K.H. Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases. Pharmacol. Ther. 2018, 183, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Batchu, S.N.; Law, E.; Brocks, D.R.; Falck, J.R.; Seubert, J.M. Epoxyeicosatrienoic acid prevents post-ischemic electrocardiogram abnormalities in an isolated heart model. J. Mol. Cell. Cardiol. 2009, 46, 67–74. [Google Scholar] [CrossRef]
- Chaudhary, K.R.; Zordoky, B.N.; Edin, M.L.; Alsaleh, N.; El-Kadi, A.O.; Zeldin, D.C.; Seubert, J.M. Differential effects of soluble epoxide hydrolase inhibition and CYP2J2 overexpression on post-ischemic cardiac function in aged mice. Prostaglandins Other Lipid Mediat. 2013, 104–105, 8–17. [Google Scholar] [CrossRef]
- Batchu, S.N.; Lee, S.B.; Qadhi, R.S.; Chaudhary, K.R.; El-Sikhry, H.; Kodela, R.; Falck, J.R.; Seubert, J.M. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br. J. Pharmacol. 2011, 162, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.L.; Samokhvalov, V.; Akhnokh, M.K.; Lee, K.; Cho, W.J.; Takawale, A.; Wang, X.; Kassiri, Z.; Seubert, J.M. Genetic deletion of soluble epoxide hydrolase provides cardioprotective responses following myocardial infarction in aged mice. Prostaglandins Other Lipid Mediat. 2017, 132, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, J.Y.; Timofeyev, V.; Qiu, H.; Hwang, S.H.; Tuteja, D.; Lu, L.; Yang, J.; Mochida, H.; Low, R.; et al. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J. Mol. Cell. Cardiol. 2009, 47, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.F.; Grant, D.F.; Cheek, J.M.; Greene, J.F.; Williamson, K.C.; Hammock, B.D. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997, 3, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Dudda, A.; Spiteller, G.; Kobelt, F. Lipid oxidation products in ischemic porcine heart tissue. Chem. Phys. Lipids 1996, 82, 39–51. [Google Scholar] [CrossRef]
- Edin, M.L.; Wang, Z.; Bradbury, J.A.; Graves, J.P.; Lih, F.B.; DeGraff, L.M.; Foley, J.F.; Torphy, R.; Ronnekleiv, O.K.; Tomer, K.B.; et al. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011, 25, 3436–3447. [Google Scholar] [CrossRef] [PubMed]
- Harrell, M.D.; Stimers, J.R. Differential effects of linoleic Acid metabolites on cardiac sodium current. J. Pharmacol. Exp. Ther. 2002, 303, 347–355. [Google Scholar] [CrossRef]
- Sisemore, M.F.; Zheng, J.; Yang, J.C.; Thompson, D.A.; Plopper, C.G.; Cortopassi, G.A.; Hammock, B.D. Cellular characterization of leukotoxin diol-induced mitochondrial dysfunction. Arch. Biochem. Biophys. 2001, 392, 32–37. [Google Scholar] [CrossRef]
- Arslan, F.; de Kleijn, D.P.; Pasterkamp, G. Innate immune signaling in cardiac ischemia. Nat. Rev. Cardiol. 2011, 8, 292–300. [Google Scholar] [CrossRef]
- Marchant, D.J.; Boyd, J.H.; Lin, D.C.; Granville, D.J.; Garmaroudi, F.S.; McManus, B.M. Inflammation in myocardial diseases. Circ. Res. 2012, 110, 126–144. [Google Scholar] [CrossRef]
- Guo, Z.; Yu, S.; Chen, X.; Ye, R.; Zhu, W.; Liu, X. NLRP3 Is Involved in Ischemia/Reperfusion Injury. CNS Neurol. Disord. Drug Targets 2016, 15, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef]
- van Hout, G.P.; Arslan, F.; Pasterkamp, G.; Hoefer, I.E. Targeting danger-associated molecular patterns after myocardial infarction. Expert Opin. Ther. Targets 2016, 20, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Arena, R.A.; Toldo, S.; Mezzaroma, E.; Azam, T.; Seropian, I.M.; Shah, K.; Canada, J.; Voelkel, N.F.; Dinarello, C.A.; et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS ONE 2012, 7, e33438. [Google Scholar] [CrossRef]
- Kumar, A.; Thota, V.; Dee, L.; Olson, J.; Uretz, E.; Parrillo, J.E. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J. Exp. Med. 1996, 183, 949–958. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Seropian, I.M.; Toldo, S.; Mezzaroma, E.; Abbate, A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm. Res. 2013, 62, 637–640. [Google Scholar] [CrossRef]
- Cain, B.S.; Meldrum, D.R.; Dinarello, C.A.; Meng, X.; Joo, K.S.; Banerjee, A.; Harken, A.H. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit. Care Med. 1999, 27, 1309–1318. [Google Scholar] [CrossRef]
- Hosenpud, J.D.; Campbell, S.M.; Mendelson, D.J. Interleukin-1-induced myocardial depression in an isolated beating heart preparation. J. Heart Transplant. 1989, 8, 460–464. [Google Scholar] [PubMed]
- Ing, D.J.; Zang, J.; Dzau, V.J.; Webster, K.A.; Bishopric, N.H. Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bak, and Bcl-x. Circ. Res. 1999, 84, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Chen, W.; Su, Y.; Rai, V.; Uche, O.U.; Li, N.; Frangogiannis, N.G. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J. Immunol. 2013, 191, 4838–4848. [Google Scholar] [CrossRef] [PubMed]
- Orn, S.; Ueland, T.; Manhenke, C.; Sandanger, O.; Godang, K.; Yndestad, A.; Mollnes, T.E.; Dickstein, K.; Aukrust, P. Increased interleukin-1beta levels are associated with left ventricular hypertrophy and remodelling following acute ST segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J. Intern. Med. 2012, 272, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, X.; Mi, S.; Li, Z.; Wang, Y.; Zhu, H.; Sun, X.; Zhao, B.; Zhao, C.; Zou, Y.; et al. Naoxintong attenuates Ischaemia/reperfusion Injury through inhibiting NLRP3 inflammasome activation. J. Cell. Mol. Med. 2017, 21, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef]
- van Hout, G.P.; Bosch, L.; Ellenbroek, G.H.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.; Robertson, A.A.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef]
- Robinson, D.R.; Urakaze, M.; Huang, R.; Taki, H.; Sugiyama, E.; Knoell, C.T.; Xu, L.; Yeh, E.T.; Auron, P.E. Dietary marine lipids suppress continuous expression of interleukin-1 beta gene transcription. Lipids 1996, 31 (Suppl. 1), S23–S31. [Google Scholar] [CrossRef]
- Inceoglu, B.; Jinks, S.L.; Schmelzer, K.R.; Waite, T.; Kim, I.H.; Hammock, B.D. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006, 79, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Darwesh, A.M.; Sosnowski, D.K.; Lee, T.Y.; Keshavarz-Bahaghighat, H.; Seubert, J.M. Insights into the cardioprotective properties of n-3 PUFAs against ischemic heart disease via modulation of the innate immune system. Chem. Biol. Interact. 2019, 308, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, T.; Duan, J.X.; Li, P.; Sun, G.Y.; Liu, Y.P.; Zhang, J.; Dong, L.; Lee, K.S.S.; Hammock, B.D.; et al. Soluble Epoxide Hydrolase Inhibitor Attenuates Lipopolysaccharide-Induced Acute Lung Injury and Improves Survival in Mice. Shock 2017, 47, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Darwesh, A.M.; Jamieson, K.L.; Wang, C.; Samokhvalov, V.; Seubert, J.M. Cardioprotective effects of CYP-derived epoxy metabolites of docosahexaenoic acid involve limiting NLRP3 inflammasome activation (1). Can. J. Physiol. Pharmacol. 2019, 97, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Camara, A.K.; Stowe, D.F.; Hoppel, C.L.; Lesnefsky, E.J. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Physiol. Cell Physiol. 2007, 292, C137–C147. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Ji, L.; Xing, W.; Zhang, W.; Zhou, H.; Qian, X.; Wang, X.; Gao, F.; Sun, X.; Zhang, H. Acute hyperglycaemia enhances oxidative stress and aggravates myocardial ischaemia/reperfusion injury: Role of thioredoxin-interacting protein. J. Cell. Mol. Med. 2013, 17, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Al-Lamki, R.; Bai, L.; Streb, J.W.; Miano, J.M.; Bradley, J.; Min, W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 2004, 94, 1483–1491. [Google Scholar] [CrossRef]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef]
- Liu, F.C.; Tsai, H.I.; Yu, H.P. Organ-Protective Effects of Red Wine Extract, Resveratrol, in Oxidative Stress-Mediated Reperfusion Injury. Oxidative Med. Cell. Longev. 2015, 2015, 568634. [Google Scholar] [CrossRef]
- Yoshioka, J.; Chutkow, W.A.; Lee, S.; Kim, J.B.; Yan, J.; Tian, R.; Lindsey, M.L.; Feener, E.P.; Seidman, C.E.; Seidman, J.G.; et al. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J. Clin. Investig. 2012, 122, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W., II; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Xu, M.; Hao, H.; Jiang, L.; Wei, Y.; Zhou, F.; Sun, J.; Zhang, J.; Ji, H.; Wang, G.; Ju, W.; et al. Cardiotonic Pill Reduces Myocardial Ischemia-Reperfusion Injury via Increasing EET Concentrations in Rats. Drug Metab. Dispos. 2016, 44, 878–887. [Google Scholar] [CrossRef]
- Darwesh, A.M.; El-Azab, M.F.; Abo-Gresha, N.M.; El-Sayed, N.M.; Moustafa, Y.M. Cardioprotective Mechanisms of Exenatide in Isoprenaline-induced Myocardial Infarction: Novel Effects on Myocardial alpha-Estrogen Receptor Expression and IGF-1/IGF-2 System. J. Cardiovasc. Pharmacol. 2018, 71, 160–173. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef]
- Seubert, J.M.; Darmon, A.J.; El-Kadi, A.O.; D’Souza, S.J.; Bend, J.R. Apoptosis in murine hepatoma hepa 1c1c7 wild-type, C12, and C4 cells mediated by bilirubin. Mol. Pharmacol. 2002, 62, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Bjornstedt, M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995, 252, 199–208. [Google Scholar] [PubMed]
- Arner, E.S.; Holmgren, A. Measurement of thioredoxin and thioredoxin reductase. In Current Protocols in Toxicology; Wiley: Hoboken, NJ, USA, 2001; Chapter 7, Unit 7.4. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Basal Respiration (nmol O2/min/mg) | ADP-Stimulated (nmol O2/min/mg) | Respiratory Control Ratio (RCR) |
|---|---|---|---|
| WT Aerobic | 0.64 ± 0.12 | 3.36 ± 0.81 | 5.26 ± 0.49 |
| sEH null Aerobic | 0.58 ± 0.15 | 2.70 ± 0.71 | 4.90 ± 0.75 |
| WT IR | 0.75 ± 0.18 | 1.02 ± 0.21 | 1.43 ± 0.11 *☨ |
| sEH null IR | 0.97 ± 0.36 | 3.95 ± 1.37 | 4.03 ± 0.31 # |
| t-AUCB IR | 0.88 ± 0.18 | 2.92 ± 0.49 | 3.87 ± 0.59 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darwesh, A.M.; Keshavarz-Bahaghighat, H.; Jamieson, K.L.; Seubert, J.M. Genetic Deletion or Pharmacological Inhibition of Soluble Epoxide Hydrolase Ameliorates Cardiac Ischemia/Reperfusion Injury by Attenuating NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2019, 20, 3502. https://doi.org/10.3390/ijms20143502
Darwesh AM, Keshavarz-Bahaghighat H, Jamieson KL, Seubert JM. Genetic Deletion or Pharmacological Inhibition of Soluble Epoxide Hydrolase Ameliorates Cardiac Ischemia/Reperfusion Injury by Attenuating NLRP3 Inflammasome Activation. International Journal of Molecular Sciences. 2019; 20(14):3502. https://doi.org/10.3390/ijms20143502
Chicago/Turabian StyleDarwesh, Ahmed M., Hedieh Keshavarz-Bahaghighat, K. Lockhart Jamieson, and John M. Seubert. 2019. "Genetic Deletion or Pharmacological Inhibition of Soluble Epoxide Hydrolase Ameliorates Cardiac Ischemia/Reperfusion Injury by Attenuating NLRP3 Inflammasome Activation" International Journal of Molecular Sciences 20, no. 14: 3502. https://doi.org/10.3390/ijms20143502
APA StyleDarwesh, A. M., Keshavarz-Bahaghighat, H., Jamieson, K. L., & Seubert, J. M. (2019). Genetic Deletion or Pharmacological Inhibition of Soluble Epoxide Hydrolase Ameliorates Cardiac Ischemia/Reperfusion Injury by Attenuating NLRP3 Inflammasome Activation. International Journal of Molecular Sciences, 20(14), 3502. https://doi.org/10.3390/ijms20143502