Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis?



Abstract
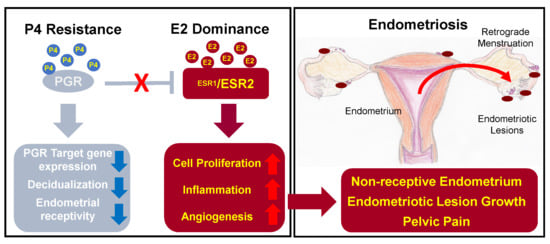
1. Introduction
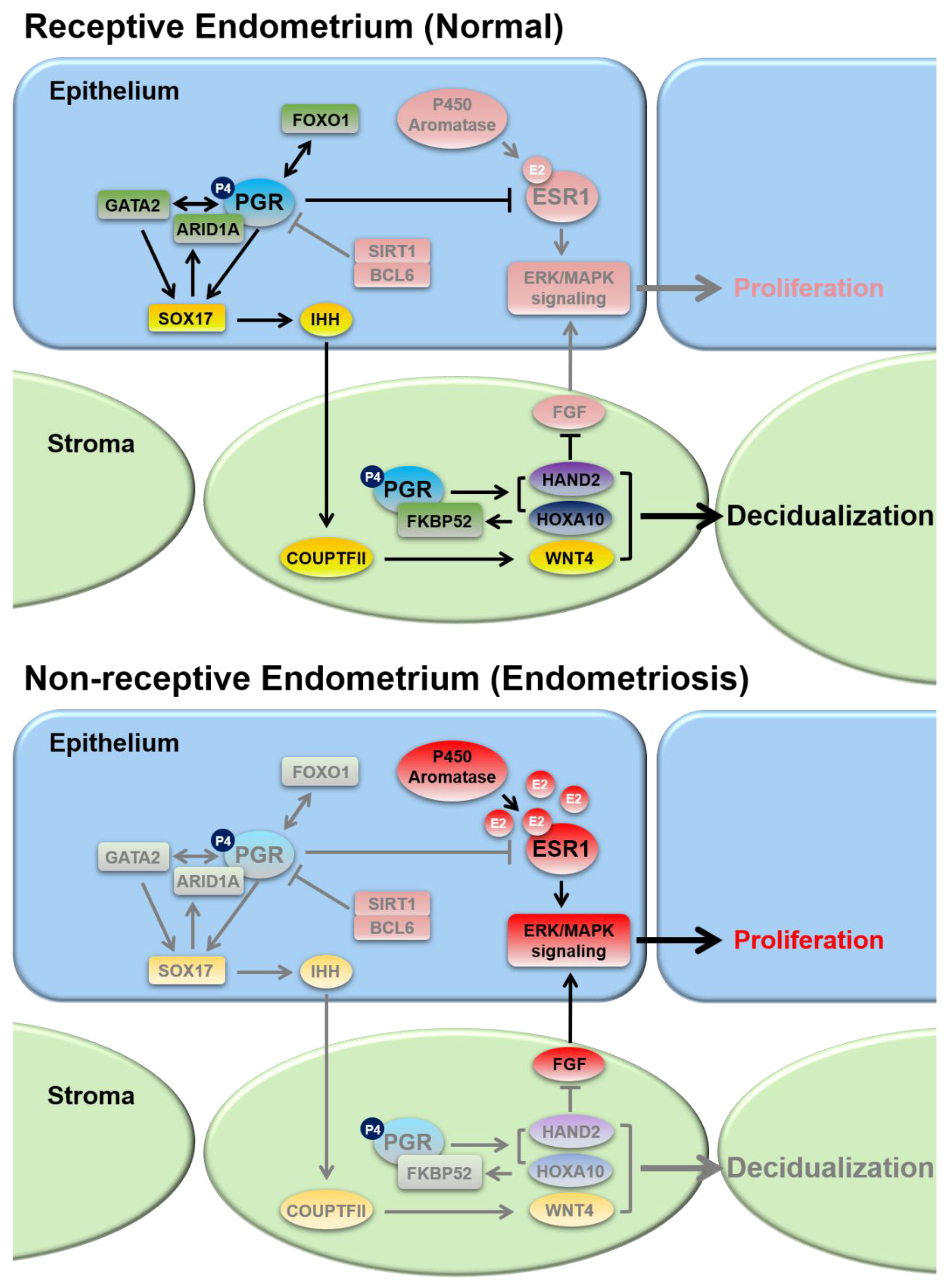
2. Steroid Hormone Regulation of Endometrial Function
2.1. Progesterone Receptors and Progesterone Signaling
2.2. Estrogen Receptors and Estrogen Signaling
2.3. Nuclear Receptor Coregulators in the Regulation of Progesterone and Estrogen Signaling
3. Dysregulation of Progesterone and Estrogen Signaling in Endometriosis
3.1. Progesterone Resistance
3.2. Estrogen Dominance and Inflammation
4. Pathologies Related to Steroid Hormone Signaling Dysregulation in Endometriosis
4.1. Infertility
4.2. Pelvic Pain
5. Hormone Therapies for Endometriosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cha, J.; Sun, X.; Dey, S.K. Mechanisms of implantation: Strategies for successful pregnancy. Nat. Med. 2012, 18, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, Y.M.; DeMayo, F.J. Role of nuclear receptors in blastocyst implantation. Semin. Cell Dev. Biol. 2013, 24, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Rubel, C.A.; Jeong, J.W.; Tsai, S.Y.; Lydon, J.P.; Demayo, F.J. Epithelial-stromal interaction and progesterone receptors in the mouse uterus. Semin. Reprod. Med. 2010, 28, 27–35. [Google Scholar] [CrossRef]
- Hantak, A.M.; Bagchi, I.C.; Bagchi, M.K. Role of uterine stromal-epithelial crosstalk in embryo implantation. Int. J. Dev. Biol. 2014, 58, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, S.P.; DeMayo, F.J. Hormone dependent uterine epithelial-stromal communication for pregnancy support. Placenta 2017, 60 (Suppl. 1), S20–S26. [Google Scholar] [CrossRef] [PubMed]
- Al-Sabbagh, M.; Lam, E.W.; Brosens, J.J. Mechanisms of endometrial progesterone resistance. Mol. Cell. Endocrinol. 2012, 358, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.G.; Rudnicki, M.; Yu, J.; Shu, Y.; Taylor, R.N. Progesterone resistance in endometriosis: Origins, consequences and interventions. Acta. Obstet. Gynecol. Scand. 2017, 96, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Vigano, P. Endometriosis. Nat. Rev. Dis. Primers 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Yilmaz, B.D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. Endometriosis. Endocr. Rev. 2019. [Google Scholar] [CrossRef]
- Wang, H.; Dey, S.K. Roadmap to embryo implantation: Clues from mouse models. Nat. Rev. Genet. 2006, 7, 185–199. [Google Scholar] [CrossRef]
- Large, M.J.; DeMayo, F.J. The regulation of embryo implantation and endometrial decidualization by progesterone receptor signaling. Mol. Cell. Endocrinol. 2012, 358, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Elguero, S.; Thakore, S.; Dahoud, W.; Bedaiwy, M.; Mesiano, S. Role of nuclear progesterone receptor isoforms in uterine pathophysiology. Hum. Reprod. Update 2015, 21, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Wetendorf, M.; DeMayo, F.J. Progesterone receptor signaling in the initiation of pregnancy and preservation of a healthy uterus. Int. J. Dev. Biol. 2014, 58, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.P.; Li, R.; DeMayo, F.J. Progesterone Receptor Regulation of Uterine Adaptation for Pregnancy. Trends Endocrinol. Metab. 2018, 29, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Krust, A.; Turcotte, B.; Stropp, U.; Tora, L.; Gronemeyer, H.; Chambon, P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. Embo J. 1990, 9, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Lydon, J.P.; DeMayo, F.J.; Funk, C.R.; Mani, S.K.; Hughes, A.R.; Montgomery, C.A., Jr.; Shyamala, G.; Conneely, O.M.; O’Malley, B.W. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995, 9, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Mulac-Jericevic, B.; Mullinax, R.A.; DeMayo, F.J.; Lydon, J.P.; Conneely, O.M. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science 2000, 289, 1751–1754. [Google Scholar] [CrossRef]
- Mulac-Jericevic, B.; Lydon, J.P.; DeMayo, F.J.; Conneely, O.M. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci. USA 2003, 100, 9744–9749. [Google Scholar] [CrossRef]
- Fleisch, M.C.; Chou, Y.C.; Cardiff, R.D.; Asaithambi, A.; Shyamala, G. Overexpression of progesterone receptor A isoform in mice leads to endometrial hyperproliferation, hyperplasia and atypia. Mol. Hum. Reprod. 2009, 15, 241–249. [Google Scholar] [CrossRef][Green Version]
- Wetendorf, M.; Wu, S.P.; Wang, X.; Creighton, C.J.; Wang, T.; Lanz, R.B.; Blok, L.; Tsai, S.Y.; Tsai, M.J.; Lydon, J.P.; et al. Decreased epithelial progesterone receptor A at the window of receptivity is required for preparation of the endometrium for embryo attachment. Biol. Reprod. 2017, 96, 313–326. [Google Scholar] [CrossRef]
- Kurita, T.; Lee, K.J.; Cooke, P.S.; Lydon, J.P.; Cunha, G.R. Paracrine regulation of epithelial progesterone receptor and lactoferrin by progesterone in the mouse uterus. Biol. Reprod. 2000, 62, 831–838. [Google Scholar] [CrossRef]
- Franco, H.L.; Rubel, C.A.; Large, M.J.; Wetendorf, M.; Fernandez-Valdivia, R.; Jeong, J.W.; Spencer, T.E.; Behringer, R.R.; Lydon, J.P.; Demayo, F.J. Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 2012, 26, 1218–1227. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, V.; Scott, M.P.; Ribon, V.; Sherman, L.; Anderson, S.M.; Maller, J.L.; Miller, W.T.; Edwards, D.P. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol. Cell 2001, 8, 269–280. [Google Scholar] [CrossRef]
- Vallejo, G.; La Greca, A.D.; Tarifa-Reischle, I.C.; Mestre-Citrinovitz, A.C.; Ballare, C.; Beato, M.; Saragueta, P. CDC2 mediates progestin initiated endometrial stromal cell proliferation: A PR signaling to gene expression independently of its binding to chromatin. PLoS ONE 2014, 9, e97311. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, K.Y.; Kwak, I.; White, L.D.; Hilsenbeck, S.G.; Lydon, J.P.; DeMayo, F.J. Identification of murine uterine genes regulated in a ligand-dependent manner by the progesterone receptor. Endocrinology 2005, 146, 3490–3505. [Google Scholar] [CrossRef]
- Rubel, C.A.; Lanz, R.B.; Kommagani, R.; Franco, H.L.; Lydon, J.P.; DeMayo, F.J. Research resource: Genome-wide profiling of progesterone receptor binding in the mouse uterus. Mol. Endocrinol. 2012, 26, 1428–1442. [Google Scholar] [CrossRef]
- Takamoto, N.; Zhao, B.; Tsai, S.Y.; DeMayo, F.J. Identification of Indian hedgehog as a progesterone-responsive gene in the murine uterus. Mol. Endocrinol. 2002, 16, 2338–2348. [Google Scholar] [CrossRef]
- Matsumoto, H.; Zhao, X.; Das, S.K.; Hogan, B.L.; Dey, S.K. Indian hedgehog as a progesterone-responsive factor mediating epithelial-mesenchymal interactions in the mouse uterus. Dev. Biol. 2002, 245, 280–290. [Google Scholar] [CrossRef]
- Lee, K.; Jeong, J.; Kwak, I.; Yu, C.T.; Lanske, B.; Soegiarto, D.W.; Toftgard, R.; Tsai, M.J.; Tsai, S.; Lydon, J.P.; et al. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat. Genet. 2006, 38, 1204–1209. [Google Scholar] [CrossRef]
- Kurihara, I.; Lee, D.K.; Petit, F.G.; Jeong, J.; Lee, K.; Lydon, J.P.; DeMayo, F.J.; Tsai, M.J.; Tsai, S.Y. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007, 3, e102. [Google Scholar] [CrossRef]
- Lee, D.K.; Kurihara, I.; Jeong, J.W.; Lydon, J.P.; DeMayo, F.J.; Tsai, M.J.; Tsai, S.Y. Suppression of ERalpha activity by COUP-TFII is essential for successful implantation and decidualization. Mol. Endocrinol. 2010, 24, 930–940. [Google Scholar] [CrossRef]
- Lee, K.Y.; Jeong, J.W.; Wang, J.; Ma, L.; Martin, J.F.; Tsai, S.Y.; Lydon, J.P.; DeMayo, F.J. Bmp2 is critical for the murine uterine decidual response. Mol. Cell. Biol. 2007, 27, 5468–5478. [Google Scholar] [CrossRef]
- Li, Q.; Kannan, A.; Wang, W.; Demayo, F.J.; Taylor, R.N.; Bagchi, M.K.; Bagchi, I.C. Bone morphogenetic protein 2 functions via a conserved signaling pathway involving Wnt4 to regulate uterine decidualization in the mouse and the human. J. Biol. Chem. 2007, 282, 31725–31732. [Google Scholar] [CrossRef]
- Franco, H.L.; Dai, D.; Lee, K.Y.; Rubel, C.A.; Roop, D.; Boerboom, D.; Jeong, J.W.; Lydon, J.P.; Bagchi, I.C.; Bagchi, M.K.; et al. WNT4 is a key regulator of normal postnatal uterine development and progesterone signaling during embryo implantation and decidualization in the mouse. FASEB J. 2011, 25, 1176–1187. [Google Scholar] [CrossRef]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, H.S.; Franco, H.L.; Broaddus, R.R.; Taketo, M.M.; Tsai, S.Y.; Lydon, J.P.; DeMayo, F.J. beta-catenin mediates glandular formation and dysregulation of beta-catenin induces hyperplasia formation in the murine uterus. Oncogene 2009, 28, 31–40. [Google Scholar] [CrossRef]
- Zhang, L.; Patterson, A.L.; Zhang, L.; Teixeira, J.M.; Pru, J.K. Endometrial stromal beta-catenin is required for steroid-dependent mesenchymal-epithelial cross talk and decidualization. Reprod. Biol. Endocrinol 2012, 10, 75. [Google Scholar] [CrossRef]
- Benson, G.V.; Lim, H.; Paria, B.C.; Satokata, I.; Dey, S.K.; Maas, R.L. Mechanisms of reduced fertility in Hoxa-10 mutant mice: Uterine homeosis and loss of maternal Hoxa-10 expression. Development 1996, 122, 2687–2696. [Google Scholar]
- Lim, H.; Ma, L.; Ma, W.G.; Maas, R.L.; Dey, S.K. Hoxa-10 regulates uterine stromal cell responsiveness to progesterone during implantation and decidualization in the mouse. Mol. Endocrinol. 1999, 13, 1005–1017. [Google Scholar] [CrossRef]
- Daikoku, T.; Song, H.; Guo, Y.; Riesewijk, A.; Mosselman, S.; Das, S.K.; Dey, S.K. Uterine Msx-1 and Wnt4 signaling becomes aberrant in mice with the loss of leukemia inhibitory factor or Hoxa-10: Evidence for a novel cytokine-homeobox-Wnt signaling in implantation. Mol. Endocrinol. 2004, 18, 1238–1250. [Google Scholar] [CrossRef]
- Li, Q.; Kannan, A.; DeMayo, F.J.; Lydon, J.P.; Cooke, P.S.; Yamagishi, H.; Srivastava, D.; Bagchi, M.K.; Bagchi, I.C. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science 2011, 331, 912–916. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Huyen, D.V.; Bany, B.M. Evidence for a conserved function of heart and neural crest derivatives expressed transcript 2 in mouse and human decidualization. Reproduction 2011, 142, 353–368. [Google Scholar] [CrossRef]
- Gao, J.; Mazella, J.; Suwanichkul, A.; Powell, D.R.; Tseng, L. Activation of the insulin-like growth factor binding protein-1 promoter by progesterone receptor in decidualized human endometrial stromal cells. Mol. Cell. Endocrinol. 1999, 153, 11–17. [Google Scholar] [CrossRef]
- Mantena, S.R.; Kannan, A.; Cheon, Y.P.; Li, Q.; Johnson, P.F.; Bagchi, I.C.; Bagchi, M.K. C/EBPbeta is a critical mediator of steroid hormone-regulated cell proliferation and differentiation in the uterine epithelium and stroma. Proc. Natl. Acad. Sci. USA 2006, 103, 1870–1875. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Bagchi, I.C.; Bagchi, M.K. The CCAAT/enhancer binding protein beta is a critical regulator of steroid-induced mitotic expansion of uterine stromal cells during decidualization. Endocrinology 2010, 151, 3929–3940. [Google Scholar] [CrossRef]
- Kommagani, R.; Szwarc, M.M.; Vasquez, Y.M.; Peavey, M.C.; Mazur, E.C.; Gibbons, W.E.; Lanz, R.B.; DeMayo, F.J.; Lydon, J.P. The Promyelocytic Leukemia Zinc Finger Transcription Factor Is Critical for Human Endometrial Stromal Cell Decidualization. PLoS Genet. 2016, 12, e1005937. [Google Scholar] [CrossRef]
- Fahnenstich, J.; Nandy, A.; Milde-Langosch, K.; Schneider-Merck, T.; Walther, N.; Gellersen, B. Promyelocytic leukaemia zinc finger protein (PLZF) is a glucocorticoid- and progesterone-induced transcription factor in human endometrial stromal cells and myometrial smooth muscle cells. Mol. Hum. Reprod. 2003, 9, 611–623. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, H.S.; Lee, K.Y.; White, L.D.; Broaddus, R.R.; Zhang, Y.W.; Vande Woude, G.F.; Giudice, L.C.; Young, S.L.; Lessey, B.A.; et al. Mig-6 modulates uterine steroid hormone responsiveness and exhibits altered expression in endometrial disease. Proc. Natl. Acad. Sci. USA 2009, 106, 8677–8682. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, T.H.; Lee, J.H.; Dunwoodie, S.L.; Ku, B.J.; Jeong, J.W. Mig-6 regulates endometrial genes involved in cell cycle and progesterone signaling. Biochem. Biophys. Res. Commun. 2015, 462, 409–414. [Google Scholar] [CrossRef][Green Version]
- Yoo, J.Y.; Shin, H.; Kim, T.H.; Choi, W.S.; Ferguson, S.D.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; Ha, U.H.; Jeong, J.W. CRISPLD2 is a target of progesterone receptor and its expression is decreased in women with endometriosis. PLoS ONE 2014, 9, e100481. [Google Scholar] [CrossRef]
- Brosens, J.J.; Gellersen, B. Death or survival--progesterone-dependent cell fate decisions in the human endometrial stroma. J. Mol. Endocrinol. 2006, 36, 389–398. [Google Scholar] [CrossRef]
- Takano, M.; Lu, Z.; Goto, T.; Fusi, L.; Higham, J.; Francis, J.; Withey, A.; Hardt, J.; Cloke, B.; Stavropoulou, A.V.; et al. Transcriptional cross talk between the forkhead transcription factor forkhead box O1A and the progesterone receptor coordinates cell cycle regulation and differentiation in human endometrial stromal cells. Mol. Endocrinol. 2007, 21, 2334–2349. [Google Scholar] [CrossRef]
- Vasquez, Y.M.; Mazur, E.C.; Li, X.; Kommagani, R.; Jiang, L.; Chen, R.; Lanz, R.B.; Kovanci, E.; Gibbons, W.E.; DeMayo, F.J. FOXO1 is required for binding of PR on IRF4, novel transcriptional regulator of endometrial stromal decidualization. Mol. Endocrinol. 2015, 29, 421–433. [Google Scholar] [CrossRef]
- Vasquez, Y.M.; Wang, X.; Wetendorf, M.; Franco, H.L.; Mo, Q.; Wang, T.; Lanz, R.B.; Young, S.L.; Lessey, B.A.; Spencer, T.E.; et al. FOXO1 regulates uterine epithelial integrity and progesterone receptor expression critical for embryo implantation. PLoS Genet. 2018, 14, e1007787. [Google Scholar] [CrossRef]
- Barent, R.L.; Nair, S.C.; Carr, D.C.; Ruan, Y.; Rimerman, R.A.; Fulton, J.; Zhang, Y.; Smith, D.F. Analysis of FKBP51/FKBP52 chimeras and mutants for Hsp90 binding and association with progesterone receptor complexes. Mol. Endocrinol. 1998, 12, 342–354. [Google Scholar] [CrossRef]
- Tranguch, S.; Cheung-Flynn, J.; Daikoku, T.; Prapapanich, V.; Cox, M.B.; Xie, H.; Wang, H.; Das, S.K.; Smith, D.F.; Dey, S.K. Cochaperone immunophilin FKBP52 is critical to uterine receptivity for embryo implantation. Proc. Natl. Acad. Sci. USA 2005, 102, 14326–14331. [Google Scholar] [CrossRef]
- Tranguch, S.; Wang, H.; Daikoku, T.; Xie, H.; Smith, D.F.; Dey, S.K. FKBP52 deficiency-conferred uterine progesterone resistance is genetic background and pregnancy stage specific. J. Clin. Investig. 2007, 117, 1824–1834. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, Y.; Edelshain, B.; Schatz, F.; Lockwood, C.J.; Taylor, H.S. FKBP4 is regulated by HOXA10 during decidualization and in endometriosis. Reproduction 2012, 143, 531–538. [Google Scholar] [CrossRef]
- Cheng, J.G.; Chen, J.R.; Hernandez, L.; Alvord, W.G.; Stewart, C.L. Dual control of LIF expression and LIF receptor function regulate Stat3 activation at the onset of uterine receptivity and embryo implantation. Proc. Natl. Acad. Sci. USA 2001, 98, 8680–8685. [Google Scholar] [CrossRef]
- Catalano, R.D.; Johnson, M.H.; Campbell, E.A.; Charnock-Jones, D.S.; Smith, S.K.; Sharkey, A.M. Inhibition of Stat3 activation in the endometrium prevents implantation: A nonsteroidal approach to contraception. Proc. Natl. Acad. Sci. USA 2005, 102, 8585–8590. [Google Scholar] [CrossRef]
- Sun, X.; Bartos, A.; Whitsett, J.A.; Dey, S.K. Uterine deletion of Gp130 or Stat3 shows implantation failure with increased estrogenic responses. Mol. Endocrinol. 2013, 27, 1492–1501. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, T.H.; Oh, S.J.; Yoo, J.Y.; Akira, S.; Ku, B.J.; Lydon, J.P.; Jeong, J.W. Signal transducer and activator of transcription-3 (Stat3) plays a critical role in implantation via progesterone receptor in uterus. FASEB J. 2013, 27, 2553–2563. [Google Scholar] [CrossRef]
- Rubel, C.A.; Franco, H.L.; Jeong, J.W.; Lydon, J.P.; DeMayo, F.J. GATA2 is expressed at critical times in the mouse uterus during pregnancy. Gene Expr. Patterns 2012, 12, 196–203. [Google Scholar] [CrossRef]
- Rubel, C.A.; Wu, S.P.; Lin, L.; Wang, T.; Lanz, R.B.; Li, X.; Kommagani, R.; Franco, H.L.; Camper, S.A.; Tong, Q.; et al. A Gata2-Dependent Transcription Network Regulates Uterine Progesterone Responsiveness and Endometrial Function. Cell Rep. 2016, 17, 1414–1425. [Google Scholar] [CrossRef]
- Hirate, Y.; Suzuki, H.; Kawasumi, M.; Takase, H.M.; Igarashi, H.; Naquet, P.; Kanai, Y.; Kanai-Azuma, M. Mouse Sox17 haploinsufficiency leads to female subfertility due to impaired implantation. Sci. Rep. 2016, 6, 24171. [Google Scholar] [CrossRef]
- Guimaraes-Young, A.; Neff, T.; Dupuy, A.J.; Goodheart, M.J. Conditional deletion of Sox17 reveals complex effects on uterine adenogenesis and function. Dev. Biol. 2016, 414, 219–227. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Wang, T.; Wu, S.P.; Jeong, J.W.; Kim, T.H.; Young, S.L.; Lessey, B.A.; Lanz, R.B.; Lydon, J.P.; et al. SOX17 regulates uterine epithelial-stromal cross-talk acting via a distal enhancer upstream of Ihh. Nat. Commun. 2018, 9, 4421. [Google Scholar] [CrossRef]
- Kim, T.H.; Yoo, J.Y.; Wang, Z.; Lydon, J.P.; Khatri, S.; Hawkins, S.M.; Leach, R.E.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; et al. ARID1A Is Essential for Endometrial Function during Early Pregnancy. PLoS Genet. 2015, 11, e1005537. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Korach, K.S. Estrogen Receptors: New Directions in the New Millennium. Endocr. Rev. 2018, 39, 664–675. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71. [Google Scholar] [CrossRef]
- Stefkovich, M.L.; Arao, Y.; Hamilton, K.J.; Korach, K.S. Experimental models for evaluating non-genomic estrogen signaling. Steroids 2018, 133, 34–37. [Google Scholar] [CrossRef]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; de Falco, A.; Bontempo, P.; Nola, E.; Auricchio, F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996, 15, 1292–1300. [Google Scholar] [CrossRef]
- O’Brien, J.E.; Peterson, T.J.; Tong, M.H.; Lee, E.J.; Pfaff, L.E.; Hewitt, S.C.; Korach, K.S.; Weiss, J.; Jameson, J.L. Estrogen-induced proliferation of uterine epithelial cells is independent of estrogen receptor alpha binding to classical estrogen response elements. J. Biol. Chem. 2006, 281, 26683–26692. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, T.H.; Lee, J.H.; Oh, S.J.; Yoo, J.Y.; Kwon, H.S.; Kim, Y.I.; Ferguson, S.D.; Ahn, J.Y.; Ku, B.J.; et al. Extracellular signal-regulated kinase 1/2 signaling pathway is required for endometrial decidualization in mice and human. PLoS ONE 2013, 8, e75282. [Google Scholar] [CrossRef]
- Krege, J.H.; Hodgin, J.B.; Couse, J.F.; Enmark, E.; Warner, M.; Mahler, J.F.; Sar, M.; Korach, K.S.; Gustafsson, J.A.; Smithies, O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc. Natl. Acad. Sci. USA 1998, 95, 15677–15682. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Deroo, B.J.; Hansen, K.; Collins, J.; Grissom, S.; Afshari, C.A.; Korach, K.S. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol. Endocrinol. 2003, 17, 2070–2083. [Google Scholar] [CrossRef]
- Wada-Hiraike, O.; Hiraike, H.; Okinaga, H.; Imamov, O.; Barros, R.P.; Morani, A.; Omoto, Y.; Warner, M.; Gustafsson, J.A. Role of estrogen receptor beta in uterine stroma and epithelium: Insights from estrogen receptor beta-/- mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18350–18355. [Google Scholar] [CrossRef]
- Lubahn, D.B.; Moyer, J.S.; Golding, T.S.; Couse, J.F.; Korach, K.S.; Smithies, O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. USA 1993, 90, 11162–11166. [Google Scholar] [CrossRef]
- Curtis, S.W.; Clark, J.; Myers, P.; Korach, K.S. Disruption of estrogen signaling does not prevent progesterone action in the estrogen receptor alpha knockout mouse uterus. Proc Natl Acad Sci USA 1999, 96, 3646–3651. [Google Scholar] [CrossRef]
- Curtis Hewitt, S.; Goulding, E.H.; Eddy, E.M.; Korach, K.S. Studies using the estrogen receptor alpha knockout uterus demonstrate that implantation but not decidualization-associated signaling is estrogen dependent. Biol. Reprod. 2002, 67, 1268–1277. [Google Scholar] [CrossRef]
- Couse, J.F.; Curtis, S.W.; Washburn, T.F.; Lindzey, J.; Golding, T.S.; Lubahn, D.B.; Smithies, O.; Korach, K.S. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol. Endocrinol. 1995, 9, 1441–1454. [Google Scholar]
- Pawar, S.; Laws, M.J.; Bagchi, I.C.; Bagchi, M.K. Uterine Epithelial Estrogen Receptor-alpha Controls Decidualization via a Paracrine Mechanism. Mol. Endocrinol. 2015, 29, 1362–1374. [Google Scholar] [CrossRef]
- Winuthayanon, W.; Hewitt, S.C.; Orvis, G.D.; Behringer, R.R.; Korach, K.S. Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc. Natl. Acad. Sci. USA 2010, 107, 19272–19277. [Google Scholar] [CrossRef]
- Cooke, P.S.; Buchanan, D.L.; Young, P.; Setiawan, T.; Brody, J.; Korach, K.S.; Taylor, J.; Lubahn, D.B.; Cunha, G.R. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc. Natl. Acad. Sci. USA 1997, 94, 6535–6540. [Google Scholar] [CrossRef]
- Winuthayanon, W.; Lierz, S.L.; Delarosa, K.C.; Sampels, S.R.; Donoghue, L.J.; Hewitt, S.C.; Korach, K.S. Juxtacrine Activity of Estrogen Receptor alpha in Uterine Stromal Cells is Necessary for Estrogen-Induced Epithelial Cell Proliferation. Sci. Rep. 2017, 7, 8377. [Google Scholar] [CrossRef]
- Zhu, L.; Pollard, J.W. Estradiol-17beta regulates mouse uterine epithelial cell proliferation through insulin-like growth factor 1 signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 15847–15851. [Google Scholar] [CrossRef]
- Adesanya, O.O.; Zhou, J.; Samathanam, C.; Powell-Braxton, L.; Bondy, C.A. Insulin-like growth factor 1 is required for G2 progression in the estradiol-induced mitotic cycle. Proc. Natl. Acad. Sci. USA 1999, 96, 3287–3291. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Lierz, S.L.; Garcia, M.; Hamilton, K.J.; Gruzdev, A.; Grimm, S.A.; Lydon, J.P.; DeMayo, F.J.; Korach, K.S. A distal super enhancer mediates estrogen-dependent mouse uterine-specific gene transcription of Insulin-like growth factor 1 (Igf1). J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Li, L.; Grimm, S.A.; Chen, Y.; Liu, L.; Li, Y.; Bushel, P.R.; Fargo, D.; Korach, K.S. Research resource: Whole-genome estrogen receptor alpha binding in mouse uterine tissue revealed by ChIP-seq. Mol. Endocrinol. 2012, 26, 887–898. [Google Scholar] [CrossRef]
- Klotz, D.M.; Hewitt, S.C.; Ciana, P.; Raviscioni, M.; Lindzey, J.K.; Foley, J.; Maggi, A.; DiAugustine, R.P.; Korach, K.S. Requirement of estrogen receptor-alpha in insulin-like growth factor-1 (IGF-1)-induced uterine responses and in vivo evidence for IGF-1/estrogen receptor cross-talk. J. Biol. Chem. 2002, 277, 8531–8537. [Google Scholar] [CrossRef]
- Richards, R.G.; Walker, M.P.; Sebastian, J.; DiAugustine, R.P. Insulin-like growth factor-1 (IGF-1) receptor-insulin receptor substrate complexes in the uterus. Altered signaling response to estradiol in the IGF-1(m/m) mouse. J. Biol. Chem. 1998, 273, 11962–11969. [Google Scholar] [CrossRef]
- Tsai, S.J.; Wu, M.H.; Chen, H.M.; Chuang, P.C.; Wing, L.Y. Fibroblast growth factor-9 is an endometrial stromal growth factor. Endocrinology 2002, 143, 2715–2721. [Google Scholar] [CrossRef]
- Surveyor, G.A.; Gendler, S.J.; Pemberton, L.; Das, S.K.; Chakraborty, I.; Julian, J.; Pimental, R.A.; Wegner, C.C.; Dey, S.K.; Carson, D.D. Expression and steroid hormonal control of Muc-1 in the mouse uterus. Endocrinology 1995, 136, 3639–3647. [Google Scholar] [CrossRef]
- Rosario, G.X.; Stewart, C.L. The Multifaceted Actions of Leukaemia Inhibitory Factor in Mediating Uterine Receptivity and Embryo Implantation. Am. J. Reprod. Immunol. 2016, 75, 246–255. [Google Scholar] [CrossRef]
- Stewart, C.L.; Kaspar, P.; Brunet, L.J.; Bhatt, H.; Gadi, I.; Kontgen, F.; Abbondanzo, S.J. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 1992, 359, 76–79. [Google Scholar] [CrossRef]
- Chen, J.R.; Cheng, J.G.; Shatzer, T.; Sewell, L.; Hernandez, L.; Stewart, C.L. Leukemia inhibitory factor can substitute for nidatory estrogen and is essential to inducing a receptive uterus for implantation but is not essential for subsequent embryogenesis. Endocrinology 2000, 141, 4365–4372. [Google Scholar] [CrossRef]
- Song, H.; Lim, H. Evidence for heterodimeric association of leukemia inhibitory factor (LIF) receptor and gp130 in the mouse uterus for LIF signaling during blastocyst implantation. Reproduction 2006, 131, 341–349. [Google Scholar] [CrossRef]
- Liang, X.H.; Deng, W.B.; Li, M.; Zhao, Z.A.; Wang, T.S.; Feng, X.H.; Cao, Y.J.; Duan, E.K.; Yang, Z.M. Egr1 protein acts downstream of estrogen-leukemia inhibitory factor (LIF)-STAT3 pathway and plays a role during implantation through targeting Wnt4. J. Biol. Chem. 2014, 289, 23534–23545. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, Y.S.; Yoon, J.A.; Lyu, S.W.; Shin, H.; Lim, H.J.; Hong, S.H.; Lee, D.R.; Song, H. Egr1 is rapidly and transiently induced by estrogen and bisphenol A via activation of nuclear estrogen receptor-dependent ERK1/2 pathway in the uterus. Reprod. Toxicol. 2014, 50, 60–67. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, Y.S.; Yoon, J.A.; Yang, S.C.; Park, M.; Seol, D.W.; Lyu, S.W.; Jun, J.H.; Lim, H.J.; Lee, D.R.; et al. Estrogen induces EGR1 to fine-tune its actions on uterine epithelium by controlling PR signaling for successful embryo implantation. FASEB J. 2018, 32, 1184–1195. [Google Scholar] [CrossRef]
- Park, M.; Kim, H.R.; Kim, Y.S.; Yang, S.C.; Yoon, J.A.; Lyu, S.W.; Lim, H.J.; Hong, S.H.; Song, H. Estrogen-induced transcription factor EGR1 regulates c-Kit transcription in the mouse uterus to maintain uterine receptivity for embryo implantation. Mol. Cell. Endocrinol. 2018, 470, 75–83. [Google Scholar] [CrossRef]
- Szwarc, M.M.; Hai, L.; Gibbons, W.E.; Mo, Q.; Lanz, R.B.; DeMayo, F.J.; Lydon, J.P. Early growth response 1 transcriptionally primes the human endometrial stromal cell for decidualization. J. Steroid Biochem. Mol. Biol. 2019, 189, 283–290. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W. An evolving understanding of nuclear receptor coregulator proteins. J. Mol. Endocrinol. 2013, 51, T23–T36. [Google Scholar] [CrossRef]
- Szwarc, M.M.; Lydon, J.P.; O’Malley, B.W. Steroid receptor coactivators as therapeutic targets in the female reproductive system. J. Steroid Biochem. Mol. Biol. 2015, 154, 32–38. [Google Scholar] [CrossRef]
- Xu, J.; Qiu, Y.; DeMayo, F.J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 1998, 279, 1922–1925. [Google Scholar] [CrossRef]
- Mukherjee, A.; Soyal, S.M.; Fernandez-Valdivia, R.; Gehin, M.; Chambon, P.; Demayo, F.J.; Lydon, J.P.; O’Malley, B.W. Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol. Cell. Biol. 2006, 26, 6571–6583. [Google Scholar] [CrossRef]
- Xu, J.; Liao, L.; Ning, G.; Yoshida-Komiya, H.; Deng, C.; O’Malley, B.W. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. USA 2000, 97, 6379–6384. [Google Scholar] [CrossRef]
- Han, S.J.; DeMayo, F.J.; Xu, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Steroid receptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol. Endocrinol. 2006, 20, 45–55. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, K.Y.; Han, S.J.; Aronow, B.J.; Lydon, J.P.; O’Malley, B.W.; DeMayo, F.J. The p160 steroid receptor coactivator 2, SRC-2, regulates murine endometrial function and regulates progesterone-independent and -dependent gene expression. Endocrinology 2007, 148, 4238–4250. [Google Scholar] [CrossRef]
- Han, S.J.; Jeong, J.; Demayo, F.J.; Xu, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Dynamic cell type specificity of SRC-1 coactivator in modulating uterine progesterone receptor function in mice. Mol. Cell. Biol. 2005, 25, 8150–8165. [Google Scholar] [CrossRef]
- Gregory, C.W.; Wilson, E.M.; Apparao, K.B.; Lininger, R.A.; Meyer, W.R.; Kowalik, A.; Fritz, M.A.; Lessey, B.A. Steroid receptor coactivator expression throughout the menstrual cycle in normal and abnormal endometrium. J. Clin. Endocrinol. Metab. 2002, 87, 2960–2966. [Google Scholar] [CrossRef]
- Balmer, N.N.; Richer, J.K.; Spoelstra, N.S.; Torkko, K.C.; Lyle, P.L.; Singh, M. Steroid receptor coactivator AIB1 in endometrial carcinoma, hyperplasia and normal endometrium: Correlation with clinicopathologic parameters and biomarkers. Mod. Pathol. 2006, 19, 1593–1605. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Fujimoto, J.; Sun, W.S.; Tamaya, T. Clinical implications of steroid receptor coactivator (SRC)-3 in uterine endometrial cancers. J. Steroid Biochem. Mol. Biol. 2007, 104, 237–240. [Google Scholar] [CrossRef]
- Kommagani, R.; Szwarc, M.M.; Kovanci, E.; Gibbons, W.E.; Putluri, N.; Maity, S.; Creighton, C.J.; Sreekumar, A.; DeMayo, F.J.; Lydon, J.P.; et al. Acceleration of the glycolytic flux by steroid receptor coactivator-2 is essential for endometrial decidualization. PLoS Genet. 2013, 9, e1003900. [Google Scholar] [CrossRef]
- Szwarc, M.M.; Hai, L.; Gibbons, W.E.; White, L.D.; Mo, Q.; Kommagani, R.; Lanz, R.B.; DeMayo, F.J.; O’Malley, B.W.; Lydon, J.P. Retinoid signaling controlled by SRC-2 in decidualization revealed by transcriptomics. Reproduction 2018, 156, 387–395. [Google Scholar] [CrossRef]
- Grimaldi, G.; Christian, M.; Steel, J.H.; Henriet, P.; Poutanen, M.; Brosens, J.J. Down-regulation of the histone methyltransferase EZH2 contributes to the epigenetic programming of decidualizing human endometrial stromal cells. Mol. Endocrinol. 2011, 25, 1892–1903. [Google Scholar] [CrossRef]
- Nanjappa, M.K.; Mesa, A.M.; Medrano, T.I.; Jefferson, W.N.; DeMayo, F.J.; Williams, C.J.; Lydon, J.P.; Levin, E.R.; Cooke, P.S. The histone methyltransferase EZH2 is required for normal uterine development and function in mice. Biol. Reprod. 2019. [Google Scholar] [CrossRef]
- Kim, T.H.; Yoo, J.Y.; Choi, K.C.; Shin, J.H.; Leach, R.E.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; Yoon, H.G.; Jeong, J.W. Loss of HDAC3 results in nonreceptive endometrium and female infertility. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Parasar, P.; Ozcan, P.; Terry, K.L. Endometriosis: Epidemiology, Diagnosis and Clinical Management. Curr. Obstet. Gynecol. Rep. 2017, 6, 34–41. [Google Scholar] [CrossRef]
- Attia, G.R.; Zeitoun, K.; Edwards, D.; Johns, A.; Carr, B.R.; Bulun, S.E. Progesterone receptor isoform A but not B is expressed in endometriosis. J. Clin. Endocrinol. Metab. 2000, 85, 2897–2902. [Google Scholar] [CrossRef]
- Yin, X.; Pavone, M.E.; Lu, Z.; Wei, J.; Kim, J.J. Increased activation of the PI3K/AKT pathway compromises decidualization of stromal cells from endometriosis. J. Clin. Endocrinol. Metab. 2012, 97, E35–E43. [Google Scholar] [CrossRef]
- Prentice, A.; Randall, B.J.; Weddell, A.; McGill, A.; Henry, L.; Horne, C.H.; Thomas, E.J. Ovarian steroid receptor expression in endometriosis and in two potential parent epithelia: Endometrium and peritoneal mesothelium. Hum. Reprod. 1992, 7, 1318–1325. [Google Scholar] [CrossRef]
- Colon-Caraballo, M.; Garcia, M.; Mendoza, A.; Flores, I. Human Endometriosis Tissue Microarray Reveals Site-specific Expression of Estrogen Receptors, Progesterone Receptor, and Ki67. Appl. Immunohistochem. Mol. Morphol. 2018. [Google Scholar] [CrossRef]
- Mousazadeh, S.; Ghaheri, A.; Shahhoseini, M.; Aflatoonian, R.; Afsharian, P. The Effect of Imbalanced Progesterone Receptor-A/-B Ratio on Gelatinase Expressions in Endometriosis. Int. J. Fertil. Steril. 2019, 13, 127–134. [Google Scholar]
- Brown, D.M.; Lee, H.C.; Liu, S.; Quick, C.M.; Fernandes, L.M.; Simmen, F.A.; Tsai, S.J.; Simmen, R.C.M. Notch-1 Signaling Activation and Progesterone Receptor Expression in Ectopic Lesions of Women With Endometriosis. J. Endocr. Soc. 2018, 2, 765–778. [Google Scholar] [CrossRef]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.W. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in endometriosis. Epigenetics 2006, 1, 106–111. [Google Scholar] [CrossRef]
- Bergqvist, A.; Ljungberg, O.; Skoog, L. Immunohistochemical analysis of oestrogen and progesterone receptors in endometriotic tissue and endometrium. Hum. Reprod. 1993, 8, 1915–1922. [Google Scholar] [CrossRef]
- Bukulmez, O.; Hardy, D.B.; Carr, B.R.; Word, R.A.; Mendelson, C.R. Inflammatory status influences aromatase and steroid receptor expression in endometriosis. Endocrinology 2008, 149, 1190–1204. [Google Scholar] [CrossRef]
- Bedaiwy, M.A.; Dahoud, W.; Skomorovska-Prokvolit, Y.; Yi, L.; Liu, J.H.; Falcone, T.; Hurd, W.W.; Mesiano, S. Abundance and Localization of Progesterone Receptor Isoforms in Endometrium in Women with and Without Endometriosis and in Peritoneal and Ovarian Endometriotic Implants. Reprod. Sci. 2015, 22, 1153–1161. [Google Scholar] [CrossRef]
- Lin, S.C.; Li, Y.H.; Wu, M.H.; Chang, Y.F.; Lee, D.K.; Tsai, S.Y.; Tsai, M.J.; Tsai, S.J. Suppression of COUP-TFII by proinflammatory cytokines contributes to the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, E427–E437. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Liu, K.; Chen, P.; Wang, D. Expression and Significance of WNT4 in Ectopic and Eutopic Endometrium of Human Endometriosis. Reprod. Sci. 2016, 23, 379–385. [Google Scholar] [CrossRef]
- Kato, N.; Iwase, A.; Ishida, C.; Nagai, T.; Mori, M.; Bayasula; Nakamura, T.; Osuka, S.; Ganiyeva, U.; Qin, Y.; et al. Upregulation of Fibroblast Growth Factors Caused by Heart and Neural Crest Derivatives Expressed 2 Suppression in Endometriotic Cells: A Possible Therapeutic Target in Endometriosis. Reprod. Sci. 2018, 26, 979–987. [Google Scholar] [CrossRef]
- Klemmt, P.A.; Carver, J.G.; Kennedy, S.H.; Koninckx, P.R.; Mardon, H.J. Stromal cells from endometriotic lesions and endometrium from women with endometriosis have reduced decidualization capacity. Fertil. Steril. 2006, 85, 564–572. [Google Scholar] [CrossRef]
- Hirota, Y.; Tranguch, S.; Daikoku, T.; Hasegawa, A.; Osuga, Y.; Taketani, Y.; Dey, S.K. Deficiency of immunophilin FKBP52 promotes endometriosis. Am. J. Pathol. 2008, 173, 1747–1757. [Google Scholar] [CrossRef]
- Dyson, M.T.; Roqueiro, D.; Monsivais, D.; Ercan, C.M.; Pavone, M.E.; Brooks, D.C.; Kakinuma, T.; Ono, M.; Jafari, N.; Dai, Y.; et al. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet. 2014, 10, e1004158. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Cheng, Y.H.; Huang, C.C.; Marsh, E.; Yin, P.; Milad, M.P.; Confino, E.; Reierstad, S.; Innes, J.; et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol. Reprod. 2007, 77, 681–687. [Google Scholar] [CrossRef]
- Smuc, T.; Pucelj, M.R.; Sinkovec, J.; Husen, B.; Thole, H.; Lanisnik Rizner, T. Expression analysis of the genes involved in estradiol and progesterone action in human ovarian endometriosis. Gynecol Endocrinol. 2007, 23, 105–111. [Google Scholar] [CrossRef]
- Yang, H.; Kang, K.; Cheng, C.; Mamillapalli, R.; Taylor, H.S. Integrative Analysis Reveals Regulatory Programs in Endometriosis. Reprod. Sci. 2015, 22, 1060–1072. [Google Scholar] [CrossRef]
- Pellegrini, C.; Gori, I.; Achtari, C.; Hornung, D.; Chardonnens, E.; Wunder, D.; Fiche, M.; Canny, G.O. The expression of estrogen receptors as well as GREB1, c-MYC, and cyclin D1, estrogen-regulated genes implicated in proliferation, is increased in peritoneal endometriosis. Fertil. Steril. 2012, 98, 1200–1208. [Google Scholar] [CrossRef]
- Wing, L.Y.; Chuang, P.C.; Wu, M.H.; Chen, H.M.; Tsai, S.J. Expression and mitogenic effect of fibroblast growth factor-9 in human endometriotic implant is regulated by aberrant production of estrogen. J. Clin. Endocrinol. Metab. 2003, 88, 5547–5554. [Google Scholar] [CrossRef]
- Han, S.J.; Hawkins, S.M.; Begum, K.; Jung, S.Y.; Kovanci, E.; Qin, J.; Lydon, J.P.; DeMayo, F.J.; O’Malley, B.W. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat. Med. 2012, 18, 1102–1111. [Google Scholar] [CrossRef]
- Kao, L.C.; Germeyer, A.; Tulac, S.; Lobo, S.; Yang, J.P.; Taylor, R.N.; Osteen, K.; Lessey, B.A.; Giudice, L.C. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology 2003, 144, 2870–2881. [Google Scholar] [CrossRef]
- Burney, R.O.; Talbi, S.; Hamilton, A.E.; Vo, K.C.; Nyegaard, M.; Nezhat, C.R.; Lessey, B.A.; Giudice, L.C. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology 2007, 148, 3814–3826. [Google Scholar] [CrossRef]
- Igarashi, T.M.; Bruner-Tran, K.L.; Yeaman, G.R.; Lessey, B.A.; Edwards, D.P.; Eisenberg, E.; Osteen, K.G. Reduced expression of progesterone receptor-B in the endometrium of women with endometriosis and in cocultures of endometrial cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fertil. Steril. 2005, 84, 67–74. [Google Scholar] [CrossRef]
- Wolfler, M.M.; Kuppers, M.; Rath, W.; Buck, V.U.; Meinhold-Heerlein, I.; Classen-Linke, I. Altered expression of progesterone receptor isoforms A and B in human eutopic endometrium in endometriosis patients. Ann. Anat 2016, 206, 1–6. [Google Scholar] [CrossRef]
- Shen, F.; Yan, C.; Liu, M.; Feng, Y.; Chen, Y. Decreased expression of mucin-1 in endometriosis endometrium correlated with progesterone receptor B involved in infertility. Arch. Gynecol. Obstet. 2015, 291, 439–445. [Google Scholar] [CrossRef]
- Pei, T.; Liu, C.; Liu, T.; Xiao, L.; Luo, B.; Tan, J.; Li, X.; Zhou, G.; Duan, C.; Huang, W. miR-194-3p Represses the Progesterone Receptor and Decidualization in Eutopic Endometrium From Women With Endometriosis. Endocrinology 2018, 159, 2554–2562. [Google Scholar] [CrossRef]
- Flores, V.A.; Vanhie, A.; Dang, T.; Taylor, H.S. Progesterone Receptor Status Predicts Response to Progestin Therapy in Endometriosis. J. Clin. Endocrinol. Metab. 2018, 103, 4561–4568. [Google Scholar] [CrossRef]
- Hou, Z.; Mamillapalli, R.; Taylor, H.S. Predictive biomarkers may allow precision therapy of endometriosis. J. Endometr. Pelvic. Pa. 2017, 9, 279–285. [Google Scholar] [CrossRef]
- Broi, M.G.D.; Rocha, C.V.J.; Meola, J.; Martins, W.P.; Carvalho, F.M.; Ferriani, R.A.; Navarro, P.A. Expression of PGR, HBEGF, ITGAV, ITGB3 and SPP1 genes in eutopic endometrium of infertile women with endometriosis during the implantation window: A pilot study. JBRA Assist. Reprod. 2017, 21, 196–202. [Google Scholar] [CrossRef]
- Zanatta, A.; Pereira, R.M.; Rocha, A.M.; Cogliati, B.; Baracat, E.C.; Taylor, H.S.; Motta, E.L.; Serafini, P.C. The relationship among HOXA10, estrogen receptor alpha, progesterone receptor, and progesterone receptor B proteins in rectosigmoid endometriosis: A tissue microarray study. Reprod. Sci. 2015, 22, 31–37. [Google Scholar] [CrossRef]
- Rocha-Junior, C.V.; Da Broi, M.G.; Miranda-Furtado, C.L.; Navarro, P.A.; Ferriani, R.A.; Meola, J. Progesterone Receptor B ( PGR-B) Is Partially Methylated in Eutopic Endometrium From Infertile Women With Endometriosis. Reprod. Sci. 2019, 1933719119828078. [Google Scholar] [CrossRef]
- Zhou, M.; Fu, J.; Xiao, L.; Yang, S.; Song, Y.; Zhang, X.; Feng, X.; Sun, H.; Xu, W.; Huang, W. miR-196a overexpression activates the MEK/ERK signal and represses the progesterone receptor and decidualization in eutopic endometrium from women with endometriosis. Hum. Reprod. 2016, 31, 2598–2608. [Google Scholar] [CrossRef]
- Smith, K.; Alnifaidy, R.; Wei, Q.; Nieman, L.K. Endometrial Indian hedgehog expression is decreased in women with endometriosis. Fertil. Steril. 2011, 95, 2738–2741.e3. [Google Scholar] [CrossRef]
- Taylor, H.S.; Bagot, C.; Kardana, A.; Olive, D.; Arici, A. HOX gene expression is altered in the endometrium of women with endometriosis. Hum. Reprod. 1999, 14, 1328–1331. [Google Scholar] [CrossRef]
- Su, R.W.; Strug, M.R.; Joshi, N.R.; Jeong, J.W.; Miele, L.; Lessey, B.A.; Young, S.L.; Fazleabas, A.T. Decreased Notch pathway signaling in the endometrium of women with endometriosis impairs decidualization. J. Clin. Endocrinol. Metab. 2015, 100, E433–E442. [Google Scholar] [CrossRef]
- Jackson, K.S.; Brudney, A.; Hastings, J.M.; Mavrogianis, P.A.; Kim, J.J.; Fazleabas, A.T. The altered distribution of the steroid hormone receptors and the chaperone immunophilin FKBP52 in a baboon model of endometriosis is associated with progesterone resistance during the window of uterine receptivity. Reprod. Sci. 2007, 14, 137–150. [Google Scholar] [CrossRef]
- Joshi, N.R.; Miyadahira, E.H.; Afshar, Y.; Jeong, J.W.; Young, S.L.; Lessey, B.A.; Serafini, P.C.; Fazleabas, A.T. Progesterone Resistance in Endometriosis Is Modulated by the Altered Expression of MicroRNA-29c and FKBP4. J. Clin. Endocrinol. Metab. 2017, 102, 141–149. [Google Scholar] [CrossRef]
- Kim, B.G.; Yoo, J.Y.; Kim, T.H.; Shin, J.H.; Langenheim, J.F.; Ferguson, S.D.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; Jeong, J.W. Aberrant activation of signal transducer and activator of transcription-3 (STAT3) signaling in endometriosis. Hum. Reprod. 2015, 30, 1069–1078. [Google Scholar] [CrossRef]
- Tsudo, T.; Harada, T.; Iwabe, T.; Tanikawa, M.; Nagano, Y.; Ito, M.; Taniguchi, F.; Terakawa, N. Altered gene expression and secretion of interleukin-6 in stromal cells derived from endometriotic tissues. Fertil. Steril. 2000, 73, 205–211. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Frank, D.A. STAT signaling in the pathogenesis and treatment of cancer. Mol. Med. 1999, 5, 432–456. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Jeong, J.W.; Fazleabas, A.T.; Tayade, C.; Young, S.L.; Lessey, B.A. Protein Inhibitor of Activated STAT3 (PIAS3) Is Down-Regulated in Eutopic Endometrium of Women with Endometriosis. Biol. Reprod. 2016, 95, 11. [Google Scholar] [CrossRef]
- Arguni, E.; Arima, M.; Tsuruoka, N.; Sakamoto, A.; Hatano, M.; Tokuhisa, T. JunD/AP-1 and STAT3 are the major enhancer molecules for high Bcl6 expression in germinal center B cells. Int. Immunol. 2006, 18, 1079–1089. [Google Scholar] [CrossRef]
- Evans-Hoeker, E.; Lessey, B.A.; Jeong, J.W.; Savaris, R.F.; Palomino, W.A.; Yuan, L.; Schammel, D.P.; Young, S.L. Endometrial BCL6 Overexpression in Eutopic Endometrium of Women With Endometriosis. Reprod. Sci. 2016, 23, 1234–1241. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, T.H.; Fazleabas, A.T.; Palomino, W.A.; Ahn, S.H.; Tayade, C.; Schammel, D.P.; Young, S.L.; Jeong, J.W.; Lessey, B.A. KRAS Activation and over-expression of SIRT1/BCL6 Contributes to the Pathogenesis of Endometriosis and Progesterone Resistance. Sci. Rep. 2017, 7, 6765. [Google Scholar] [CrossRef]
- Lessey, B.A.; Kim, J.J. Endometrial receptivity in the eutopic endometrium of women with endometriosis: It is affected, and let me show you why. Fertil. Steril. 2017, 108, 19–27. [Google Scholar] [CrossRef]
- Han, S.J.; O’Malley, B.W. The dynamics of nuclear receptors and nuclear receptor coregulators in the pathogenesis of endometriosis. Hum. Reprod. Update 2014, 20, 467–484. [Google Scholar] [CrossRef]
- Noble, L.S.; Simpson, E.R.; Johns, A.; Bulun, S.E. Aromatase expression in endometriosis. J. Clin. Endocrinol. Metab. 1996, 81, 174–179. [Google Scholar]
- Zeitoun, K.; Takayama, K.; Sasano, H.; Suzuki, T.; Moghrabi, N.; Andersson, S.; Johns, A.; Meng, L.; Putman, M.; Carr, B.; et al. Deficient 17beta-hydroxysteroid dehydrogenase type 2 expression in endometriosis: Failure to metabolize 17beta-estradiol. J. Clin. Endocrinol. Metab. 1998, 83, 4474–4480. [Google Scholar]
- Lessey, B.A.; Palomino, W.A.; Apparao, K.B.; Young, S.L.; Lininger, R.A. Estrogen receptor-alpha (ER-alpha) and defects in uterine receptivity in women. Reprod. Biol. Endocrinol. 2006, 4 (Suppl. 1), S9. [Google Scholar] [CrossRef]
- Osinski, M.; Wirstlein, P.; Wender-Ozegowska, E.; Mikolajczyk, M.; Jagodzinski, P.P.; Szczepanska, M. HSD3B2, HSD17B1, HSD17B2, ESR1, ESR2 and AR expression in infertile women with endometriosis. Ginekol. Pol. 2018, 89, 125–134. [Google Scholar] [CrossRef]
- Juhasz-Boss, I.; Fischer, C.; Lattrich, C.; Skrzypczak, M.; Malik, E.; Ortmann, O.; Treeck, O. Endometrial expression of estrogen receptor beta and its splice variants in patients with and without endometriosis. Arch. Gynecol. Obstet. 2011, 284, 885–891. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Murakami, T.; Uehara, S.; Canis, M.; Sasano, H.; Okamura, K. Expression of estrogen receptor alpha and beta in peritoneal and ovarian endometriosis. Fertil. Steril. 2001, 75, 1198–1205. [Google Scholar] [CrossRef]
- Tamura, M.; Deb, S.; Sebastian, S.; Okamura, K.; Bulun, S.E. Estrogen up-regulates cyclooxygenase-2 via estrogen receptor in human uterine microvascular endothelial cells. Fertil. Steril. 2004, 81, 1351–1356. [Google Scholar] [CrossRef]
- Wu, M.H.; Lu, C.W.; Chuang, P.C.; Tsai, S.J. Prostaglandin E2: The master of endometriosis? Exp. Biol. Med. (Maywood) 2010, 235, 668–677. [Google Scholar] [CrossRef]
- Monsivais, D.; Dyson, M.T.; Yin, P.; Coon, J.S.; Navarro, A.; Feng, G.; Malpani, S.S.; Ono, M.; Ercan, C.M.; Wei, J.J.; et al. ERbeta- and prostaglandin E2-regulated pathways integrate cell proliferation via Ras-like and estrogen-regulated growth inhibitor in endometriosis. Mol. Endocrinol. 2014, 28, 1304–1315. [Google Scholar] [CrossRef]
- Han, S.J.; Jung, S.Y.; Wu, S.P.; Hawkins, S.M.; Park, M.J.; Kyo, S.; Qin, J.; Lydon, J.P.; Tsai, S.Y.; Tsai, M.J.; et al. Estrogen Receptor beta Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell 2015, 163, 960–974. [Google Scholar] [CrossRef]
- Symons, L.K.; Miller, J.E.; Kay, V.R.; Marks, R.M.; Liblik, K.; Koti, M.; Tayade, C. The Immunopathophysiology of Endometriosis. Trends Mol. Med. 2018, 24, 748–762. [Google Scholar] [CrossRef]
- Montagna, P.; Capellino, S.; Villaggio, B.; Remorgida, V.; Ragni, N.; Cutolo, M.; Ferrero, S. Peritoneal fluid macrophages in endometriosis: Correlation between the expression of estrogen receptors and inflammation. Fertil. Steril. 2008, 90, 156–164. [Google Scholar] [CrossRef]
- Greaves, E.; Temp, J.; Esnal-Zufiurre, A.; Mechsner, S.; Horne, A.W.; Saunders, P.T. Estradiol is a critical mediator of macrophage-nerve cross talk in peritoneal endometriosis. Am. J. Pathol. 2015, 185, 2286–2297. [Google Scholar] [CrossRef]
- Kumagami, A.; Ito, A.; Yoshida-Komiya, H.; Fujimori, K.; Sato, A. Expression patterns of the steroid receptor coactivator family in human ovarian endometriosis. J. Obstet. Gynaecol. Res. 2011, 37, 1269–1276. [Google Scholar] [CrossRef]
- Suzuki, A.; Horiuchi, A.; Oka, K.; Miyamoto, T.; Kashima, H.; Shiozawa, T. Immunohistochemical detection of steroid receptor cofactors in ovarian endometriosis: Involvement of down-regulated SRC-1 expression in the limited growth activity of the endometriotic epithelium. Virchows. Arch. 2010, 456, 433–441. [Google Scholar] [CrossRef]
- Cho, Y.J.; Lee, J.E.; Park, M.J.; O’Malley, B.W.; Han, S.J. Bufalin suppresses endometriosis progression by inducing pyroptosis and apoptosis. J. Endocrinol. 2018, 237, 255–269. [Google Scholar] [CrossRef]
- Dimitriadis, E.; Stoikos, C.; Stafford-Bell, M.; Clark, I.; Paiva, P.; Kovacs, G.; Salamonsen, L.A. Interleukin-11, IL-11 receptoralpha and leukemia inhibitory factor are dysregulated in endometrium of infertile women with endometriosis during the implantation window. J. Reprod. Immunol. 2006, 69, 53–64. [Google Scholar] [CrossRef]
- Arici, A.; Engin, O.; Attar, E.; Olive, D.L. Modulation of leukemia inhibitory factor gene expression and protein biosynthesis in human endometrium. J. Clin. Endocrinol. Metab. 1995, 80, 1908–1915. [Google Scholar]
- Kelleher, A.M.; Peng, W.; Pru, J.K.; Pru, C.A.; DeMayo, F.J.; Spencer, T.E. Forkhead box a2 (FOXA2) is essential for uterine function and fertility. Proc. Natl. Acad. Sci. USA 2017, 114, E1018–E1026. [Google Scholar] [CrossRef]
- Lin, A.; Yin, J.; Cheng, C.; Yang, Z.; Yang, H. Decreased expression of FOXA2 promotes eutopic endometrial cell proliferation and migration in patients with endometriosis. Reprod. Biomed. Online 2018, 36, 181–187. [Google Scholar] [CrossRef]
- Holoch, K.J.; Lessey, B.A. Endometriosis and infertility. Clin. Obstet. Gynecol. 2010, 53, 429–438. [Google Scholar] [CrossRef]
- De Ziegler, D.; Borghese, B.; Chapron, C. Endometriosis and infertility: Pathophysiology and management. Lancet 2010, 376, 730–738. [Google Scholar] [CrossRef]
- Macer, M.L.; Taylor, H.S. Endometriosis and infertility: A review of the pathogenesis and treatment of endometriosis-associated infertility. Obstet. Gynecol. Clin. N. Am. 2012, 39, 535–549. [Google Scholar] [CrossRef]
- Haydardedeoglu, B.; Zeyneloglu, H.B. The impact of endometriosis on fertility. Womens Health (Lond.) 2015, 11, 619–623. [Google Scholar] [CrossRef]
- Tomassetti, C.; D’Hooghe, T. Endometriosis and infertility: Insights into the causal link and management strategies. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 51, 25–33. [Google Scholar] [CrossRef]
- Moberg, C.; Bourlev, V.; Ilyasova, N.; Olovsson, M. Levels of oestrogen receptor, progesterone receptor and alphaB-crystallin in eutopic endometrium in relation to pregnancy in women with endometriosis. Hum. Fertil. (Camb.) 2015, 18, 30–37. [Google Scholar] [CrossRef]
- Lessey, B.A.; Castelbaum, A.J.; Sawin, S.W.; Buck, C.A.; Schinnar, R.; Bilker, W.; Strom, B.L. Aberrant integrin expression in the endometrium of women with endometriosis. J. Clin. Endocrinol. Metab. 1994, 79, 643–649. [Google Scholar]
- Daftary, G.S.; Troy, P.J.; Bagot, C.N.; Young, S.L.; Taylor, H.S. Direct regulation of beta3-integrin subunit gene expression by HOXA10 in endometrial cells. Mol. Endocrinol. 2002, 16, 571–579. [Google Scholar]
- Kim, J.J.; Taylor, H.S.; Lu, Z.; Ladhani, O.; Hastings, J.M.; Jackson, K.S.; Wu, Y.; Guo, S.W.; Fazleabas, A.T. Altered expression of HOXA10 in endometriosis: Potential role in decidualization. Mol. Hum. Reprod. 2007, 13, 323–332. [Google Scholar] [CrossRef]
- Moberg, C.; Bourlev, V.; Ilyasova, N.; Olovsson, M. Endometrial expression of LIF and its receptor and peritoneal fluid levels of IL-1alpha and IL-6 in women with endometriosis are associated with the probability of pregnancy. Arch. Gynecol. Obstet. 2015, 292, 429–437. [Google Scholar] [CrossRef]
- Carter, J.E. Combined hysteroscopic and laparoscopic findings in patients with chronic pelvic pain. J. Am. Assoc. Gynecol. Laparosc. 1994, 2, 43–47. [Google Scholar] [CrossRef]
- Morotti, M.; Vincent, K.; Becker, C.M. Mechanisms of pain in endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 209, 8–13. [Google Scholar] [CrossRef]
- Berkley, K.J.; Rapkin, A.J.; Papka, R.E. The pains of endometriosis. Science 2005, 308, 1587–1589. [Google Scholar] [CrossRef]
- Morotti, M.; Vincent, K.; Brawn, J.; Zondervan, K.T.; Becker, C.M. Peripheral changes in endometriosis-associated pain. Hum. Reprod. Update 2014, 20, 717–736. [Google Scholar] [CrossRef]
- Bjorling, D.E.; Beckman, M.; Clayton, M.K.; Wang, Z.Y. Modulation of nerve growth factor in peripheral organs by estrogen and progesterone. Neuroscience 2002, 110, 155–167. [Google Scholar] [CrossRef]
- McLaren, J.; Prentice, A.; Charnock-Jones, D.S.; Millican, S.A.; Muller, K.H.; Sharkey, A.M.; Smith, S.K. Vascular endothelial growth factor is produced by peritoneal fluid macrophages in endometriosis and is regulated by ovarian steroids. J. Clin. Investig. 1996, 98, 482–489. [Google Scholar] [CrossRef]
- Krizsan-Agbas, D.; Pedchenko, T.; Hasan, W.; Smith, P.G. Oestrogen regulates sympathetic neurite outgrowth by modulating brain derived neurotrophic factor synthesis and release by the rodent uterus. Eur. J. Neurosci. 2003, 18, 2760–2768. [Google Scholar] [CrossRef]
- Tokushige, N.; Markham, R.; Russell, P.; Fraser, I.S. Effects of hormonal treatment on nerve fibers in endometrium and myometrium in women with endometriosis. Fertil. Steril. 2008, 90, 1589–1598. [Google Scholar] [CrossRef]
- Greaves, E.; Collins, F.; Esnal-Zufiaurre, A.; Giakoumelou, S.; Horne, A.W.; Saunders, P.T. Estrogen receptor (ER) agonists differentially regulate neuroangiogenesis in peritoneal endometriosis via the repellent factor SLIT3. Endocrinology 2014, 155, 4015–4026. [Google Scholar] [CrossRef]
- Rocha, M.G.; e Silva, J.C.; Ribeiro da Silva, A.; Candido Dos Reis, F.J.; Nogueira, A.A.; Poli-Neto, O.B. TRPV1 expression on peritoneal endometriosis foci is associated with chronic pelvic pain. Reprod. Sci. 2011, 18, 511–515. [Google Scholar] [CrossRef]
- Greaves, E.; Grieve, K.; Horne, A.W.; Saunders, P.T. Elevated peritoneal expression and estrogen regulation of nociceptive ion channels in endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, E1738–E1743. [Google Scholar] [CrossRef]
- Vercellini, P.; Buggio, L.; Frattaruolo, M.P.; Borghi, A.; Dridi, D.; Somigliana, E. Medical treatment of endometriosis-related pain. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 51, 68–91. [Google Scholar] [CrossRef]
- Tosti, C.; Biscione, A.; Morgante, G.; Bifulco, G.; Luisi, S.; Petraglia, F. Hormonal therapy for endometriosis: From molecular research to bedside. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 209, 61–66. [Google Scholar] [CrossRef]
- Ferrero, S.; Evangelisti, G.; Barra, F. Current and emerging treatment options for endometriosis. Expert Opin. Pharm. 2018, 19, 1109–1125. [Google Scholar] [CrossRef]
- Brown, J.; Pan, A.; Hart, R.J. Gonadotrophin-releasing hormone analogues for pain associated with endometriosis. Cochrane Database Syst. Rev. 2010, CD008475. [Google Scholar]
- Cetel, N.S.; Rivier, J.; Vale, W.; Yen, S.S. The dynamics of gonadotropin inhibition in women induced by an antagonistic analog of gonadotropin-releasing hormone. J. Clin. Endocrinol. Metab. 1983, 57, 62–65. [Google Scholar] [CrossRef]
- Taylor, H.S.; Giudice, L.C.; Lessey, B.A.; Abrao, M.S.; Kotarski, J.; Archer, D.F.; Diamond, M.P.; Surrey, E.; Johnson, N.P.; Watts, N.B.; et al. Treatment of Endometriosis-Associated Pain with Elagolix, an Oral GnRH Antagonist. N. Engl. J. Med. 2017, 377, 28–40. [Google Scholar] [CrossRef]
- Dunselman, G.A.; Vermeulen, N.; Becker, C.; Calhaz-Jorge, C.; D’Hooghe, T.; De Bie, B.; Heikinheimo, O.; Horne, A.W.; Kiesel, L.; Nap, A.; et al. ESHRE guideline: Management of women with endometriosis. Hum. Reprod. 2014, 29, 400–412. [Google Scholar] [CrossRef]
- Bilotas, M.; Meresman, G.; Stella, I.; Sueldo, C.; Baranao, R.I. Effect of aromatase inhibitors on ectopic endometrial growth and peritoneal environment in a mouse model of endometriosis. Fertil. Steril. 2010, 93, 2513–2518. [Google Scholar] [CrossRef]
- Verma, A.; Konje, J.C. Successful treatment of refractory endometriosis-related chronic pelvic pain with aromatase inhibitors in premenopausal patients. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 143, 112–115. [Google Scholar] [CrossRef]
- Kulak, J., Jr.; Fischer, C.; Komm, B.; Taylor, H.S. Treatment with bazedoxifene, a selective estrogen receptor modulator, causes regression of endometriosis in a mouse model. Endocrinology 2011, 152, 3226–3232. [Google Scholar] [CrossRef]
- Yao, Z.; Shen, X.; Capodanno, I.; Donnelly, M.; Fenyk-Melody, J.; Hausamann, J.; Nunes, C.; Strauss, J.; Vakerich, K. Validation of rat endometriosis model by using raloxifene as a positive control for the evaluation of novel SERM compounds. J. Invest. Surg. 2005, 18, 177–183. [Google Scholar] [CrossRef]
- Stratton, P.; Sinaii, N.; Segars, J.; Koziol, D.; Wesley, R.; Zimmer, C.; Winkel, C.; Nieman, L.K. Return of chronic pelvic pain from endometriosis after raloxifene treatment: A randomized controlled trial. Obstet. Gynecol. 2008, 111, 88–96. [Google Scholar] [CrossRef]
- Vercellini, P.; Somigliana, E.; Daguati, R.; Vigano, P.; Meroni, F.; Crosignani, P.G. Postoperative oral contraceptive exposure and risk of endometrioma recurrence. Am. J. Obstet. Gynecol. 2008, 198, 504.e1–504.e5. [Google Scholar] [CrossRef]
- Harada, T.; Momoeda, M.; Taketani, Y.; Hoshiai, H.; Terakawa, N. Low-dose oral contraceptive pill for dysmenorrhea associated with endometriosis: A placebo-controlled, double-blind, randomized trial. Fertil. Steril. 2008, 90, 1583–1588. [Google Scholar] [CrossRef]
- Harada, T.; Kosaka, S.; Elliesen, J.; Yasuda, M.; Ito, M.; Momoeda, M. Ethinylestradiol 20 mug/drospirenone 3 mg in a flexible extended regimen for the management of endometriosis-associated pelvic pain: A randomized controlled trial. Fertil. Steril. 2017, 108, 798–805. [Google Scholar] [CrossRef]
- Muneyyirci-Delale, O.; Karacan, M. Effect of norethindrone acetate in the treatment of symptomatic endometriosis. Int. J. Fertil. Womens Med. 1998, 43, 24–27. [Google Scholar]
- Crosignani, P.G.; Luciano, A.; Ray, A.; Bergqvist, A. Subcutaneous depot medroxyprogesterone acetate versus leuprolide acetate in the treatment of endometriosis-associated pain. Hum. Reprod. 2006, 21, 248–256. [Google Scholar] [CrossRef]
- Lockhat, F.B.; Emembolu, J.O.; Konje, J.C. The efficacy, side-effects and continuation rates in women with symptomatic endometriosis undergoing treatment with an intra-uterine administered progestogen (levonorgestrel): A 3 year follow-up. Hum. Reprod. 2005, 20, 789–793. [Google Scholar] [CrossRef]
- Sroyraya, M.; Songkoomkrong, S.; Changklungmoa, N.; Poljaroen, J.; Weerakiet, S.; Sophonsritsuk, A.; Wongkularb, A.; Lertvikool, S.; Tingthanatikul, Y.; Sobhon, P. Differential expressions of estrogen and progesterone receptors in endometria and cyst walls of ovarian endometrioma from women with endometriosis and their responses to depo-medroxyprogesterone acetate treatment. Mol. Cell Probes. 2018, 40, 27–36. [Google Scholar] [CrossRef]
- Selak, V.; Farquhar, C.; Prentice, A.; Singla, A. Danazol for pelvic pain associated with endometriosis. Cochrane Database Syst. Rev. 2007, CD000068. [Google Scholar]
- Strowitzki, T.; Faustmann, T.; Gerlinger, C.; Seitz, C. Dienogest in the treatment of endometriosis-associated pelvic pain: A 12-week, randomized, double-blind, placebo-controlled study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 151, 193–198. [Google Scholar] [CrossRef]
- Harada, T.; Momoeda, M.; Taketani, Y.; Aso, T.; Fukunaga, M.; Hagino, H.; Terakawa, N. Dienogest is as effective as intranasal buserelin acetate for the relief of pain symptoms associated with endometriosis--a randomized, double-blind, multicenter, controlled trial. Fertil. Steril. 2009, 91, 675–681. [Google Scholar] [CrossRef]
- Fu, L.; Osuga, Y.; Morimoto, C.; Hirata, T.; Hirota, Y.; Yano, T.; Taketani, Y. Dienogest inhibits BrdU uptake with G0/G1 arrest in cultured endometriotic stromal cells. Fertil. Steril. 2008, 89 (Suppl. 5), 1344–1347. [Google Scholar] [CrossRef]
- Hayashi, A.; Tanabe, A.; Kawabe, S.; Hayashi, M.; Yuguchi, H.; Yamashita, Y.; Okuda, K.; Ohmichi, M. Dienogest increases the progesterone receptor isoform B/A ratio in patients with ovarian endometriosis. J. Ovarian Res. 2012, 5, 31. [Google Scholar] [CrossRef]
- Shimizu, Y.; Mita, S.; Takeuchi, T.; Notsu, T.; Mizuguchi, K.; Kyo, S. Dienogest, a synthetic progestin, inhibits prostaglandin E2 production and aromatase expression by human endometrial epithelial cells in a spheroid culture system. Steroids 2011, 76, 60–67. [Google Scholar] [CrossRef]
- Kettel, L.M.; Murphy, A.A.; Morales, A.J.; Yen, S.S. Clinical efficacy of the antiprogesterone RU486 in the treatment of endometriosis and uterine fibroids. Hum. Reprod. 1994, 9 (Suppl. 1), 116–120. [Google Scholar] [CrossRef]
- Kettel, L.M.; Murphy, A.A.; Morales, A.J.; Ulmann, A.; Baulieu, E.E.; Yen, S.S. Treatment of endometriosis with the antiprogesterone mifepristone (RU486). Fertil. Steril. 1996, 65, 23–28. [Google Scholar] [CrossRef]
- Casper, R.F. Progestin-only pills may be a better first-line treatment for endometriosis than combined estrogen-progestin contraceptive pills. Fertil. Steril. 2017, 107, 533–536. [Google Scholar] [CrossRef]
- Vercellini, P.; Cortesi, I.; Crosignani, P.G. Progestins for symptomatic endometriosis: A critical analysis of the evidence. Fertil. Steril. 1997, 68, 393–401. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Molecule | Symbol | Function | Dysregulation | Reference | |
|---|---|---|---|---|---|
| P4 Signaling Factors | Progesterone Receptor | PGR | Nuclear receptor | Decreased | [121,122,123,124,125,126,127,128,129,130] |
| Chicken ovalbumin upstream promoter-transcription factor II | COUPTFII | Transcription factor | Decreased | [131] | |
| Wnt family member 4 | WNT4 | Secreted signaling protein | Decreased | [132] | |
| Heart and neural crest derivatives expressed 2 | HAND2 | Transcription factor | Decreased | [133] | |
| Insulin-like growth factor binding protein 1 | IGFBP1 | Circulating growth factor binding protein | Decreased | [134] | |
| Forkhead box O1 | FOXO1 | Transcription factor | Decreased | [122] | |
| FK506 binding protein prolyl isomerase 4 | FKBP52 | Immunophilin | Decreased | [135] | |
| GATA binding protein 2 | GATA2 | Transcription factor | Decreased | [136] | |
| E2 Signaling Factors | Estrogen receptor 1 | ESR1 | Nuclear receptor | Decreased | [136,137,138,139] |
| Estrogen receptor 2 | ESR2 | Nuclear receptor | Increased | [136,137,138,139] | |
| Myc proto-oncogene protein | c-MYC | Transcription factor | Increased | [140] | |
| Cyclin D1 | CCND1 | Cell cycle regulator | Increased | [140] | |
| Growth regulating estrogen receptor binding 1 | GREB | Growth regulator | Increased | [140] | |
| Fibroblast growth factor 9 | FGF-9 | Secreted growth factor | Increased | [141] | |
| Steroid receptor coactivator-1 | SRC-1 | Transcriptional co-activator | Increased | [142] |
| Treatment Type | Molecular Action | Therapeutic Effect | Reference | |
|---|---|---|---|---|
| Estrogen (E2) Signaling Modifiers | Gonadotropin-releasing hormone (GnRH) agonists | Decrease E2 production through negative feedback | Reduce endometriosis-related pain | [213,214] |
| GnRH antagonists | Decrease E2 production by competing for GnRH receptors | Reduce endometriosis-related pain | [215,216] | |
| Aromatase inhibitors | Decrease E2 production by inhibiting conversion of androgens to E2 | Reduce endometriosis-related pain and lesion size | [217,218,219] | |
| Selective estrogen receptor modulators (SERMs) | Decrease estrogen receptor 1 (ESR1) action through direct inhibition | Reduce endometriotic lesions | [213,220,221,222] | |
| Progesterone (P4) Signaling Modifiers | Combined oral contraceptives (COCs) | Suppress ovarian steroid production and supplement P4 levels | Reduce endometriosis-related pain and recurrence after surgery | [217,223,224,225] |
| Progestins | Supplement P4 levels | Reduce endometriosis-related pain and lesions | [212,217,226,227,228,229,230,231,232,233,234,235] | |
| Selective progesterone receptor modulators (SPRMs) | Interact with progesterone receptor (PGR) to enhance downstream effects | Reduce endometriosis-related pain and lesions | [212,236,237] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquardt, R.M.; Kim, T.H.; Shin, J.-H.; Jeong, J.-W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. https://doi.org/10.3390/ijms20153822
Marquardt RM, Kim TH, Shin J-H, Jeong J-W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? International Journal of Molecular Sciences. 2019; 20(15):3822. https://doi.org/10.3390/ijms20153822
Chicago/Turabian StyleMarquardt, Ryan M., Tae Hoon Kim, Jung-Ho Shin, and Jae-Wook Jeong. 2019. "Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis?" International Journal of Molecular Sciences 20, no. 15: 3822. https://doi.org/10.3390/ijms20153822
APA StyleMarquardt, R. M., Kim, T. H., Shin, J.-H., & Jeong, J.-W. (2019). Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? International Journal of Molecular Sciences, 20(15), 3822. https://doi.org/10.3390/ijms20153822