The Observation of Ligand-Binding-Relevant Open States of Fatty Acid Binding Protein by Molecular Dynamics Simulations and a Markov State Model


Abstract
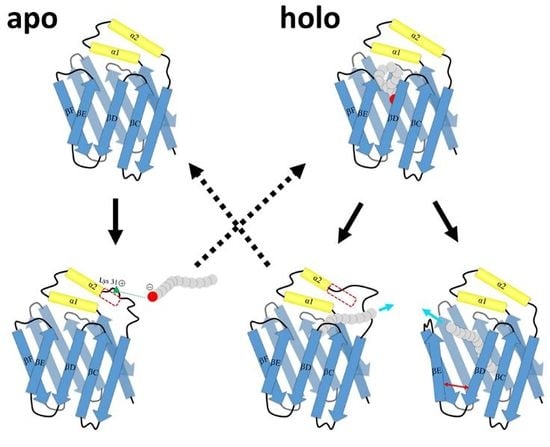
1. Introduction
2. Results and Discussion
2.1. Structural Properties
2.2. Intermediate States of Apo-FABP
2.3. Intermediate States of Ligand-Binding HFABP
2.4. Ligand Binding Model
3. Materials and Methods
3.1. Molecular Dynamics (MD) Simulation
3.2. Markov State Model Analysis
3.3. Apparent Free Energy Barrier
3.4. Mean First Passage Time
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Storch, J.; Thumser, A.E. Tissue-specific functions in the fatty acid-binding protein family. J. Biol. Chem. 2010, 285, 32679–32683. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Bernlohr, D.A. Metabolic functions of fabps--mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015, 11, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Sugiyama, S.; Ishida, H.; Niiyama, M.; Matsuoka, D.; Hara, T.; Mizohata, E.; Murakami, S.; Inoue, T.; Matsuoka, S.; et al. Structure of the human-heart fatty-acid-binding protein 3 in complex with the fluorescent probe 1-anilinonaphthalene-8-sulphonic acid. J. Synchrotron Radiat. 2013, 20, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.D.; He, Y.; Wang, S.; Wong, G.T.; Irwin, M.G.; Xia, Z. Heart-type fatty acid binding protein (h-fabp) as a biomarker for acute myocardial injury and long-term post-ischemic prognosis. Acta Pharmacol. Sin. 2018, 39, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.W.; Veerkamp, J.H. New insights into the structure and function of fatty acid-binding proteins. Cell Mol. Life Sci. 2002, 59, 1096–1116. [Google Scholar] [CrossRef]
- Hodsdon, M.E.; Cistola, D.P. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15n nmr relaxation and 1h exchange. Biochemistry 1997, 36, 2278–2290. [Google Scholar] [CrossRef] [PubMed]
- Richieri, G.V.; Low, P.J.; Ogata, R.T.; Kleinfeld, A.M. Binding kinetics of engineered mutants provide insight about the pathway for entering and exiting the intestinal fatty acid binding protein. Biochemistry 1999, 38, 5888–5895. [Google Scholar] [CrossRef]
- He, Y.; Yang, X.; Wang, H.; Estephan, R.; Francis, F.; Kodukula, S.; Storch, J.; Stark, R.E. Solution-state molecular structure of apo and oleate-liganded liver fatty acid-binding protein. Biochemistry 2007, 46, 12543–12556. [Google Scholar] [CrossRef]
- Sacchettini, J.C.; Gordon, J.I.; Banaszak, L.J. Crystal structure of rat intestinal fatty-acid-binding protein. Refinement and analysis of the escherichia coli-derived protein with bound palmitate. J. Mol. Biol 1989, 208, 327–339. [Google Scholar] [CrossRef]
- Cistola, D.P.; Kim, K.; Rogl, H.; Frieden, C. Fatty acid interactions with a helix-less variant of intestinal fatty acid-binding protein. Biochemistry 1996, 35, 7559–7565. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Mu, Y.; Yang, D. Molecular dynamics simulation of ligand dissociation from liver fatty acid binding protein. PLoS ONE 2009, 4, e6081. [Google Scholar] [CrossRef] [PubMed]
- Sacchettini, J.C.; Gordon, J.I.; Banaszak, L.J. Refined apoprotein structure of rat intestinal fatty acid binding protein produced in escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 7736–7740. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Long, D.; Yang, D. Rapid data collection for protein structure determination by nmr spectroscopy. J. Am. Chem. Soc. 2007, 129, 7722–7723. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Fan, J.S.; Zhou, H.; Lin, Q.; Yang, D. Local unfolding of fatty acid binding protein to allow ligand entry for binding. Angew Chem. Int. Ed. Engl. 2016, 55, 6869–6872. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, D. Coexistence of multiple minor states of fatty acid binding protein and their functional relevance. Sci. Rep. 2016, 6, 34171. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Yang, D. Millisecond timescale dynamics of human liver fatty acid binding protein: Testing of its relevance to the ligand entry process. Biophys. J. 2010, 98, 3054–3061. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hunter, N.H.; Bakula, B.C.; Bruce, C.D. Molecular dynamics simulations of apo and holo forms of fatty acid binding protein 5 and cellular retinoic acid binding protein ii reveal highly mobile protein, retinoic acid ligand, and water molecules. J. Biomol. Struct. Dyn. 2018, 36, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Bakowies, D.; van Gunsteren, W.F. Simulations of apo and holo-fatty acid binding protein: Structure and dynamics of protein, ligand and internal water. J. Mol. Biol. 2002, 315, 713–736. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.; Nachliel, E.; Gutman, M. Fatty acid binding proteins: Same structure but different binding mechanisms? Molecular dynamics simulations of intestinal fatty acid binding protein. Biophys. J. 2006, 90, 1535–1545. [Google Scholar] [CrossRef]
- Tsfadia, Y.; Friedman, R.; Kadmon, J.; Selzer, A.; Nachliel, E.; Gutman, M. Molecular dynamics simulations of palmitate entry into the hydrophobic pocket of the fatty acid binding protein. FEBS Lett. 2007, 581, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, D.; Sugiyama, S.; Murata, M.; Matsuoka, S. Molecular dynamics simulations of heart-type fatty acid binding protein in apo and holo forms, and hydration structure analyses in the binding cavity. J. Phys. Chem. B 2015, 119, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Dong, Z. Exploration of gated ligand binding recognizes an allosteric site for blocking fabp4-protein interaction. Phys. Chem. Chem. Phys. 2015, 17, 32257–32267. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, A.; Gierasch, L.M.; Pastore, A.; Radford, S.E. Energy landscapes of functional proteins are inherently risky. Nat. Chem. Biol. 2014, 10, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Hummer, G.; Eaton, W.A. Native contacts determine protein folding mechanisms in atomistic simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 17874–17879. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.J.; Liu, H.Z.; Li, M.H.; Huo, S.H. Network representation of conformational transitions between hidden intermediates of rd-apocytochrome b(562). J. Chem. Phys. 2015, 143. [Google Scholar] [CrossRef] [PubMed]
- Young, A.C.M.; Scapin, G.; Kromminga, A.; Patel, S.B.; Veerkamp, J.H.; Sacchettini, J.C. Structural studies on human muscle fatty-acid-binding protein at 1.4-angstrom resolution—binding interactions with 3 c18 fatty-acids. Structure 1994, 2, 523–534. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J. Small molecule allosteric modulators of g-protein-coupled receptors: Drug-target interactions. J. Med. Chem. 2019, 62, 24–45. [Google Scholar] [CrossRef]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric methods and their applications: Facilitating the discovery of allosteric drugs and the investigation of allosteric mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef]
- Lu, S.; Ji, M.; Ni, D.; Zhang, J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today 2018, 23, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the amber ff99sb protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Lambrakos, S.G.; Boris, J.P.; Oran, E.S.; Chandrasekhar, I.; Nagumo, M. A modified shake algorithm for maintaining rigid bonds in molecular-dynamics simulations of large molecules. J. Comput. Phys. 1989, 85, 473–486. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with amber on gpus. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Beauchamp, K.A.; Bowman, G.R.; Lane, T.J.; Maibaum, L.; Haque, I.S.; Pande, V.S. Msmbuilder2: Modeling conformational dynamics on the picosecond to millisecond scale. J. Chem. Theory Comput. 2011, 7, 3412–3419. [Google Scholar] [CrossRef]
- Perez-Hernandez, G.; Paul, F.; Giorgino, T.; De Fabritiis, G.; Noe, F. Identification of slow molecular order parameters for markov model construction. J. Chem. Phys. 2013, 139. [Google Scholar] [CrossRef]
- Bowman, G.R.; Huang, X.; Pande, V.S. Using generalized ensemble simulations and markov state models to identify conformational states. Methods 2009, 49, 197–201. [Google Scholar] [CrossRef]
- Li, M.; Duan, M.; Fan, J.; Han, L.; Huo, S. Graph representation of protein free energy landscape. J. Chem. Phys. 2013, 139, 185101. [Google Scholar] [CrossRef]
- Hunter, J.J. The computation of key properties of markov chains via perturbations. Linear Algebra Appl. 2016, 511, 176–202. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A1 | A2 | A3 | A4 | A5 | |
|---|---|---|---|---|---|
| A1 | 0 | 124.87 | 63.66 | 124.81 | 25.84 |
| A2 | 36.11 | 0 | 13.54 | 76.22 | 60.47 |
| A3 | 22.58 | 61.22 | 0 | 62.91 | 46.94 |
| A4 | 24.82 | 64.98 | 4.0 | 0 | 49.08 |
| A5 | 2.51 | 125.91 | 64.69 | 125.75 | 0 |
| H1 | H2 | H3 | H4 | H5 | H6 | |
|---|---|---|---|---|---|---|
| H1 | 0 | 639.15 | 144.61 | 616.00 | 199.05 | 671.48 |
| H2 | 74.08 | 0 | 41.95 | 48.47 | 92.98 | 103.68 |
| H3 | 32.12 | 494.54 | 0 | 471.39 | 54.44 | 526.87 |
| H4 | 73.52 | 71.07 | 41.40 | 0 | 92.07 | 55.67 |
| H5 | 33.48 | 492.49 | 1.36 | 468.98 | 0 | 524.47 |
| H6 | 74.54 | 71.81 | 42.42 | 1.20 | 93.10 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Duan, M.; Yang, M. The Observation of Ligand-Binding-Relevant Open States of Fatty Acid Binding Protein by Molecular Dynamics Simulations and a Markov State Model. Int. J. Mol. Sci. 2019, 20, 3476. https://doi.org/10.3390/ijms20143476
Guo Y, Duan M, Yang M. The Observation of Ligand-Binding-Relevant Open States of Fatty Acid Binding Protein by Molecular Dynamics Simulations and a Markov State Model. International Journal of Molecular Sciences. 2019; 20(14):3476. https://doi.org/10.3390/ijms20143476
Chicago/Turabian StyleGuo, Yue, Mojie Duan, and Minghui Yang. 2019. "The Observation of Ligand-Binding-Relevant Open States of Fatty Acid Binding Protein by Molecular Dynamics Simulations and a Markov State Model" International Journal of Molecular Sciences 20, no. 14: 3476. https://doi.org/10.3390/ijms20143476
APA StyleGuo, Y., Duan, M., & Yang, M. (2019). The Observation of Ligand-Binding-Relevant Open States of Fatty Acid Binding Protein by Molecular Dynamics Simulations and a Markov State Model. International Journal of Molecular Sciences, 20(14), 3476. https://doi.org/10.3390/ijms20143476