Lipid Reshaping and Lipophagy Are Induced in a Modeled Ischemia-Reperfusion Injury of Blood Brain Barrier

 , , , ,
, , , ,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. OGD/ogR Rearranges Rat Brain Endothelial Cells (RBE4) Total FA Profile
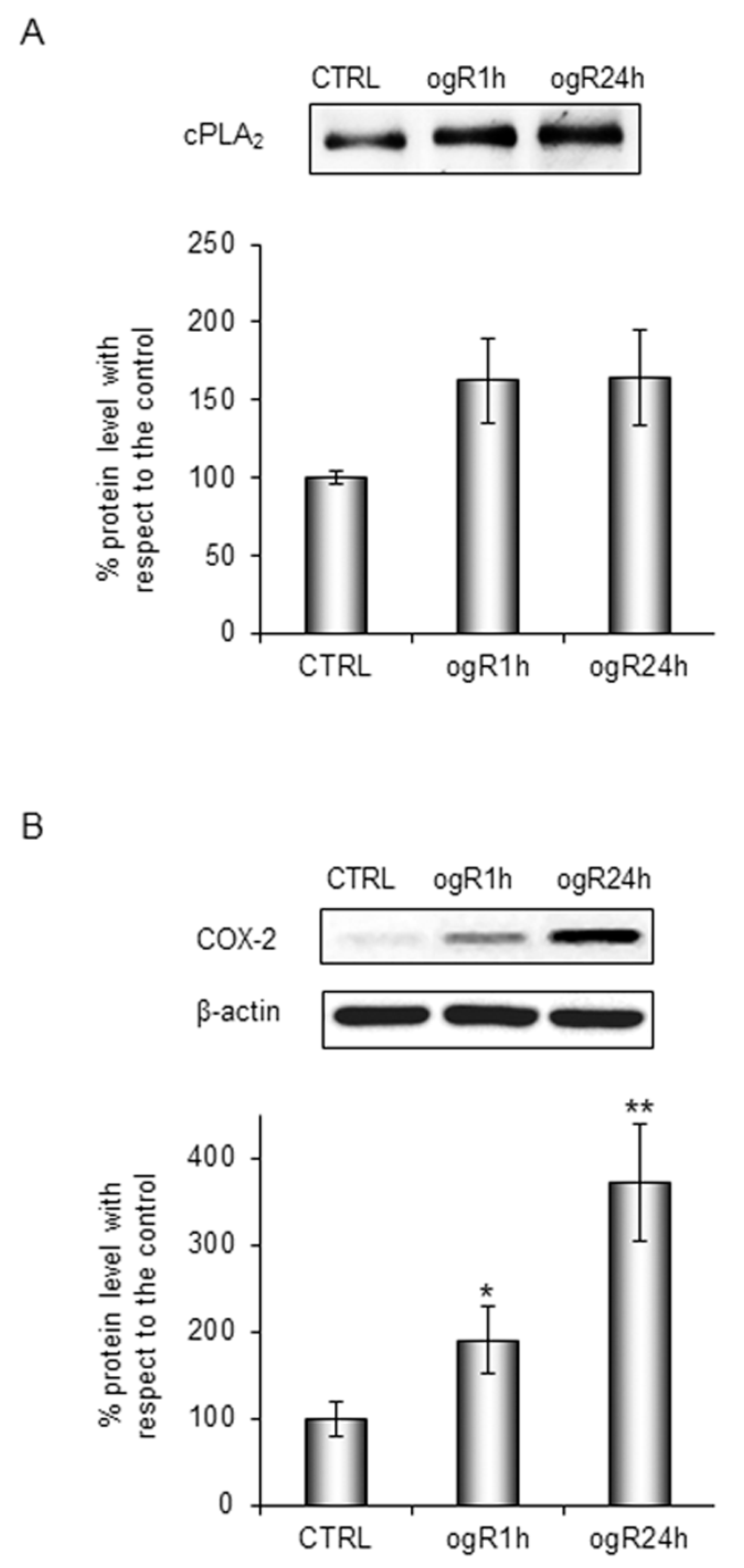
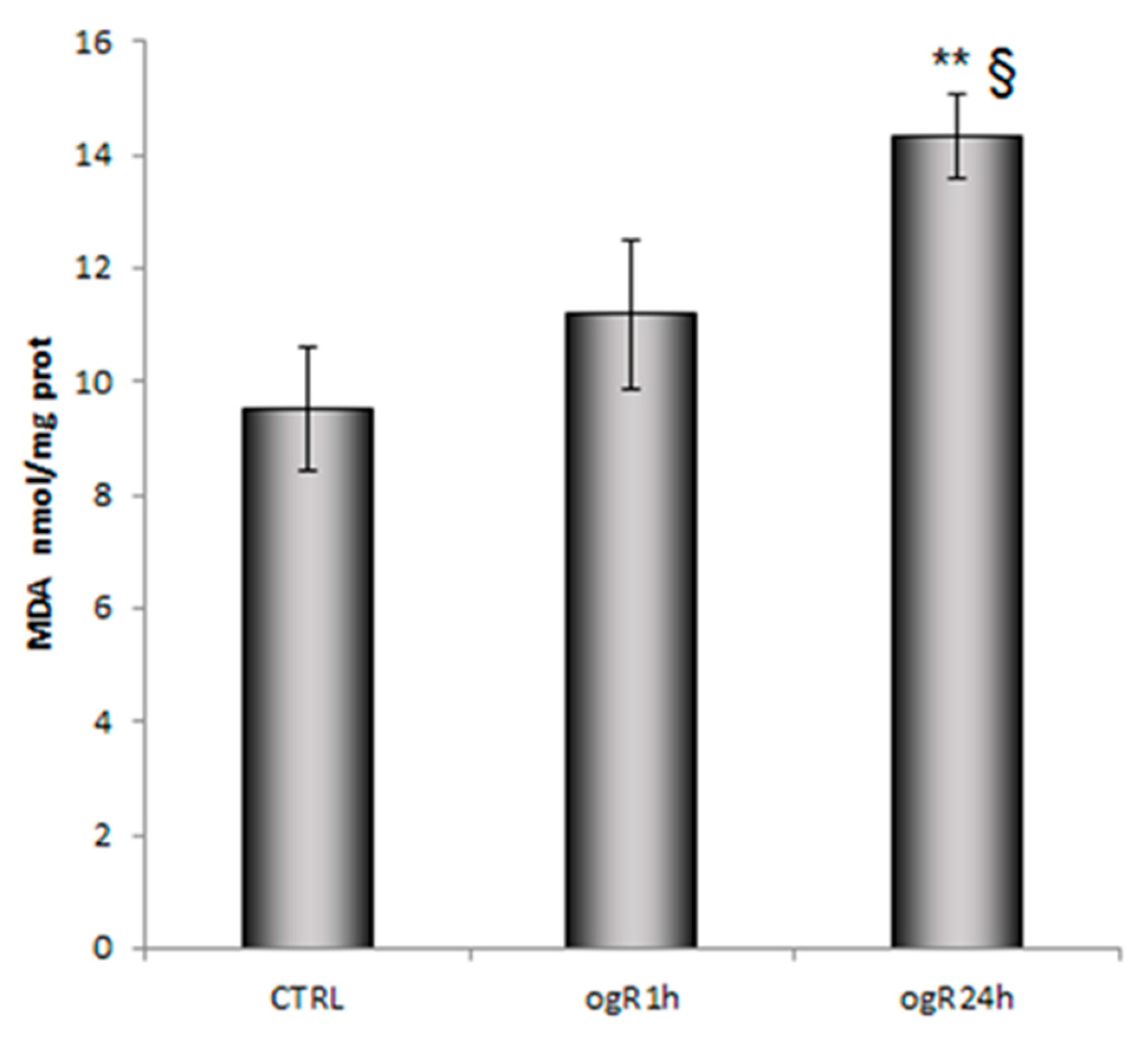
2.2. Lipid Peroxidation and AA Cascade Activation Under OGD/ogR
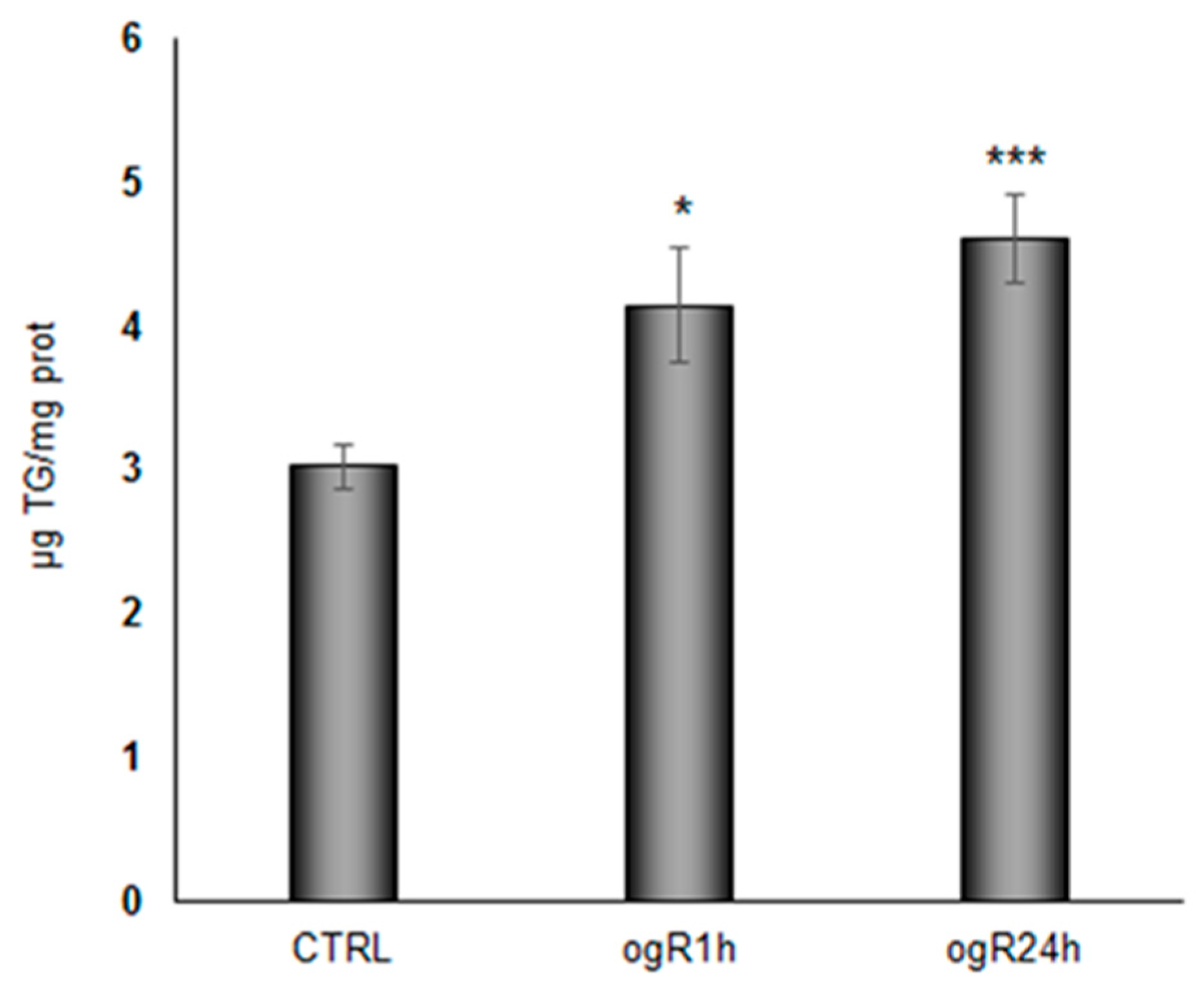
2.3. OGD/ogR Induces TG Increase and LD Formation
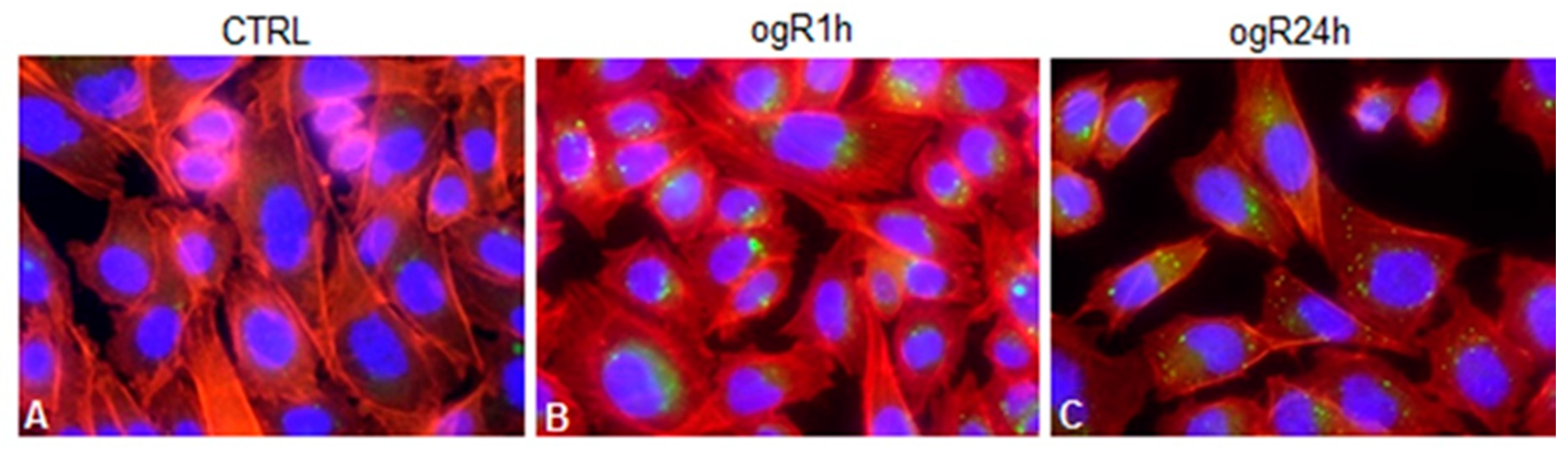
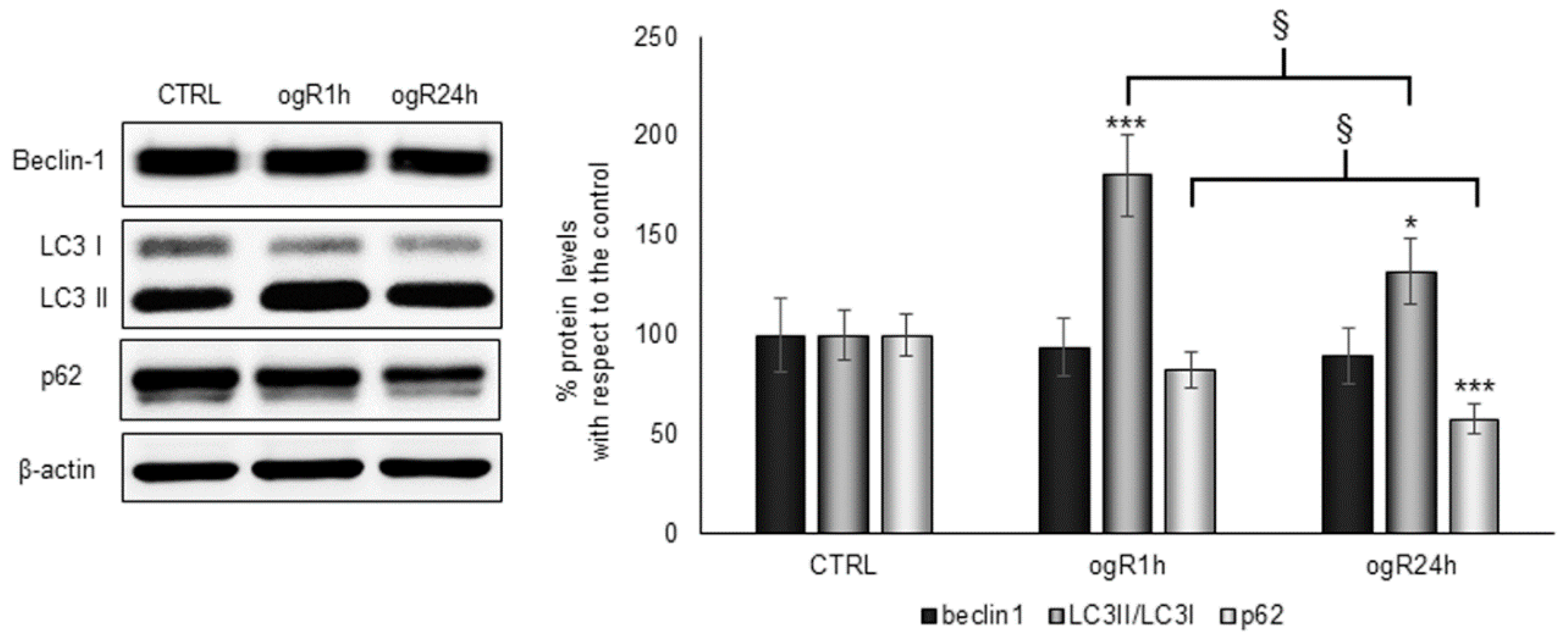
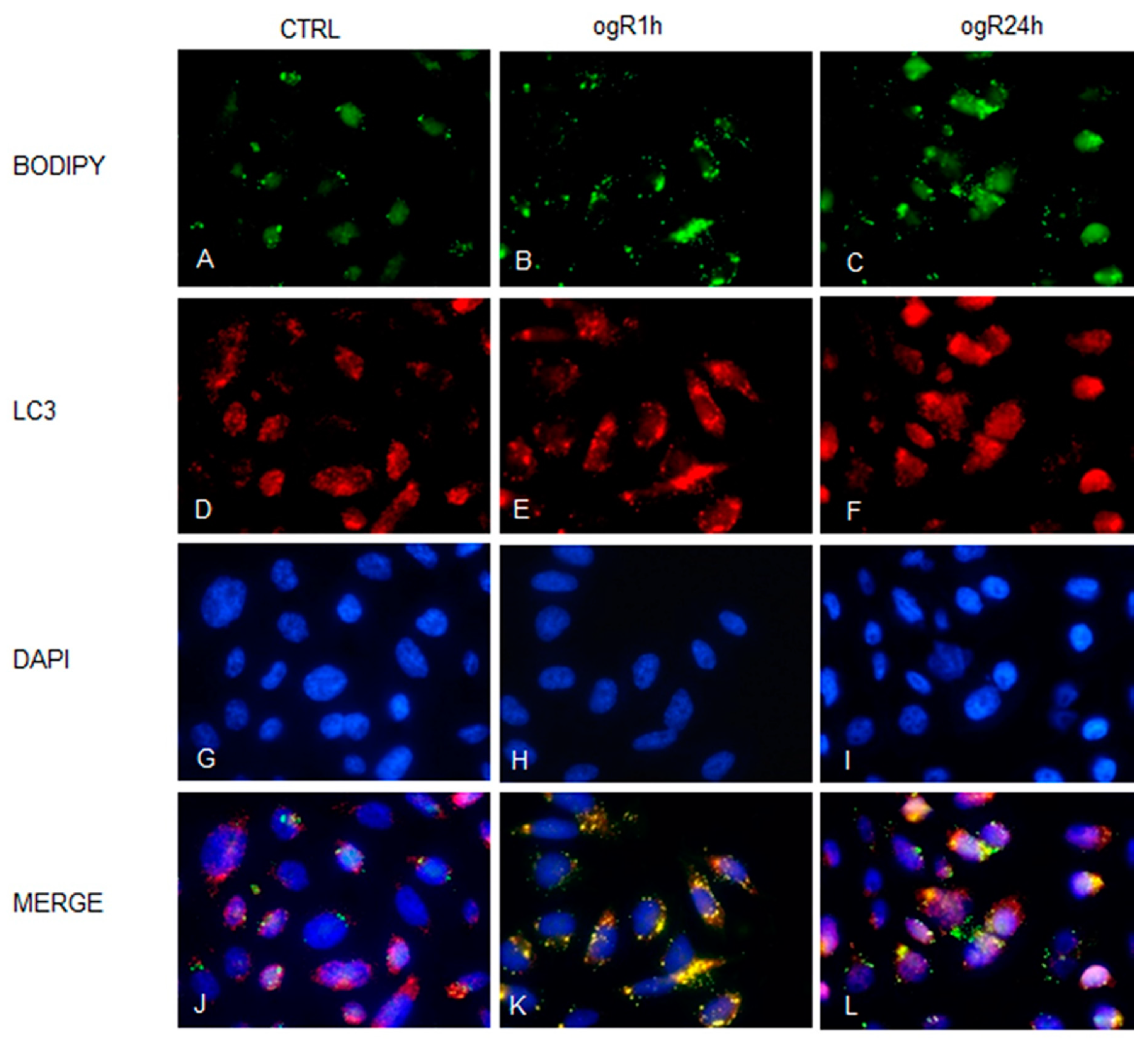
2.4. OGD/ogR-Induced Lipophagy
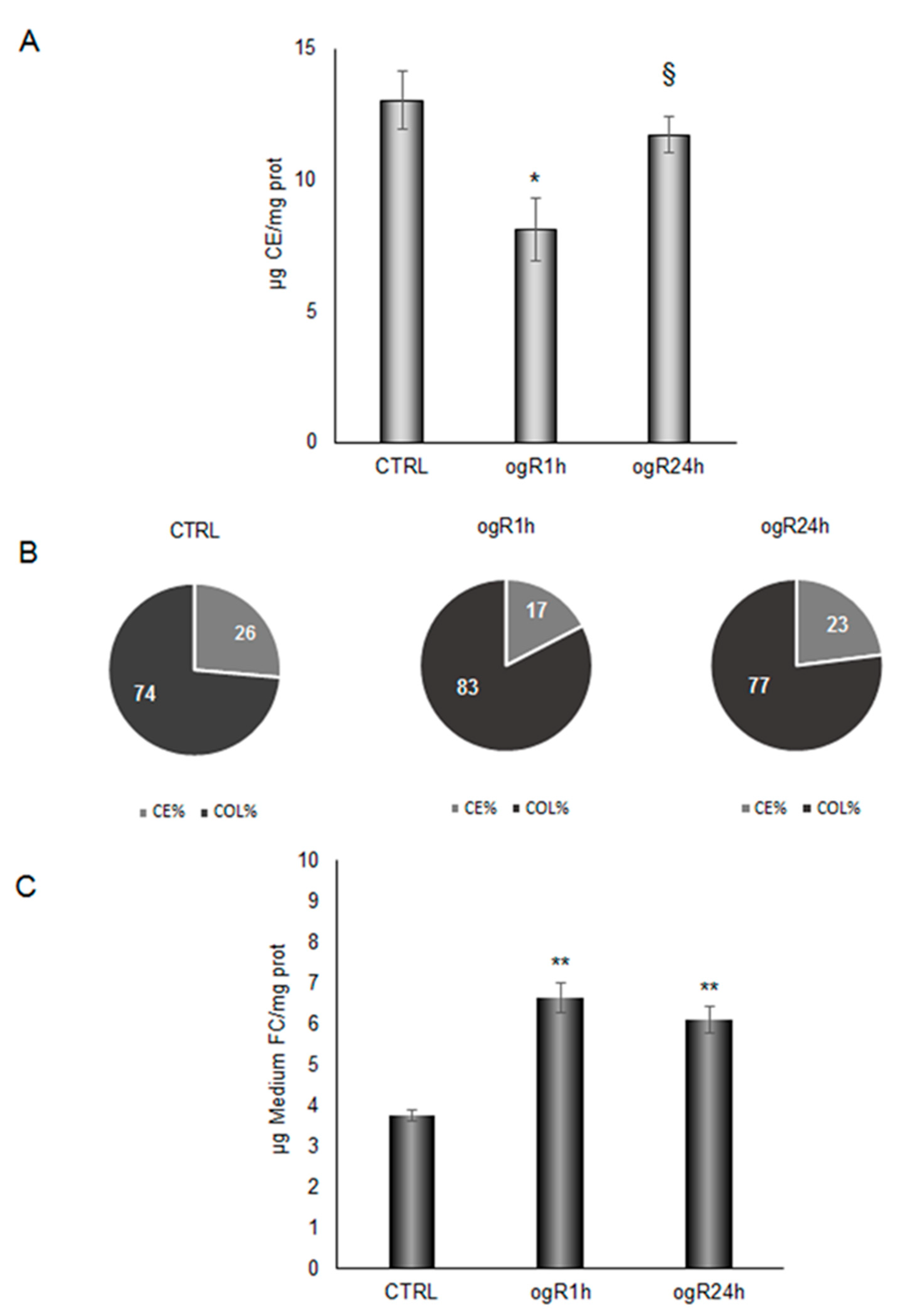
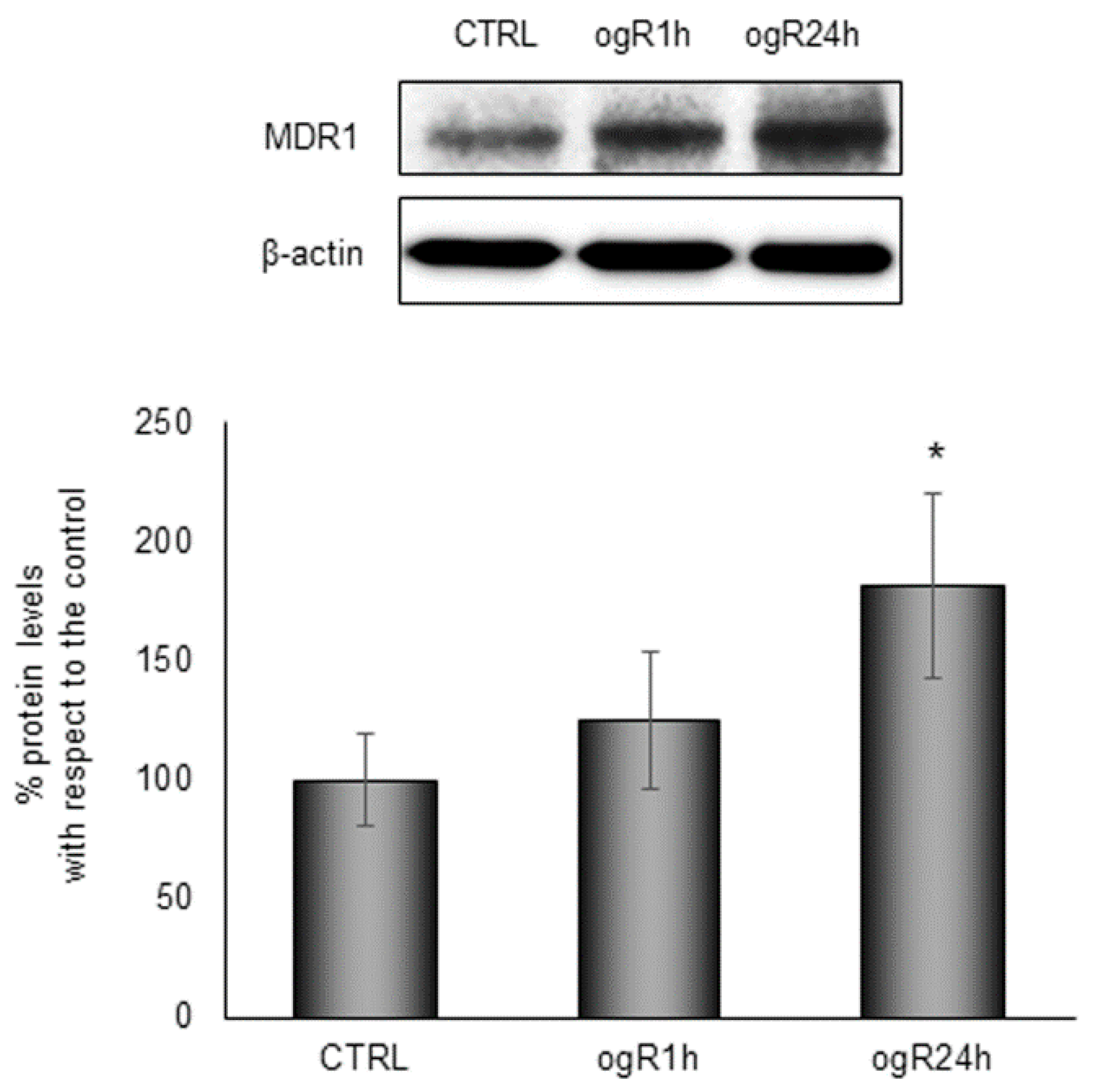
2.5. OGD/ogR-Induced CE Hydrolysis and FC Release in Extracellular Milieu
3. Materials and Methods
3.1. Materials
3.2. Cell Cultures
3.3. OGD Treatment
3.4. Homogenate Obtainment and MEFs Isolation
3.5. Electrophoresis and Immunoblotting
3.6. Lipid Extraction and Quantification
3.7. MDA Analysis
3.8. LD and LC3 Immunostaining
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharm. Rev. 2005, 5, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Prediction of blood–brain barrier permeation in drug discovery from in vivo, in vitro and in silico models. Drug Discov. Today Technol. 2004, 1, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Seelig, A. The role of size and charge for blood-brain barrier permeation of drugs and fatty acids. J. Mol. Neurosci. 2007, 33, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood–brainbarrier in health and disease. Epilepsia 2012, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Badaut, J.; Tang, J.; Obenaus, A.; Hartman, R.; Pearce, W.J. The vascular neural network-A new paradigm in stroke pathophysiology. Nat. Rev. Neurol. 2012, 8, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell Neurosci. 2016, 4, 56. [Google Scholar] [CrossRef]
- Tian, X.; Peng, J.; Zhong, J.; Yang, M.; Pang, J.N.J.; Li, M.; An, R.; Zhang, Q.; Xu, L.; Dong, Z. β-Caryophyllene Protects In Vitro Neurovascular Unit against Oxygen-Glucose Deprivation and Re-oxygenation-Induced Injury. J. Neurochem. 2016, 139, 757–768. [Google Scholar] [CrossRef]
- Boström, P.; Magnusson, B.; Svensson, P.A.; Wiklund, O.; Borén, J.; Carlsson, L.M.; Ståhlman, M.; Olofsson, S.O.; Hultén, L.M. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arter. Thromb. Vasc. Biol. 2006, 26, 1871–1876. [Google Scholar] [CrossRef]
- Lam, T.; Harmancey, R.; Vasquez, H.; Gilbert, B.; Patel, N.; Hariharan, V.; Lee, A.; Covey, M.; Taegtmeyer, H. Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Cell Death Discov. 2016, 2, 16061. [Google Scholar] [CrossRef]
- Unger, R.H.; Orci, L. Lipoapoptosis: Its mechanism and its diseases. Biochim. Biophys. Acta 2002, 1585, 202–212. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, I.M.; Cojocaru, M.; Sapira, V.; Ionescu, A. Evaluation of oxidative stress in patients with acute ischemic stroke. Rom. J. Intern. Med. 2013, 51, 97–106. [Google Scholar] [PubMed]
- Kirac, E.; Özcan, F.; Tuzcu, H.; Elpek, G.O.; Aslan, M. Analysis of polyunsaturated fatty acids and the omega-6 inflammatory pathway in hepatic ischemia/re-perfusion injury. Mol. Med. Rep. 2015, 12, 4149–4156. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, A.; Lonati, E.; Milani, C.; Rizzo, A.M.; Farina, F.; Botto, L.; Masserini, M.; Palestini, P.; Bulbarelli, A. Ischemic conditions and β-secretase activation: The impact of membrane cholesterol enrichment as triggering factor in rat brain endothelial cells. Int. J. Biochem. Cell Biol. 2015, 69, 95–104. [Google Scholar] [CrossRef]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Nito, C.; Kamada, H.; Endo, H.; Niizuma, K.; Myer, D.J.; Chan, P.H. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J. Cereb Blood Flow Metab. 2008, 28, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Dennis, E.A. Phospholipase A2 catalysis and lipid mediator lipidomics. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 766–771. [Google Scholar] [CrossRef]
- Donkor, J.; Zhang, P.; Wong, S.; O’Loughlin, L.; Dewald, J.; Kok, B.P.; Brindley, D.N.; Reue, K. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J. Biol. Chem. 2009, 284, 29968–29978. [Google Scholar] [CrossRef]
- Bulbarelli, A.; Lonati, E.; Brambilla, A.; Orlando, A.; Cazzaniga, E.; Piazza, F.; Ferrarese, C.; Masserini, M.; Sancini, G. Aβ production in brain capillary endothelial cells after oxygen and glucose deprivation. Mol. Cell Neurosci. 2012, 49, 415–422. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Z.; Zhao, C.; Wang, Y.; Wu, G.; Xiao, J.; McClain, C.J.; Li, X.; Feng, W. HIF-1α and HIF-2α are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1α-mediated fatty acid β-oxidation. Toxicol. Lett. 2014, 226, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lossinsky, A.S.; Shivers, R.R. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol. Histopathol. 2004, 19, 535–564. [Google Scholar] [PubMed]
- Moore, S.A. Polyunsaturated fatty acid synthesis and release by brain-derived cells in vitro. J. Mol. Neurosci. 2001, 16, 195–200. [Google Scholar] [CrossRef]
- Shi, Z.; Ren, H.; Luo, C.; Yao, X.; Li, P.; He, C.; Kang, J.X.; Wan, J.B.; Yuan, T.F.; Su, H. Enriched Endogenous Omega-3 Polyunsaturated Fatty Acids Protect Cortical Neurons from Experimental Ischemic Injury. Mol. Neurobiol. 2016, 53, 6482–6488. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef]
- Gubern, A.; Barceló-Torns, M.; Casas, J.; Barneda, D.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Lipid droplet biogenesis induced by stress involves triacylglycerol synthesis that depends on group VIA phospholipase A2. J. Biol. Chem. 2009, 284, 5697–5708. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef]
- Lonati, E.; Sala, G.; Tresoldi, V.; Coco, S.; Salerno, D.; Milani, C.; Losurdo, M.; Farina, F.; Botto, L.; Ferrarese, C.; et al. Ischemic Conditions Affect Rerouting of Tau Protein Levels: Evidences for Alteration in Tau Processing and Secretion in Hippocampal Neurons. J. Mol. Neurosci. 2018, 66, 604–616. [Google Scholar] [CrossRef]
- Ye, F.; Kaneko, H.; Hayashi, Y.; Takayama, K.; Hwang, S.J.; Nishizawa, Y.; Kimoto, R.; Nagasaka, Y.; Tsunekawa, T.; Matsuura, T.; et al. Malondialdehyde induces autophagy dysfunction and VEGF secretion in the retinal pigment epithelium in age-related macular degeneration. Free Radic. Biol. Med. 2016, 94, 121–134. [Google Scholar] [CrossRef]
- Shibata, M.; Yoshimura, K.; Furuya, N.; Koike, M.; Ueno, T.; Komatsu, M.; Arai, H.; Tanaka, K.; Kominami, E.; Uchiyama, Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys Res. Commun. 2009, 382, 419–423. [Google Scholar] [CrossRef]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Simmons, C.; Ingham, V.; Williams, A.; Bate, C. Platelet-activating factor antagonists enhance intracellular degradation of amyloid-β42 in neurons via regulation of cholesterol ester hydrolases. Alzheimers Res. Ther. 2014, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Zhao, B.; Bie, J.; Song, J. Macrophage cholesteryl ester mobilization and atherosclerosis. Vasc. Pharm. 2010, 52, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Marcel, Y.L. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arter. Thromb. Vasc. Biol. 2012, 32, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yu, X.H.; Zheng, X.L.; Ou, X.; Tang, C.K. Interferon-stimulated gene 15 promotes cholesterol efflux by activating autophagy via the miR-17-5p/Beclin-1 pathway in THP-1 macrophage-derived foam cells. Eur. J. Pharm. 2018, 827, 13–21. [Google Scholar] [CrossRef]
- ElAli, A.; Hermann, D.M. Liver X receptor activation enhances blood-brain barrier integrity in the ischemic brain and increases the abundance of ATP-binding cassette transporters ABCB1 and ABCC1 on brain capillary cells. Brain Pathol. 2012, 22, 175–187. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Vitale, N. Cholesterol and the journey of extracellular vesicles. J. Lipid Res. 2018, 59, 2255–2261. [Google Scholar] [CrossRef]
- Gao, Q.; Goodman, J.M. The lipid droplet-a well-connected organelle. Front. Cell Dev. Biol. 2015, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Fitsanakis, V.A.; dos Santos, A.P.; Olivi, L.; Bressler, J.P. Blood-brain barrier and cell-cell interactions: Methods for establishing in vitro models of the blood-brain barrier and transport measurements. Methods Mol. Biol. 2006, 341, 1–15. [Google Scholar]
- Balbuena, P.; Li, W.; Ehrich, M. Assessments of tight junction proteins occludin, claudin 5 and scaffold proteins ZO1 and ZO2 in endothelial cells of the rat blood-brain barrier: Cellular responses to neurotoxicants malathion and lead acetate. Neurotoxicology 2011, 32, 58–67. [Google Scholar] [CrossRef]
- Faria, A.; Pestana, D.; Teixeira, D.; Couraud, P.O.; Romero, I.; Weksler, B.; de Freitas, V.; Mateus, N.; Calhau, C. Insights into the putative catechin and epicatechin transport across blood-brain barrier. Food Funct. 2011, 2, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Roux, F.; Couraud, P.O. Rat brain endothelial cell lines for the study of blood-brain barrier permeability and transport functions. Cell Mol. Neurobiol. 2005, 25, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Fazakas, C.; Krizbai, I.A. In vitro models of the blood-brain barrier. Acta Neurobiol. Exp. 2011, 71, 113–128. [Google Scholar]
- Cimarosti, H.; Ashikaga, E.; Jaafari, N.; Dearden, L.; Rubin, P.; Wilkinson, K.A.; Henley, J.M. Enhanced SUMOylation and SENP-1 protein levels following oxygen and glucose deprivation in neurons. J. Cereb Blood Flow Metab. 2011, 32, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Chigurupati, S.; Arumugam, T.V.; Jo, D.G.; Li, H.; Chan, S.L. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke 2011, 42, 2589–2594. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.; Botto, L.; Chinello, C.; Cunati, D.; Magni, F.; Masserini, M.; Palestini, P. Characterization of prion protein-enriched domains, isolated from rat cerebellar granule cells in culture. J. Neurochem. 2009, 110, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.; Winston, S.E.; Fuller, S.A.; Hurrell, J.G. Immunoblotting and immunodetection. Curr. Protoc. Cell Biol. 2011, 83, 10–18. [Google Scholar] [CrossRef]
- Serini, S.; Cassano, R.; Corsetto, P.A.; Rizzo, A.M.; Calviello, G.; Trombino, S. Omega-3 PUFA Loaded in Resveratrol-Based Solid Lipid Nanoparticles: Physicochemical Properties and Antineoplastic Activities in Human Colorectal Cancer Cells In Vitro. Int. J. Mol. Sci. 2018, 19, 586. [Google Scholar] [CrossRef]
- Aoun, M.; Corsetto, P.A.; Nugue, G.; Montorfano, G.; Ciusani, E.; Crouzier, D.; Hogarth, P.; Gregory, A.; Hayflick, S.; Zorzi, G.; et al. Changes in Red Blood Cell membrane lipid composition: A new perspective into the pathogenesis of PKAN. Mol Genet. Metab. 2017, 121, 180–189. [Google Scholar] [CrossRef]
- Rizzo, A.M.; Corsetto, P.A.; Montorfano, G.; Opizzi, A.; Faliva, M.; Giacosa, A.; Ricevuti, G.; Pelucchi, C.; Berra, B.; Rondanelli, M. Comparison between the AA/EPA ratio in depressed and non depressed elderly females: Omega-3 fatty acid supplementation correlates with improved symptoms but does not change immunological parameters. Nutr. J. 2012, 11, 82. [Google Scholar] [CrossRef]
- Christie, W.W. Rapid separation and quantification of lipid classes by high performance liquid chromatography and mass (light-scattering) detection. J. Lipid Res. 1985, 26, 507–512. [Google Scholar] [PubMed]
- Ungaro, F.; Tacconi, C.; Massimino, L.; Corsetto, P.A.; Correale, C.; Fonteyne, P.; Piontini, A.; Garzarelli, V.; Calcaterra, F.; Della Bella, S.; et al. MFSD2A Promotes Endothelial Generation of Inflammation-Resolving Lipid Mediators and Reduces Colitis in Mice. Gastroenterology 2017, 153, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Karatas, F.; Karatepe, M.; Baysar, A. Determination of Free Malondialdehyde in Human Serum by High-Performance Liquid Chromatography. Anal. Biochem. 2002, 311, 76–79. [Google Scholar] [CrossRef]
- Layé, S.; Nadjar, A.; Joffre, C.; Bazinet, R.P. Anti-Inflammatory Effects of Omega-3 Fatty Acids in the Brain: Physiological Mechanisms and Relevance to Pharmacology. Pharm. Rev. 2018, 70, 12–38. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, F.; Jouin, M.; Alessandri, J.M.; Roux, F.; Perrière, N.; Langelier, B.; Lavialle, M.; Cunnane, S.; Guesnet, P. n-3 long-chain fatty acids and regulation of glucose transport in two models of rat brain endothelial cells. Neurochem Int. 2010, 56, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Simak, J.; Gelderman, M.P.; Yu, H.; Wright, V.; Baird, A.E. Circulating endothelial microparticles in acute ischemic stroke: A link to severity, lesion volume and outcome. J. Thromb. Haemost. 2006, 4, 1296–1302. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % | ||||||
|---|---|---|---|---|---|---|
| CTRL | ogR1h | ogR24h | ||||
| Mean ± S.E. | Mean ± S.E. | Mean ± S.E. | ||||
| C16:0 | 19.308 | 1.025 | 18.683 | 0.634 | 18.820 | 0.098 |
| C16:1 | 6.219 | 0.474 | 6.288 | 0.600 | 5.579 | 0.720 |
| C18:0 | 11.560 | 1.490 | 9.620 | 0.432 | 10.354 | 1.141 |
| C18:1 | 40.927 | 2,685 | 43.309 | 0.981 | 48.557 | 1.557 |
| C18:2 | 9.002 | 1.297 | 7.996 | 0.448 | 5.982 | 0.379 |
| C18:3 n-3 | 0.942 | 0.181 | 1.102 | 0.252 | 1.265 | 0.118 |
| C20:3 | 1.173 | 0.406 | 0.674 | 0.385 | 0.593 | 0.315 |
| C20:4 n-6 | 4.294 | 0.066 | 3.962 | 0.127 | 3.217 | 0.107 |
| C20:5 | 4,425 | 0.621 | 5.386 | 0.319 | 3.794 | 0.756 |
| C22:5 | 1.147 | 0.040 | 1.774 | 0.356 | 0.625 | 0.083 |
| C22:6 | 1.002 | 0.140 | 1.206 | 0.069 | 1.214 | 0.267 |
| PUFA | 21.985 | 1.414 | 22.099 | 1.143 | 16.690 | 0.848 |
| n-6 PUFA | 14.469 | 1.425 | 12.631 | 0.854 | 9.791 | 0.647 |
| n-3 PUFA | 6.979 | 0.342 | 9.467 | 0.387 | 6.899 | 0.965 |
| Total FA (µg/mg prot) | 20.634 | 2.497 | 16.929 | 0.998 | 23.904 | 0.699 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lonati, E.; Corsetto, P.A.; Montorfano, G.; Zava, S.; Carrozzini, T.; Brambilla, A.; Botto, L.; Palestini, P.; Rizzo, A.M.; Bulbarelli, A. Lipid Reshaping and Lipophagy Are Induced in a Modeled Ischemia-Reperfusion Injury of Blood Brain Barrier. Int. J. Mol. Sci. 2019, 20, 3752. https://doi.org/10.3390/ijms20153752
Lonati E, Corsetto PA, Montorfano G, Zava S, Carrozzini T, Brambilla A, Botto L, Palestini P, Rizzo AM, Bulbarelli A. Lipid Reshaping and Lipophagy Are Induced in a Modeled Ischemia-Reperfusion Injury of Blood Brain Barrier. International Journal of Molecular Sciences. 2019; 20(15):3752. https://doi.org/10.3390/ijms20153752
Chicago/Turabian StyleLonati, Elena, Paola Antonia Corsetto, Gigliola Montorfano, Stefania Zava, Tatiana Carrozzini, Anna Brambilla, Laura Botto, Paola Palestini, Angela Maria Rizzo, and Alessandra Bulbarelli. 2019. "Lipid Reshaping and Lipophagy Are Induced in a Modeled Ischemia-Reperfusion Injury of Blood Brain Barrier" International Journal of Molecular Sciences 20, no. 15: 3752. https://doi.org/10.3390/ijms20153752
APA StyleLonati, E., Corsetto, P. A., Montorfano, G., Zava, S., Carrozzini, T., Brambilla, A., Botto, L., Palestini, P., Rizzo, A. M., & Bulbarelli, A. (2019). Lipid Reshaping and Lipophagy Are Induced in a Modeled Ischemia-Reperfusion Injury of Blood Brain Barrier. International Journal of Molecular Sciences, 20(15), 3752. https://doi.org/10.3390/ijms20153752