A Systems Biology Approach for Personalized Medicine in Refractory Epilepsy

 ,
,  ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Heterogeneity in Pharmacoresistant Epilepsy
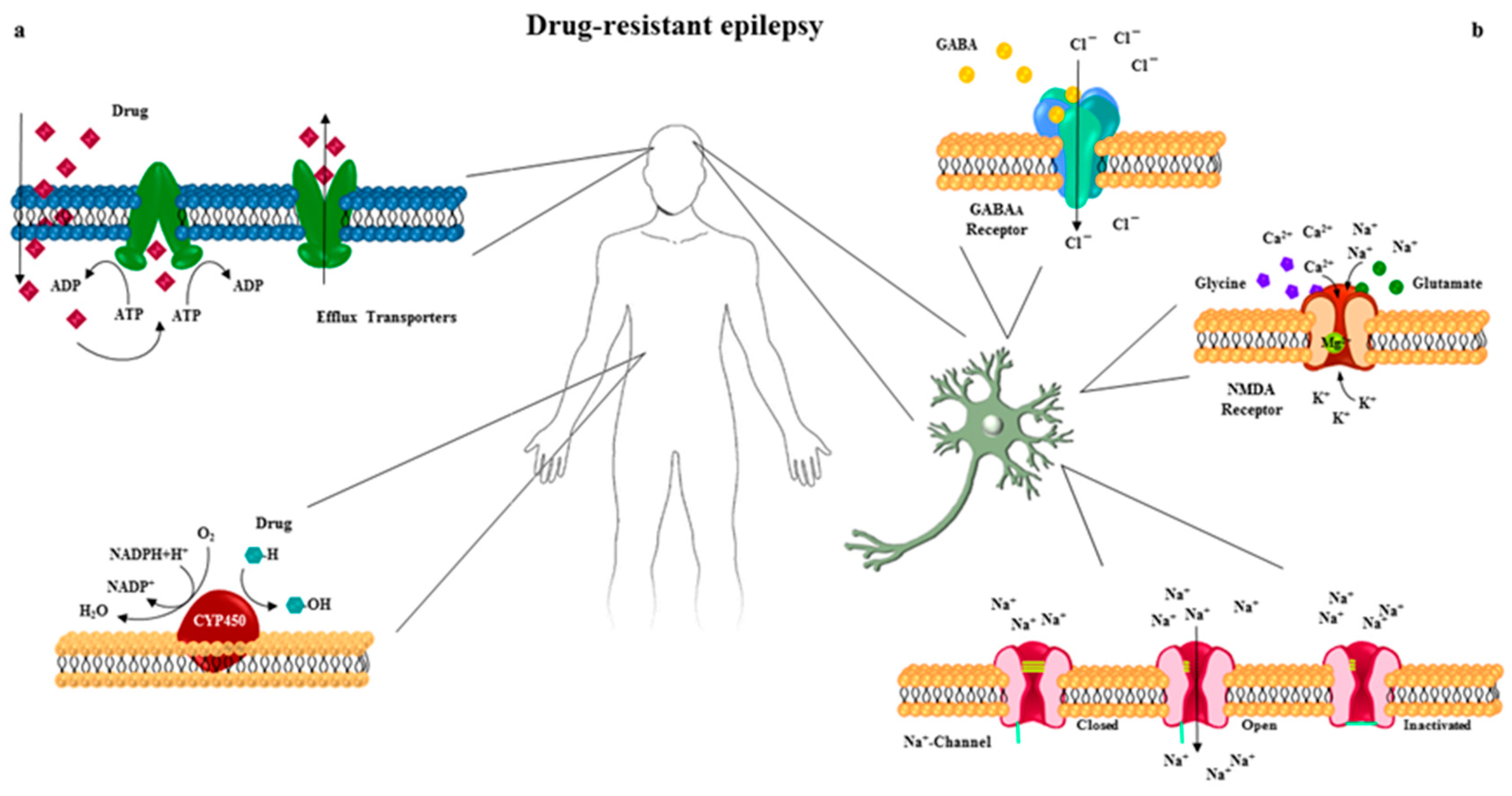
3. Drug Transporters in Pharmacoresistant Epilepsy
4. Cytochrome P450 Superfamily in Pharmacoresistant Epilepsy
5. Drug Target Sensitivity in Pharmacoresistant Epilepsy
5.1. The Voltage-Gated Na+ Channel
5.2. GABAA Receptors
5.3. Glutamate Receptors
5.4. GABAA and NMDA Receptors Trafficking
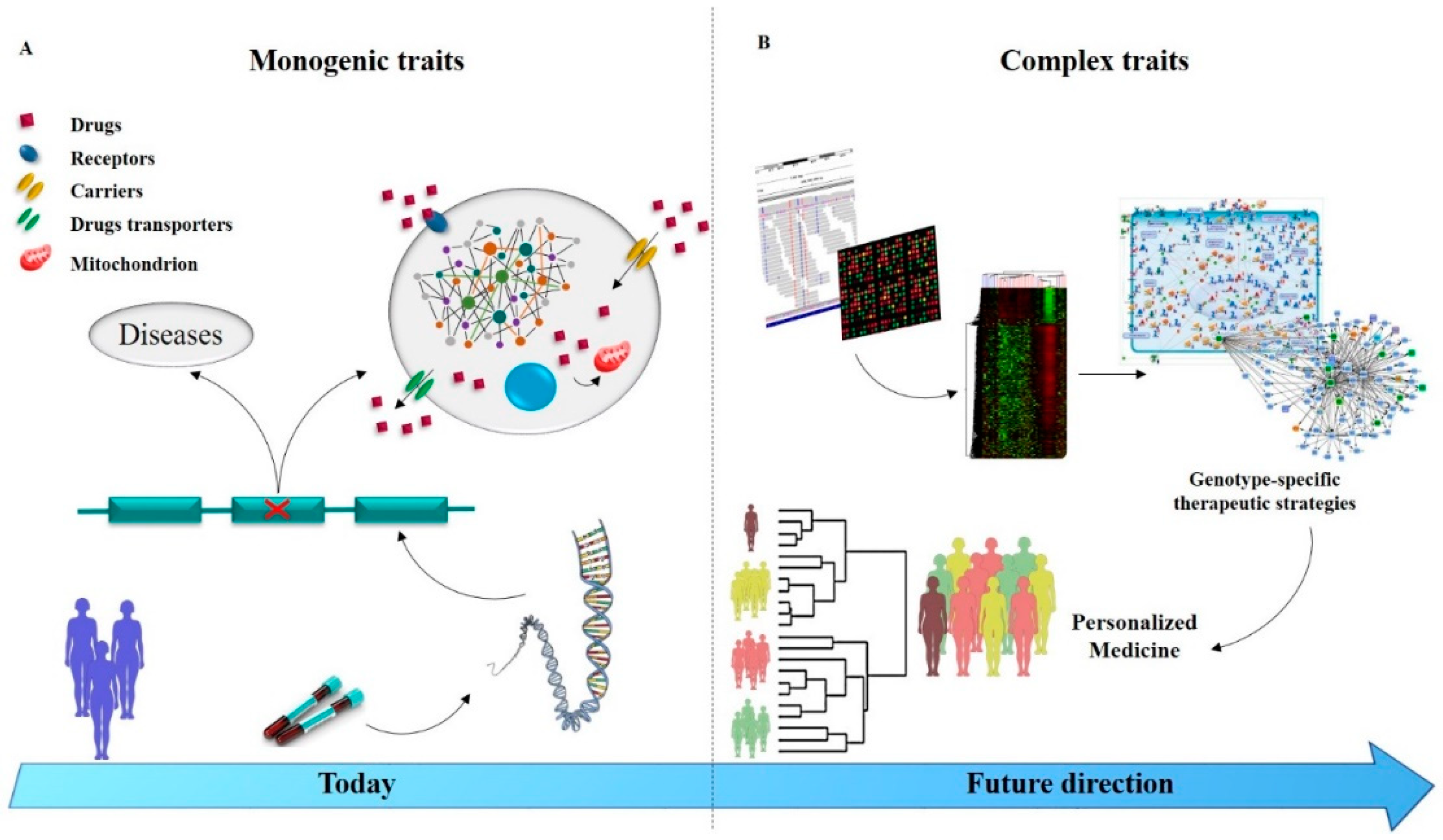
6. Managing Drug Resistance Epilepsy in the Genomic Era
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ILAE | International League Against Epilepsy |
| ABC | Adenosine triphosphate-Binding Cassette |
| P-gp | P-glycoprotein |
| GABA | Gamma Amino-Butyric Acid |
| CNS | Central Nervous System |
| NMDA | N-methyl-d-aspartate |
| aCGH | Array-Comparative Genomic Hybridization |
| NGS | Next Generation Sequencing |
| CNVs | Copy Number Variations |
| GLUT-1 | Glucose Transporter Type 1 |
| IGEs | Idiopathic Generalized Epilepsies |
| CAE | Childhood Absence Epilepsy |
References
- Tate, S.K.; Sisodiya, S.M. Multidrug resistance in epilepsy: a pharmacogenomic update. Expert Opin. Pharmacother. 2007, 8, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Refractory epilepsy: a progressive, intractable but preventable condition? Seizure-Eur. J. Epilepsy 2002, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.T.; Kelly, M.M. Defining intractability: comparisons among published definitions. Epilepsia 2006, 47, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshe, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- French, J.A. Refractory epilepsy: clinical overview. Epilepsia 2007, 48, 3–7. [Google Scholar] [CrossRef]
- Tang, F.; Hartz, A.; Bauer, B. Drug-resistant epilepsy: multiple hypotheses, few answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef]
- Greenfield, S.; Kravitz, R.; Duan, N.; Kaplan, S.H. Heterogeneity of treatment effects: implications for guidelines, payment, and quality assessment. Am. J. Med. 2007, 120, S3–S9. [Google Scholar] [CrossRef]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2005, 129, 18–35. [Google Scholar] [CrossRef]
- Löscher, W.; Klotz, U.; Zimprich, F.; Schmidt, D. The clinical impact of pharmacogenetics on the treatment of epilepsy. Epilepsia 2009, 50, 1–23. [Google Scholar] [CrossRef]
- Mirza, N.; Vasieva, O.; Marson, A.G.; Pirmohamed, M. Exploring the genomic basis of pharmacoresistance in epilepsy: an integrative analysis of large-scale gene expression profiling studies on brain tissue from epilepsy surgery. Hum. Mol. Genet. 2011, 20, 4381–4394. [Google Scholar] [CrossRef]
- Goldstein, D.B.; Need, A.C.; Singh, R.; Sisodiya, S.M. Potential genetic causes of heterogeneity of treatment effects. Am. J. Med. 2007, 120, S21–S25. [Google Scholar] [CrossRef] [PubMed]
- Cascorbi, I. Significance of Pharmacogenomics in Precision Medicine. Clin. Pharmacol. Ther. 2018, 103, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.J.; French, J.A. New antiepileptic drugs—Discovery, development, and update. Neurotherapeutics 2007, 4, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Muller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; A, L.L.; et al. Clinical Pharmacogenetics Implementation, C. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin. Pharm. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Snyder, M. Systems biology: personalized medicine for the future? Curr Opin Pharm. 2012, 12, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, D.G. Systems biology, complexity, and the impact on antiepileptic drug discovery. Epilepsy Behav. 2014, 38, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Scheffer, I.E.; Kiley, M. The management of epilepsy in children and adults. Med. J. Aust. 2018, 208, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef]
- Falco-Walter, J.J.; Scheffer, I.E.; Fisher, R.S. The new definition and classification of seizures and epilepsy. Epilepsy Res. 2018, 139, 73–79. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Lee, Y.J.; Hwang, S.K.; Kwon, S. The Clinical Spectrum of Benign Epilepsy with Centro-Temporal Spikes: a Challenge in Categorization and Predictability. J. Epilepsy Res. 2017, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Carmant, L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef] [PubMed]
- Beretta, S.; Carone, D.; Zanchi, C.; Bianchi, E.; Pirovano, M.; Trentini, C.; Padovano, G.; Colombo, M.; Cereda, D.; Scanziani, S.; et al. Long-term applicability of the new ILAE definition of epilepsy. Results from the PRO-LONG study. Epilepsia 2017, 58, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.J. Approaches to refractory epilepsy. Ann. Indian Acad. Neurol. 2014, 17, S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Kerb, R.; Hoffmeyer, S.; Brinkmann, U. ABC drug transporters: hereditary polymorphisms and pharmacological impact in MDR1, MRP1 and MRP2. Pharmacogenomics 2001, 2, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—an update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J. Pharmacol. Exp. Ther. 2002, 301, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Sisodiya, S.M. Mechanisms of antiepileptic drug resistance. Curr. Opin. Neurol. 2003, 16, 197–201. [Google Scholar] [CrossRef]
- Tishler, D.M.; Weinberg, K.I.; Hinton, D.R.; Barbaro, N.; Annett, G.M.; Raffel, C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia 1995, 36, 1–6. [Google Scholar] [CrossRef]
- Hoffmeyer, S.; Burk, O.; Von Richter, O.; Arnold, H.P.; Brockmöller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. 2000, 97, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Kerb, R.; Weale, M.E.; Brinkmann, U.; Smith, A.; Goldstein, D.B.; Wood, N.W.; Sisodiya, S.M. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N. Engl. J. Med. 2003, 348, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-C.; Jen Tai, J.; Kao, P.-J.; Lin, M.-S.; Liou, H.-H. Association of polymorphisms in NR1I2 and ABCB1 genes with epilepsy treatment responses. Pharmacogenomics 2007, 7, 551–561. [Google Scholar]
- Kwan, P.; Baum, L.; Wong, V.; Ng, P.W.; Lui, C.H.; Sin, N.C.; Hui, A.C.; Yu, E.; Wong, L.K. Association between ABCB1 C3435T polymorphism and drug-resistant epilepsy in Han Chinese. Epilepsy Behav. 2007, 11, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Keangpraphun, T.; Towanabut, S.; Chinvarun, Y.; Kijsanayotin, P. Association of ABCB 1 C3435T polymorphism with phenobarbital resistance in Thai patients with epilepsy. J. Clin. Pharm. Ther. 2015, 40, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Seo, T.; Ishitsu, T.; Ueda, N.; Nakada, N.; Yurube, K.; Ueda, K.; Nakagawa, K. ABCB1 polymorphisms influence the response to antiepileptic drugs in Japanese epilepsy patients. Pharmacogenomics 2006. [Google Scholar] [CrossRef]
- Subenthiran, S.; Abdullah, N.R.; Joseph, J.P.; Muniandy, P.K.; Mok, B.T.; Kee, C.C.; Ismail, Z.; Mohamed, Z. Linkage disequilibrium between polymorphisms of ABCB1 and ABCC2 to predict the treatment outcome of Malaysians with complex partial seizures on treatment with carbamazepine mono-therapy at the Kuala Lumpur Hospital. PloS ONE 2013, 8, e64827. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Cao, B.Q.; Wang, B.; Wu, S.Q.; Jiang, D.H. Association between ABCB1 genetic polymorphism and the effect on epilepsy following phenytoin treatment. Exp. Ther. Med. 2016, 12, 1780–1784. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maleki, M.; Sayyah, M.; Kamgarpour, F.; Karimipoor, M.; Arab, A.; Rajabi, A.; Gharagozli, K.; Shamshiri, A.R.; Ananloo, E.S. Association between ABCB1-T1236C polymorphism and drug-resistant epilepsy in Iranian female patients. Iran. Biomed. J. 2010, 14, 89. [Google Scholar]
- Vahab, S.A.; Sen, S.; Ravindran, N.; Mony, S.; Mathew, A.; Vijayan, N.; Nayak, G.; Bhaskaranand, N.; Banerjee, M.; Satyamoorthy, K. Analysis of genotype and haplotype effects of ABCB1 (MDR1) polymorphisms in the risk of medically refractory epilepsy in an Indian population. Drug Metab. Pharm. 2009, 24, 255–260. [Google Scholar] [CrossRef]
- Alpman, A.; Ozkinay, F.; Tekgul, H.; Gokben, S.; Pehlivan, S.; Schalling, M.; Ozkinay, C. Multidrug resistance 1 (MDR1) gene polymorphisms in childhood drug-resistant epilepsy. J. Child. Neurol. 2010, 25, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Haerian, B.S.; Lim, K.S.; Mohamed, E.H.; Tan, H.J.; Tan, C.T.; Raymond, A.A.; Wong, C.P.; Wong, S.W.; Mohamed, Z. Lack of association of ABCB1 haplotypes on five loci with response to treatment in epilepsy. Seizure 2011, 20, 546–553. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, D.W.; Lee, S.K.; Chu, K.; Jang, I.J.; Yu, K.S.; Cho, J.Y.; Kim, S.J. Lack of association between ABCB1, ABCG2, and ABCC2 genetic polymorphisms and multidrug resistance in partial epilepsy. Epilepsy Res. 2009, 84, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-C.; Tai, J.J.; Lin, C.-J.; Lee, M.-J.; Liou, H.-H. Complex haplotypic effects of the ABCB1 gene on epilepsy treatment response. Pharmacogenomics 2005, 6, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wu, L.; Jin, L.; Xu, Q.; Shen, Y. Association analysis of a polymorphism of MDR1 gene and refractory temporal lobe epilepsy in a Chinese population. Neurol. Asia 2007, 12, 94–95. [Google Scholar]
- Zimprich, F.; Sunder-Plassmann, R.; Stogmann, E.; Gleiss, A.; Dal-Bianco, A.; Zimprich, A.; Plumer, S.; Baumgartner, C.; Mannhalter, C. Association of an ABCB1 gene haplotype with pharmacoresistance in temporal lobe epilepsy. Neurology 2004, 63, 1087–1089. [Google Scholar] [CrossRef]
- Ufer, M.; Mosyagin, I.; Muhle, H.; Jacobsen, T.; Haenisch, S.; Häsler, R.; Faltraco, F.; Remmler, C.; von Spiczak, S.; Kroemer, H.K. Non-response to antiepileptic pharmacotherapy is associated with the ABCC2− 24C> T polymorphism in young and adult patients with epilepsy. Pharm. Genom. 2009, 19, 353–362. [Google Scholar] [CrossRef]
- Lakhan, R.; Kumari, R.; Singh, K.; Kalita, J.; Misra, U.K.; Mittal, B. Possible role of CYP2C9 & CYP2C19 single nucleotide polymorphisms in drug refractory epilepsy. Indian J. Med. Res. 2011, 134, 295–301. [Google Scholar]
- Lopez-Garcia, M.A.; Feria-Romero, I.A.; Serrano, H.; Rayo-Mares, D.; Fagiolino, P.; Vazquez, M.; Escamilla-Nunez, C.; Grijalva, I.; Escalante-Santiago, D.; Orozco-Suarez, S. Influence of genetic variants of CYP2D6, CYP2C9, CYP2C19 and CYP3A4 on antiepileptic drug metabolism in pediatric patients with refractory epilepsy. Pharm. Rep. 2017, 69, 504–511. [Google Scholar] [CrossRef]
- Saruwatari, J.; Ishitsu, T.; Nakagawa, K. Update on the Genetic Polymorphisms of Drug-Metabolizing Enzymes in Antiepileptic Drug Therapy. Pharmaceuticals 2010, 3, 2709–2732. [Google Scholar] [CrossRef]
- Talwar, P.; Kanojia, N.; Mahendru, S.; Baghel, R.; Grover, S.; Arora, G.; Grewal, G.K.; Parween, S.; Srivastava, A.; Singh, M.; et al. Genetic contribution of CYP1A1 variant on treatment outcome in epilepsy patients: a functional and interethnic perspective. Pharm. J. 2017, 17, 242–251. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome p450 system. J. Adv. Pr. Oncol. 2013, 4, 263–268. [Google Scholar]
- Seven, M.; Batar, B.; Unal, S.; Yesil, G.; Yuksel, A.; Guven, M. The effect of genetic polymorphisms of cytochrome P450 CYP2C9, CYP2C19, and CYP2D6 on drug-resistant epilepsy in Turkish children. Mol. Diagn. Ther. 2014, 18, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [PubMed]
- Pilgrim, J.L.; Gerostamoulos, D.; Drummer, O.H. Review: Pharmacogenetic aspects of the effect of cytochrome P450 polymorphisms on serotonergic drug metabolism, response, interactions, and adverse effects. Forensic Sci. Med. Pathol. 2011, 7, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Dresser, G.K.; Spence, J.D.; Bailey, D.G. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin. Pharm. 2000, 38, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. How to explain multidrug resistance in epilepsy? Epilepsy Curr. 2005, 5, 107–112. [Google Scholar] [CrossRef]
- Remy, S.; Gabriel, S.; Urban, B.W.; Dietrich, D.; Lehmann, T.N.; Elger, C.E.; Heinemann, U.; Beck, H. A novel mechanism underlying drug resistance in chronic epilepsy. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2003, 53, 469–479. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Löscher, W.; Rho, J.M. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb. Perspect. Med. 2016, a022780. [Google Scholar] [CrossRef]
- Hitiris, N.; Brodie, M.J. Modern antiepileptic drugs: guidelines and beyond. Curr. Opin. Neurol. 2006, 19, 175–180. [Google Scholar] [CrossRef]
- de Lera Ruiz, M.; Kraus, R.L. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; Avoli, M. Sodium channels as molecular targets for antiepileptic drugs. Brain Res. Rev. 1998, 26, 16–28. [Google Scholar] [CrossRef]
- Catterall, W.A. Molecular properties of brain sodium channels: an important target for anticonvulsant drugs. Adv. Neurol. 1999, 79, 441–456. [Google Scholar] [PubMed]
- Köhling, R. Voltage-gated sodium channels in epilepsy. Epilepsia 2002, 43, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Lucas, P.T.; Meadows, L.S.; Nicholls, J.; Ragsdale, D.S. An epilepsy mutation in the β1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin. Epilepsy Res. 2005, 64, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Yankaya, B.; Troost, D.; Van Vliet, E.A.; Da Silva, F.H.L.; Gorter, J.A. Induction of neonatal sodium channel II and III α-isoform mRNAs in neurons and microglia after status epilepticus in the rat hippocampus. Eur. J. Neurosci. 2001, 13, 1261–1266. [Google Scholar] [CrossRef]
- Ellerkmann, R.; Remy, S.; Chen, J.; Sochivko, D.; Elger, C.; Urban, B.; Becker, A.; Beck, H. Molecular and functional changes in voltage-dependent Na+ channels following pilocarpine-induced status epilepticus in rat dentate granule cells. Neuroscience 2003, 119, 323–333. [Google Scholar] [CrossRef]
- Kwan, P.; Poon, W.S.; Ng, H.-K.; Kang, D.E.; Wong, V.; Ng, P.W.; Lui, C.H.; Sin, N.C.; Wong, K.S.; Baum, L. Multidrug resistance in epilepsy and polymorphisms in the voltage-gated sodium channel genes SCN1A, SCN2A, and SCN3A: correlation among phenotype, genotype, and mRNA expression. Pharm. Genom. 2008, 18, 989–998. [Google Scholar] [CrossRef]
- Kumari, R.; Lakhan, R.; Garg, R.; Kalita, J.; Misra, U.; Mittal, B. Pharmacogenomic association study on the role of drug metabolizing, drug transporters and drug target gene polymorphisms in drug-resistant epilepsy in a north Indian population. Indian J. Hum. Genet. 2011, 17, S32. [Google Scholar]
- Lakhan, R.; Kumari, R.; Misra, U.K.; Kalita, J.; Pradhan, S.; Mittal, B. Differential role of sodium channels SCN1A and SCN2A gene polymorphisms with epilepsy and multiple drug resistance in the north Indian population. Br. J. Clin. Pharmacol. 2009, 68, 214–220. [Google Scholar] [CrossRef]
- Abe, T.; Seo, T.; Ishitsu, T.; Nakagawa, T.; Hori, M.; Nakagawa, K. Association between SCN1A polymorphism and carbamazepine-resistant epilepsy. Br. J. Clin. Pharmacol. 2008, 66, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-L.; Wu, X.-Y.; Zheng, J.; Wu, Z.-Y.; Hong, Z.; Zhong, M.-K. Association of SCN1A, SCN2A and ABCC2 gene polymorphisms with the response to antiepileptic drugs in Chinese Han patients with epilepsy. Pharmacogenomics 2014, 15, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Margari, L.; Legrottaglie, A.R.; Vincenti, A.; Coppola, G.; Operto, F.F.; Buttiglione, M.; Cassano, A.; Bartolomeo, N.; Mariggiò, M.A. Association between SCN1A gene polymorphisms and drug resistant epilepsy in pediatric patients. Seizure 2018, 55, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: phasic and tonic activation of GABA A receptors. Nat. Rev. Neurosci. 2005, 6, 215. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABA A receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Chua, H.C.; Chebib, M. GABAA Receptors and the Diversity in their Structure and Pharmacology. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 79, pp. 1–34. [Google Scholar]
- Bethmann, K.; Fritschy, J.-M.; Brandt, C.; Löscher, W. Antiepileptic drug resistant rats differ from drug responsive rats in GABAA receptor subunit expression in a model of temporal lobe epilepsy. Neurobiol. Dis. 2008, 31, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Granger, P.; Biton, B.; Faure, C.; Vige, X.; Depoortere, H.; Graham, D.; Langer, S.Z.; Scatton, B.; Avenet, P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol. Pharmacol. 1995, 47, 1189–1196. [Google Scholar]
- Sieghart, W. Structure and pharmacology of gamma-aminobutyric acidA receptor subtypes. Pharmacol. Rev. 1995, 47, 181–234. [Google Scholar]
- Simeone, T.A.; Wilcox, K.S.; White, H.S. Subunit selectivity of topiramate modulation of heteromeric GABAA receptors. Neuropharmacology 2006, 50, 845–857. [Google Scholar] [CrossRef]
- Brooks-Kayal, A.; Shumate, M.; Jin, H.; Rikhter, T.; Coulter, D. Selective changes in single cell GABAA receptor expression and functionin temporal lobe epilepsy. Nat. Med. 1999, 5, 590. [Google Scholar] [CrossRef]
- Bowser, D.N.; Wagner, D.A.; Czajkowski, C.; Cromer, B.A.; Parker, M.W.; Wallace, R.H.; Harkin, L.A.; Mulley, J.C.; Marini, C.; Berkovic, S.F. Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [γ2 (R43Q)] found in human epilepsy. Proc. Natl. Acad. Sci. USA 2002, 99, 15170–15175. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Lakhan, R.; Kalita, J.; Misra, U.; Mittal, B. Association of alpha subunit of GABAA receptor subtype gene polymorphisms with epilepsy susceptibility and drug resistance in north Indian population. Seizure 2010, 19, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-C.; Chen, P.-L.; Huang, W.-M.; Tai, J.J.; Hsieh, T.-J.; Ding, S.-T.; Hsieh, Y.-W.; Liou, H.-H. Gene-wide tagging study of the effects of common genetic polymorphisms in the α subunits of the GABAA receptor on epilepsy treatment response. Pharmacogenomics 2013, 14, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef]
- Hackos, D.H.; Hanson, J.E. Diverse modes of NMDA receptor positive allosteric modulation: Mechanisms and consequences. Neuropharmacology 2017, 112, 34–45. [Google Scholar] [CrossRef]
- Addis, L.; Virdee, J.; Vidler, L.; Collier, D.; Pal, D.; Ursu, D. Epilepsy-associated GRIN2A mutations reduce NMDA receptor trafficking and agonist potency–molecular profiling and functional rescue. Sci. Rep. 2017, 7, 66. [Google Scholar] [CrossRef]
- Chen, W.; Tankovic, A.; Burger, P.B.; Kusumoto, H.; Traynelis, S.F.; Yuan, H. Functional evaluation of a de novo GRIN2A mutation identified in a patient with profound global developmental delay and refractory epilepsy. Mol. Pharmacol. 2017, 91, 317–330. [Google Scholar] [CrossRef]
- Yuan, H.; Hansen, K.B.; Zhang, J.; Pierson, T.M.; Markello, T.C.; Fajardo, K.V.F.; Holloman, C.M.; Golas, G.; Adams, D.R.; Boerkoel, C.F. Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat. Commun. 2014, 5, 3251. [Google Scholar] [CrossRef]
- Hung, C.-C.; Ho, J.-L.; Chang, W.-L.; Tai, J.J.; Hsieh, T.-J.; Hsieh, Y.-W.; Liou, H.-H. Association of genetic variants in six candidate genes with valproic acid therapy optimization. Pharmacogenomics 2011, 12, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, H.P.; Sun, C.; Yeh, J.L.; Mangan, P.S.; Kapur, J. GABAA receptor internalization during seizures. Epilepsia 2007, 48, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.E.; Liu, H.; Wasterlain, C.G. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 2005, 25, 7724–7733. [Google Scholar] [CrossRef] [PubMed]
- Niquet, J.; Baldwin, R.; Suchomelova, L.; Lumley, L.; Naylor, D.; Eavey, R.; Wasterlain, C.G. Benzodiazepine-refractory status epilepticus: pathophysiology and principles of treatment. Ann. N. Y. Acad. Sci. 2016, 1378, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, H.P.; Yeh, J.-L.; Kapur, J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J. Neurosci. 2005, 25, 5511–5520. [Google Scholar] [CrossRef]
- Naylor, D.E.; Liu, H.; Niquet, J.; Wasterlain, C.G. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis. 2013, 54, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-J.; Kim, H.; Kim, W.-J.; Chung, S.; Kim, Y.-H.; Cho, I.; Lee, B.I.; Heo, K. Trafficking patterns of NMDA and GABAA receptors in a Mg2+-free cultured hippocampal neuron model of status epilepticus. Epilepsy Res. 2017, 136, 143–148. [Google Scholar] [CrossRef]
- Wasterlain, C.G.; Baldwin, R.; Naylor, D.E.; Thompson, K.W.; Suchomelova, L.; Niquet, J. Rational polytherapy in the treatment of acute seizures and status epilepticus. Epilepsia 2011, 52, 70–71. [Google Scholar] [CrossRef]
- Glauser, T.; Shinnar, S.; Gloss, D.; Alldredge, B.; Arya, R.; Bainbridge, J.; Bare, M.; Bleck, T.; Dodson, W.E.; Garrity, L.; et al. Evidence-Based Guideline: Treatment of Convulsive Status Epilepticus in Children and Adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016, 16, 48–61. [Google Scholar] [CrossRef]
- Poduri, A. When Should Genetic Testing Be Performed in Epilepsy Patients? Epilepsy Curr. 2017, 17, 16–22. [Google Scholar] [CrossRef]
- Handel, A.E.; Disanto, G.; Ramagopalan, S.V. Next-generation sequencing in understanding complex neurological disease. Expert Rev. Neurother. 2013, 13, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Pong, A.W.; Geary, B.R.; Engelstad, K.M.; Natarajan, A.; Yang, H.; De Vivo, D.C. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia 2012, 53, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Helbig, I.; Mefford, H.C.; Sharp, A.J.; Guipponi, M.; Fichera, M.; Franke, A.; Muhle, H.; De Kovel, C.; Baker, C.; Von Spiczak, S. 15q13. 3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009, 41, 160. [Google Scholar] [CrossRef] [PubMed]
- Dibbens, L.M.; Mullen, S.; Helbig, I.; Mefford, H.C.; Bayly, M.A.; Bellows, S.; Leu, C.; Trucks, H.; Obermeier, T.; Wittig, M. Familial and sporadic 15q13. 3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum. Mol. Genet. 2009, 18, 3626–3631. [Google Scholar] [CrossRef] [PubMed]
- De Kovel, C.G.; Trucks, H.; Helbig, I.; Mefford, H.C.; Baker, C.; Leu, C.; Kluck, C.; Muhle, H.; Von Spiczak, S.; Ostertag, P. Recurrent microdeletions at 15q11. 2 and 16p13. 11 predispose to idiopathic generalized epilepsies. Brain 2009, 133, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Muhle, H.; Ostertag, P.; von Spiczak, S.; Buysse, K.; Baker, C.; Franke, A.; Malafosse, A.; Genton, P.; Thomas, P. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010, 6, e1000962. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Mortazavi, A. The common ground of genomics and systems biology. BMC Syst. Biol. 2014, 8, S1. [Google Scholar] [CrossRef]
- Delles, C.; Husi, H. Systems biology approach in hypertension research. In Hypertension; Springer: Heidelberg, Germany, 2017; pp. 69–79. [Google Scholar]
- Noell, G.; Faner, R.; Agusti, A. From systems biology to P4 medicine: applications in respiratory medicine. Eur. Respir Rev. 2018, 27, 147. [Google Scholar] [CrossRef]
- Wang, L. Pharmacogenomics: a systems approach. Wiley Interdiscip Rev. Syst. Biol. Med. 2010, 2, 3–22. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Huang, R.S.; Cox, N.J. SCAN: a systems biology approach to pharmacogenomic discovery. Methods Mol. Biol. 2013, 1015, 213–224. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naimo, G.D.; Guarnaccia, M.; Sprovieri, T.; Ungaro, C.; Conforti, F.L.; Andò, S.; Cavallaro, S. A Systems Biology Approach for Personalized Medicine in Refractory Epilepsy. Int. J. Mol. Sci. 2019, 20, 3717. https://doi.org/10.3390/ijms20153717
Naimo GD, Guarnaccia M, Sprovieri T, Ungaro C, Conforti FL, Andò S, Cavallaro S. A Systems Biology Approach for Personalized Medicine in Refractory Epilepsy. International Journal of Molecular Sciences. 2019; 20(15):3717. https://doi.org/10.3390/ijms20153717
Chicago/Turabian StyleNaimo, Giuseppina Daniela, Maria Guarnaccia, Teresa Sprovieri, Carmine Ungaro, Francesca Luisa Conforti, Sebastiano Andò, and Sebastiano Cavallaro. 2019. "A Systems Biology Approach for Personalized Medicine in Refractory Epilepsy" International Journal of Molecular Sciences 20, no. 15: 3717. https://doi.org/10.3390/ijms20153717
APA StyleNaimo, G. D., Guarnaccia, M., Sprovieri, T., Ungaro, C., Conforti, F. L., Andò, S., & Cavallaro, S. (2019). A Systems Biology Approach for Personalized Medicine in Refractory Epilepsy. International Journal of Molecular Sciences, 20(15), 3717. https://doi.org/10.3390/ijms20153717