CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities

 , and
, and
Abstract
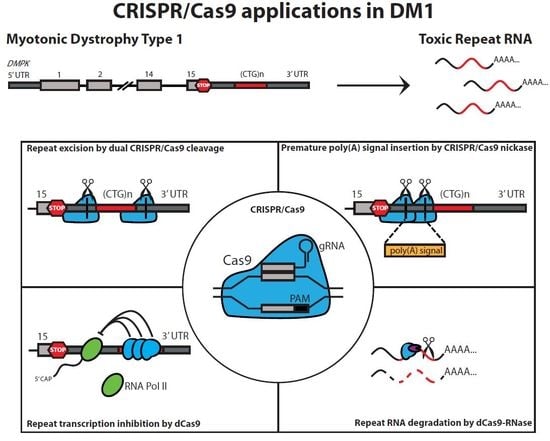
1. Introduction
1.1. Basic Principles of CRISPR/Cas Technology
1.2. Clinical Genetics and Disease Mechanisms in DM1
2. CRISPR/Cas-Mediated Genome Editing in the DM1 Locus
2.1. Excision of the Expanded (CTG•CAG)n Repeat
2.2. Reduction of the (CTG•CAG)n Repeat Length
2.3. Allele-Specific Gene Editing
2.4. Insertion of a Premature Polyadenylation Signal in DMPK
3. CRISPR/dCas9 as a Guide to the DM1 Locus
4. CRISPR/dCas9-Mediated Targeting and Elimination of Expanded Repeat RNA
5. Therapeutic Outlook for CRISPR/Cas-Mediated Ex Vivo and In Vivo Approaches in DM1
5.1. Ex Vivo Cell Therapy
5.2. In Vivo Gene Editing
6. Conclusions
Funding
Conflicts of Interest
References
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qi, L.S. A CRISPR-dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Doudna, J.A. Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol. Cell 2015, 58, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Batra, R.; Nelles, D.A.; Pirie, E.; Blue, S.M.; Marina, R.J.; Wang, H.; Chaim, I.A.; Thomas, J.D.; Zhang, N.; Nguyen, V.; et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 2017, 170, 899–912. [Google Scholar] [CrossRef] [PubMed]
- De Antonio, M.; Dogan, C.; Hamroun, D.; Mati, M.; Zerrouki, S.; Eymard, B.; Katsahian, S.; Bassez, G.; French Myotonic Dystrophy Clinical, N. Unravelling the myotonic dystrophy type 1 clinical spectrum: A systematic registry-based study with implications for disease classification. Rev. Neurol. 2016, 172, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Sicot, G.; Gomes-Pereira, M. RNA toxicity in human disease and animal models: From the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta 2013, 1832, 1390–1409. [Google Scholar] [CrossRef]
- Thomas, J.D.; Oliveira, R.; Sznajder, L.J.; Swanson, M.S. Myotonic Dystrophy and Developmental Regulation of RNA Processing. Compr. Physiol. 2018, 8, 509–553. [Google Scholar] [CrossRef]
- Andre, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy. Front. Neurol. 2018, 9, 368. [Google Scholar] [CrossRef]
- Cumming, S.A.; Hamilton, M.J.; Robb, Y.; Gregory, H.; McWilliam, C.; Cooper, A.; Adam, B.; McGhie, J.; Hamilton, G.; Herzyk, P.; et al. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur. J. Hum. Genet. 2018, 26, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Braida, C.; Stefanatos, R.K.; Adam, B.; Mahajan, N.; Smeets, H.J.; Niel, F.; Goizet, C.; Arveiler, B.; Koenig, M.; Lagier-Tourenne, C.; et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2010, 19, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Banez-Coronel, M.; Ranum, L.P.W. Repeat-associated non-AUG (RAN) translation: Insights from pathology. Lab. Investig. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Gagnon, C.; Groh, W.J.; Gutmann, L.; Johnson, N.E.; Meola, G.; Moxley, R., 3rd; Pandya, S.; Rogers, M.T.; Simpson, E.; et al. Consensus-based care recommendations for adults with myotonic dystrophy type 1. Neurol. Clin. Pract. 2018, 8, 507–520. [Google Scholar] [CrossRef] [PubMed]
- van der Bent, M.L.; van Cruchten, R.T.P.; Wansink, D.G. CHAPTER 7 Targeting Toxic Repeats. In Advances in Nucleic Acid Therapeutics; Agrawal, S., Gait, M.J., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2019; pp. 126–150. [Google Scholar]
- Lopez-Morato, M.; Brook, J.D.; Wojciechowska, M. Small Molecules Which Improve Pathogenesis of Myotonic Dystrophy Type 1. Front. Neurol. 2018, 9, 349. [Google Scholar] [CrossRef]
- Overby, S.J.; Cerro-Herreros, E.; Llamusi, B.; Artero, R. RNA-mediated therapies in myotonic dystrophy. Drug Discov. Today 2018, 23, 2013–2022. [Google Scholar] [CrossRef] [PubMed]
- van Agtmaal, E.L.; Andre, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef]
- Gudde, A.; van Heeringen, S.J.; de Oude, A.I.; van Kessel, I.D.G.; Estabrook, J.; Wang, E.T.; Wieringa, B.; Wansink, D.G. Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol. 2017, 14, 1374–1388. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef]
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol. Ther. Nucleic Acids 2017, 9, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, L.; Wang, H.; Santostefano, K.; Thapa, A.; Cleary, J.; Li, H.; Guo, X.; Terada, N.; Ashizawa, T.; et al. Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9. Mol. Ther. 2018, 26, 2617–2630. [Google Scholar] [CrossRef] [PubMed]
- Yanovsky-Dagan, S.; Bnaya, E.; Diab, M.A.; Handal, T.; Zahdeh, F.; van den Broek, W.J.A.A.; Epsztejn-Litman, S.; Wansink, D.G.; Eiges, R. Deletion of the CTG Expansion in Myotonic Dystrophy Type 1 Reverses DMPK Aberrant Methylation in Human Embryonic Stem Cells but not Affected Myoblasts. bioRxiv 2019, 631457. [Google Scholar] [CrossRef]
- Pinto, B.S.; Saxena, T.; Oliveira, R.; Mendez-Gomez, H.R.; Cleary, J.D.; Denes, L.T.; McConnell, O.; Arboleda, J.; Xia, G.; Swanson, M.S.; et al. Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol. Cell 2017, 68, 479–490. [Google Scholar] [CrossRef]
- Petruska, J.; Arnheim, N.; Goodman, M.F. Stability of intrastrand hairpin structures formed by the CAG/CTG class of DNA triplet repeats associated with neurological diseases. Nucleic Acids Res. 1996, 24, 1992–1998. [Google Scholar] [CrossRef]
- Axford, M.M.; Wang, Y.H.; Nakamori, M.; Zannis-Hadjopoulos, M.; Thornton, C.A.; Pearson, C.E. Detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type I disease locus in patient tissues. PLoS Genet. 2013, 9, e1003866. [Google Scholar] [CrossRef]
- Liu, G.; Chen, X.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat. Chem. Biol 2010, 6, 652–659. [Google Scholar] [CrossRef]
- Mosbach, V.; Poggi, L.; Richard, G.F. Trinucleotide repeat instability during double-strand break repair: From mechanisms to gene therapy. Curr. Genet. 2019, 65, 17–28. [Google Scholar] [CrossRef]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef]
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S.T. Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS ONE 2016, 11, e0165499. [Google Scholar] [CrossRef]
- Ouellet, D.L.; Cherif, K.; Rousseau, J.; Tremblay, J.P. Deletion of the GAA repeats from the human frataxin gene using the CRISPR-Cas9 system in YG8R-derived cells and mouse models of Friedreich ataxia. Gene Ther. 2017, 24, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Xie, Y.; Xiong, Z.; Yang, Y.; Xian, Y.; Ou, Z.; Song, B.; Chen, Y.; Xie, Y.; Li, H.; et al. CRISPR/Cas9-Targeted Deletion of Polyglutamine in Spinocerebellar Ataxia Type 3-Derived Induced Pluripotent Stem Cells. Stem Cells Dev. 2018, 27, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, M.; Juzwa, W.; Krzyzosiak, W.J.; Olejniczak, M. Precise Excision of the CAG Tract from the Huntingtin Gene by Cas9 Nickases. Front. Neurosci. 2018, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Marthaler, A.G.; Schmid, B.; Tubsuwan, A.; Poulsen, U.B.; Engelbrecht, A.F.; Mau-Holzmann, U.A.; Hyttel, P.; Nielsen, J.E.; Nielsen, T.T.; Holst, B. Generation of an isogenic, gene-corrected control cell line of the spinocerebellar ataxia type 2 patient-derived iPSC line H271. Stem Cell Res. 2016, 16, 180–183. [Google Scholar] [CrossRef]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Richard, G.F.; Viterbo, D.; Khanna, V.; Mosbach, V.; Castelain, L.; Dujon, B. Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS ONE 2014, 9, e95611. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, D.; Moye, C.; Morton, J.; Sykoudis, K.; Lin, Y.; Carroll, D.; Wilson, J.H. Zinc-finger directed double-strand breaks within CAG repeat tracts promote repeat instability in human cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9607–9612. [Google Scholar] [CrossRef] [PubMed]
- Mosbach, V.; Poggi, L.; Viterbo, D.; Charpentier, M.; Richard, G.F. TALEN-Induced Double-Strand Break Repair of CTG Trinucleotide Repeats. Cell Rep. 2018, 22, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Cinesi, C.; Aeschbach, L.; Yang, B.; Dion, V. Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat. Commun. 2016, 7, 13272. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Kim, K.H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef] [PubMed]
- Carrell, S.T.; Carrell, E.M.; Auerbach, D.; Pandey, S.K.; Bennett, C.F.; Dirksen, R.T.; Thornton, C.A. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet. 2016, 25, 4328–4338. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Guo, X.; Santostefano, K.; Wang, Y.; Reid, T.; Zeng, D.; Terada, N.; Ashizawa, T.; Xia, G. Genome Therapy of Myotonic Dystrophy Type 1 iPS Cells for Development of Autologous Stem Cell Therapy. Mol. Ther. 2016, 24, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Gao, Y.; Jin, S.; Subramony, S.H.; Terada, N.; Ranum, L.P.; Swanson, M.S.; Ashizawa, T. Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 2015, 33, 1829–1838. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Haenfler, J.M.; Skariah, G.; Rodriguez, C.M.; Monteiro da Rocha, A.; Parent, J.M.; Smith, G.D.; Todd, P.K. Targeted Reactivation of FMR1 Transcription in Fragile X Syndrome Embryonic Stem Cells. Front. Mol. Neurosci. 2018, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Alshareef, S.; Mahfouz, M.M. CRISPR base editors: Genome editing without double-stranded breaks. Biochem. J. 2018, 475, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Strutt, S.C.; Torrez, R.M.; Kaya, E.; Negrete, O.A.; Doudna, J.A. RNA-dependent RNA targeting by CRISPR-Cas9. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009, 325, 336–339. [Google Scholar] [CrossRef]
- Mulders, S.A.; van den Broek, W.J.; Wheeler, T.M.; Croes, H.J.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef]
- Birbrair, A.; Delbono, O. Pericytes are Essential for Skeletal Muscle Formation. Stem Cell Rev. 2015, 11, 547–548. [Google Scholar] [CrossRef]
- Kostallari, E.; Baba-Amer, Y.; Alonso-Martin, S.; Ngoh, P.; Relaix, F.; Lafuste, P.; Gherardi, R.K. Pericytes in the myovascular niche promote post-natal myofiber growth and satellite cell quiescence. Development 2015, 142, 1242–1253. [Google Scholar] [CrossRef]
- Dellavalle, A.; Maroli, G.; Covarello, D.; Azzoni, E.; Innocenzi, A.; Perani, L.; Antonini, S.; Sambasivan, R.; Brunelli, S.; Tajbakhsh, S.; et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2011, 2, 499. [Google Scholar] [CrossRef]
- Xia, G.; Terada, N.; Ashizawa, T. Human iPSC Models to Study Orphan Diseases: Muscular Dystrophies. Curr. Stem Cell Rep. 2018, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.W.; Suk, J.S.; Kim, A.J.; Hsiang, Y.H.; Klibanov, A.L.; Hanes, J.; Price, R.J. Markedly enhanced skeletal muscle transfection achieved by the ultrasound-targeted delivery of non-viral gene nanocarriers with microbubbles. J. Control. Release 2012, 162, 414–421. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Wang, J.; Satterle, A.; Wu, Q.; Wang, J.; Liu, F. Gene transfer to skeletal muscle by site-specific delivery of electroporation and ultrasound. Biochem. Biophys. Res. Commun. 2012, 424, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Crawford, R.W.; Meuse, L.; Miller, D.G.; Russell, D.W.; Chamberlain, J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004, 10, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Arruda, V.R.; Stedman, H.H.; Nichols, T.C.; Haskins, M.E.; Nicholson, M.; Herzog, R.W.; Couto, L.B.; High, K.A. Regional intravascular delivery of AAV-2-F.IX to skeletal muscle achieves long-term correction of hemophilia B in a large animal model. Blood 2005, 105, 3458–3464. [Google Scholar] [CrossRef]
- Monckton, D.G. Manage risk of accidental gene editing of germline. Nature 2019, 568, 458. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Baylis, F.; Zhang, F.; Charpentier, E.; Berg, P.; Bourgain, C.; Friedrich, B.; Joung, J.K.; Li, J.; Liu, D.; et al. Adopt a moratorium on heritable genome editing. Nature 2019, 567, 165–168. [Google Scholar] [CrossRef]
- Ponnazhagan, S.; Weigel, K.A.; Raikwar, S.P.; Mukherjee, P.; Yoder, M.C.; Srivastava, A. Recombinant human parvovirus B19 vectors: Erythroid cell-specific delivery and expression of transduced genes. J. Virol. 1998, 72, 5224–5230. [Google Scholar]
- Chao, H.; Mao, L.; Bruce, A.T.; Walsh, C.E. Sustained expression of human factor VIII in mice using a parvovirus-based vector. Blood 2000, 95, 1594–1599. [Google Scholar]
- Zhang, L.; Wang, D.; Fischer, H.; Fan, P.D.; Widdicombe, J.H.; Kan, Y.W.; Dong, J.Y. Efficient expression of CFTR function with adeno-associated virus vectors that carry shortened CFTR genes. Proc. Natl. Acad. Sci. USA 1998, 95, 10158–10163. [Google Scholar] [CrossRef]
- Sun, L.; Li, J.; Xiao, X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat. Med. 2000, 6, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Bragg, L.; Meggiolaro, L.; Rossi, M.; Caffarini, M.; Naz, N.; Santoleri, S.; Cossu, G. Gene and Cell Therapy for Muscular Dystrophies: Are We Getting There? Hum. Gene Ther. 2018, 29, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.E.; Duan, D. Perspective on Adeno-Associated Virus Capsid Modification for Duchenne Muscular Dystrophy Gene Therapy. Hum. Gene Ther. 2015, 26, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Yuan, Z.; Cao, Z.; Wang, B.; Qiao, C.; Li, J.; Xiao, X. A muscle-targeting peptide displayed on AAV2 improves muscle tropism on systemic delivery. Gene Ther. 2009, 16, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef]
- Calcedo, R.; Morizono, H.; Wang, L.; McCarter, R.; He, J.; Jones, D.; Batshaw, M.L.; Wilson, J.M. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin. Vaccine Immunol. 2011, 18, 1586–1588. [Google Scholar] [CrossRef]
- Conboy, I.; Murthy, N.; Etienne, J.; Robinson, Z. Making gene editing a therapeutic reality. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Monahan, P.E.; Samulski, R.J. AAV vectors: Is clinical success on the horizon? Gene Ther. 2000, 7, 24–30. [Google Scholar] [CrossRef]
- Russell, D.W.; Kay, M.A. Adeno-associated virus vectors and hematology. Blood 1999, 94, 864–874. [Google Scholar] [PubMed]
- Vakulskas, C.A.; Behlke, M.A. Evaluation and Reduction of CRISPR Off-Target Cleavage Events. Nucleic Acid Ther. 2019. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Study | CRISPR/Cas Strategy, Cas Type, (CTG•CAG)n Length | sgRNA 1 | Delivery | DM1 Biomarkers Examined | Cell Types Used |
|---|---|---|---|---|---|
| (CTG•CAG)n Excision | |||||
| Van Agtmaal et al., 2017 [18] | (CTG•CAG)n excision hSpCas9 Unaffected: n = 5/13 DM1: n = ~550/2600 | Repeat-flanking sgRNAs Upstream: 11 bp Downstream: 51 bp | Nucleofection: Cas9 and sgRNA expression plasmids | DMPK, SIX5 and DM1-AS RNA expression DMPK protein expression DMPK RNA subcellular distribution (CUG)n RNA foci and MBNL1 foci BIN-1 ex11 and DMD ex78 splicing Myogenic differentiation | Human: Immortalized unaffected and DM1 myoblasts Murine: Immortalized DM500 myoblasts |
| Provenzano et al., 2017 [22] | (CTG•CAG)n excision eSpCas9 Unaffected: n = 5/13 DM1: n = 300–1000 | Repeat-flanking sgRNAs Upstream: 189 bp Downstream: 305 bp | Lipofection and nucleofection: Cas9 and sgRNA expression plasmids | DMPK RNA expression (CUG)n RNA foci and MBNL1 foci SERCA1 ex22 and INSR ex11 splicing Myogenic differentiation DMPK protein expression | Human: HEK293FT cells Immortalized unaffected and DM1 inducible MYOD1-expressing fibroblasts |
| Dastidar et al., 2018 [21] | (CTG•CAG)n excision hSpCas9 Unaffected: n = 5/13 DM1: n = 1000–1700 | Repeat-flanking sgRNAs Upstream: 24 bp Downstream: 51 bp | Lentiviral transduction: CMV-hspCas9-EF1-GFP virus Nucleofection: Cas9/sgRNA RNP complexes | DMPK RNA expression (CUG)n RNA foci andMBNL1 foci SERCA1 ex22 splicing | Human: DM1 iPSC-derived myogenic cells DM1 primary myoblasts DM1 iPSCs |
| Wang et al., 2018 [23] | (CTG•CAG)n excision SpCas9 and SaCas9 Unaffected: n = 5 DM1: n ≥ 2000 | Repeat-flanking sgRNAs Upstream: <220 bp Downstream: <220 bp | Lipofection: Cas9 and sgRNA expression plasmids | (CUG)n and (CAG)n RNA foci | Human: HEK293FT cells DM1 iPSC-derived neural stem cells |
| Yanovsky-Dagan et al., 2019 [24] | (CTG•CAG)n excision SpCas9 Unaffected: n = 5 DM1: n = 2000 | Repeat-flanking sgRNAs Upstream: 11 bp Downstream: 47 bp | Transfection: Cas9 and sgRNA expression plasmids | SIX5 expression DM1 locus CpG hypermethylation H3K9me3 enrichment | Human: HEK293T cells DM1 hESCs |
| Insertion Poly(A) Signal Cassette | |||||
| Wang et al., 2018 [23] | Insertion poly(A) signal cassette SpCas9n Unaffected: n = 5 DM1: n ≥ 2000 | Paired sgRNAs between stop codon and repeat Donor template: 5′ HR arm: 97 bp 3′ HR arm: 184 bp | Lipofection: Cas9n and sgRNA expression plasmids and donor template | (CUG)n RNA foci DMPK RNA subcellular distribution MAPT ex3, MBNL1/2 ex7, SERCA1 ex22 and INSR ex11 splicing Myogenic differentiation | Human: DM1 iPSCs DM1 iPSC-derived neural stem cells |
| dCas9-Mediated Repeat Transcription Inhibition, Repeat RNA Visualization and Degradation | |||||
| Pinto et al., 2017 [25] | (CTG)n transcription block dSpCas9 and dSaCas9 (CTG•CAG)n plasmids n = 0/12/40/240/480/960 (interrupted) DM1: n ≥ 2000 | (CAG)6 sgRNA | Transfection: plasmids expressing dCas9 and sgRNAs Transduction: AAV2/6-dSaCas9-sgRNA and AAV2/9-dSaCas9-sgRNA | (CUG)n RNA foci (CUG)n RNA expression Multiple splice modes (RNA-seq) RAN translation Expression of (CUG)n and (CAG)n repeat-containing transcripts Myotonia | Human: Transiently transfected HEK293T and HeLa cells DM1 primary myoblasts Murine: HSALR mice (EDL muscle ex vivo, tibialis anterior and gastrocnemius in vivo) |
| Batra et al., 2017 [6] | (CUG)n RNA visualization and degradation 666dSpCas9, dSpCas9-GFP and PIN-dSpCas9 (CTG•CAG)n plasmids n = 105 n = 960 (interrupted) DM1: n ≥ 2700 | (CAG)n sgRNA | Lipofection: Cas9 and sgRNA expression plasmids Lentiviral transduction: U6-sgRNA and EFS-PIN-dCas9 | (CUG)n RNA expression (CUG)n RNA foci MBNL1 foci Multiple splice modes (RNA-seq) Expression of (CUG)n and (CAG)n repeat-containing transcripts | Human: DM1 primary myoblasts Primate: COS-M6 cells |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raaijmakers, R.H.L.; Ripken, L.; Ausems, C.R.M.; Wansink, D.G. CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities. Int. J. Mol. Sci. 2019, 20, 3689. https://doi.org/10.3390/ijms20153689
Raaijmakers RHL, Ripken L, Ausems CRM, Wansink DG. CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities. International Journal of Molecular Sciences. 2019; 20(15):3689. https://doi.org/10.3390/ijms20153689
Chicago/Turabian StyleRaaijmakers, Renée H.L., Lise Ripken, C. Rosanne M. Ausems, and Derick G. Wansink. 2019. "CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities" International Journal of Molecular Sciences 20, no. 15: 3689. https://doi.org/10.3390/ijms20153689
APA StyleRaaijmakers, R. H. L., Ripken, L., Ausems, C. R. M., & Wansink, D. G. (2019). CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities. International Journal of Molecular Sciences, 20(15), 3689. https://doi.org/10.3390/ijms20153689