Battle between Host Immune Cellular Responses and HCMV Immune Evasion



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Human Cytomegalovirus (HCMV)
2. Implications of HCMV in Hematopoietic Stem Cell Transplantation (HSCT)
3. Bidirectional Relationship between Host Strategies and HCMV Immune Escape Mechanisms
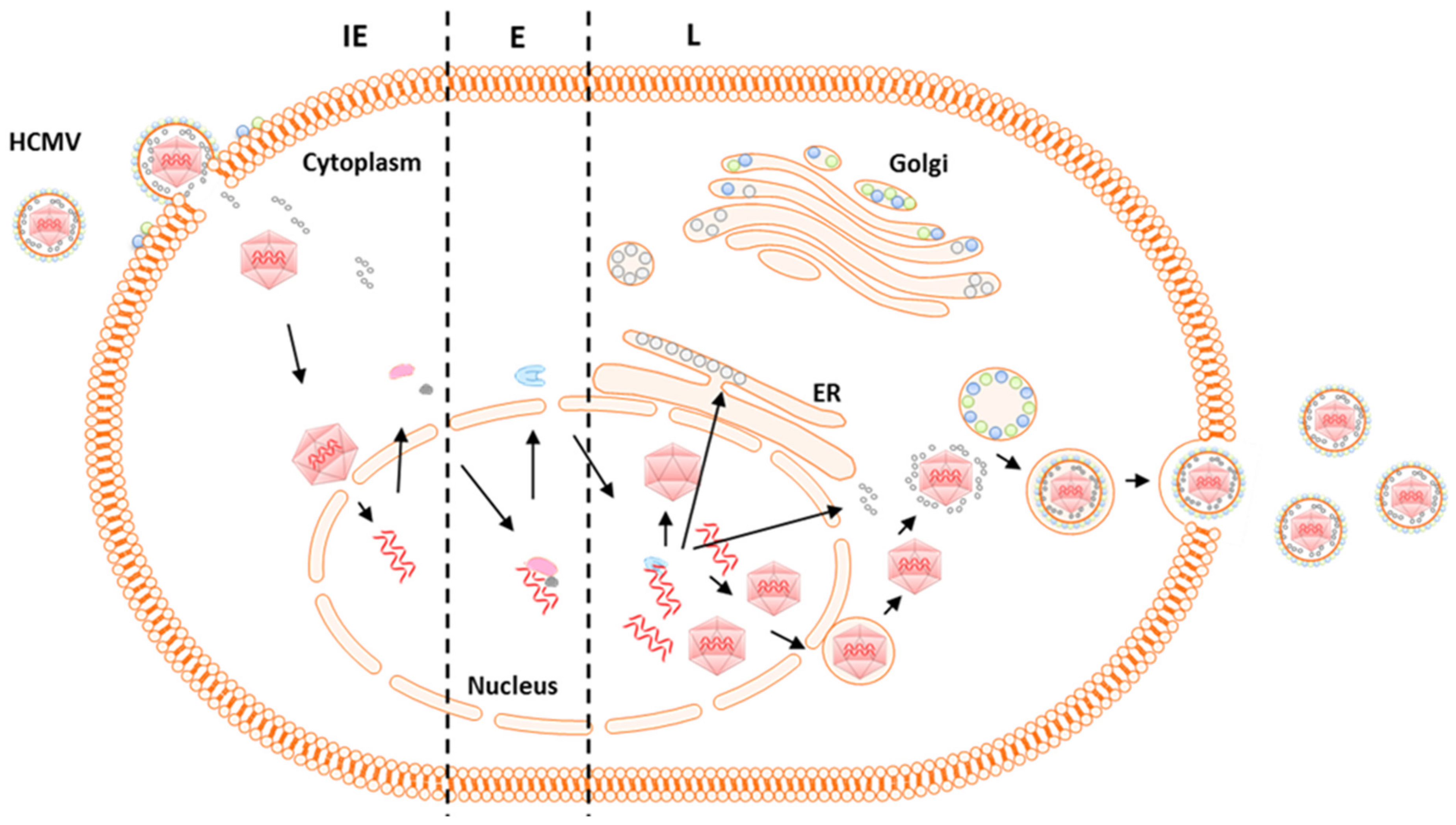
4. HCMV Virus Genome and Gene Expression
5. Host Responses to HCMV Infection
5.1. Innate Immunity
5.2. Adaptive Immunity
5.2.1. Humoral Immunity
5.2.2. Cellular Immunity
HCMV-Specific CD8+ T Cell Responses
HCMV-Specific CD4+ T Cell Responses
HCMV-Induced Regulatory T Cells (iTreg)
6. HCMV Escape Mechanisms
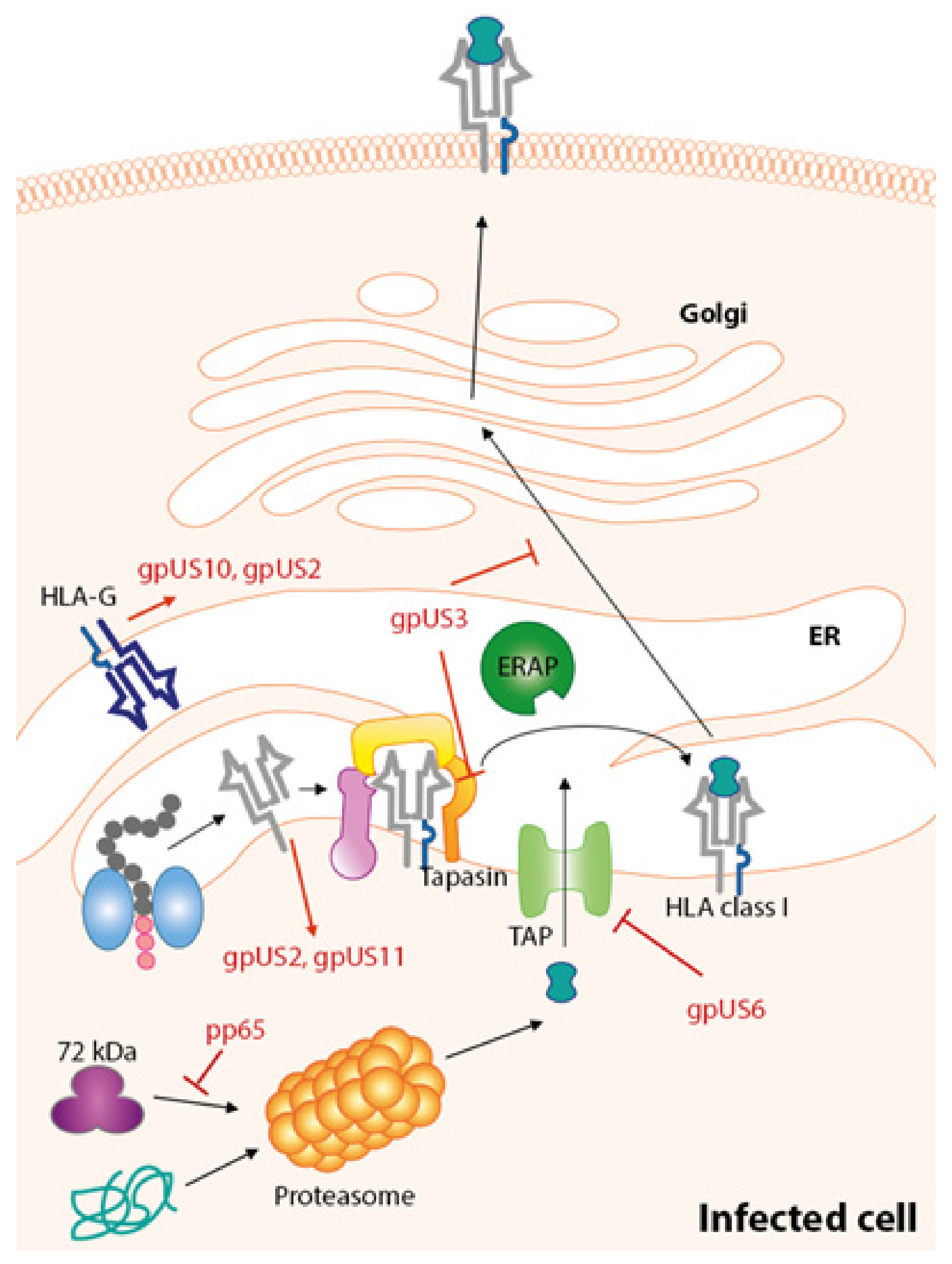
7. Immune Evasion Proteins Targeting HLA-Class I Pathway
7.1. gpUS3
7.2. gpUS2
7.3. US11
7.4. US6
7.5. US10
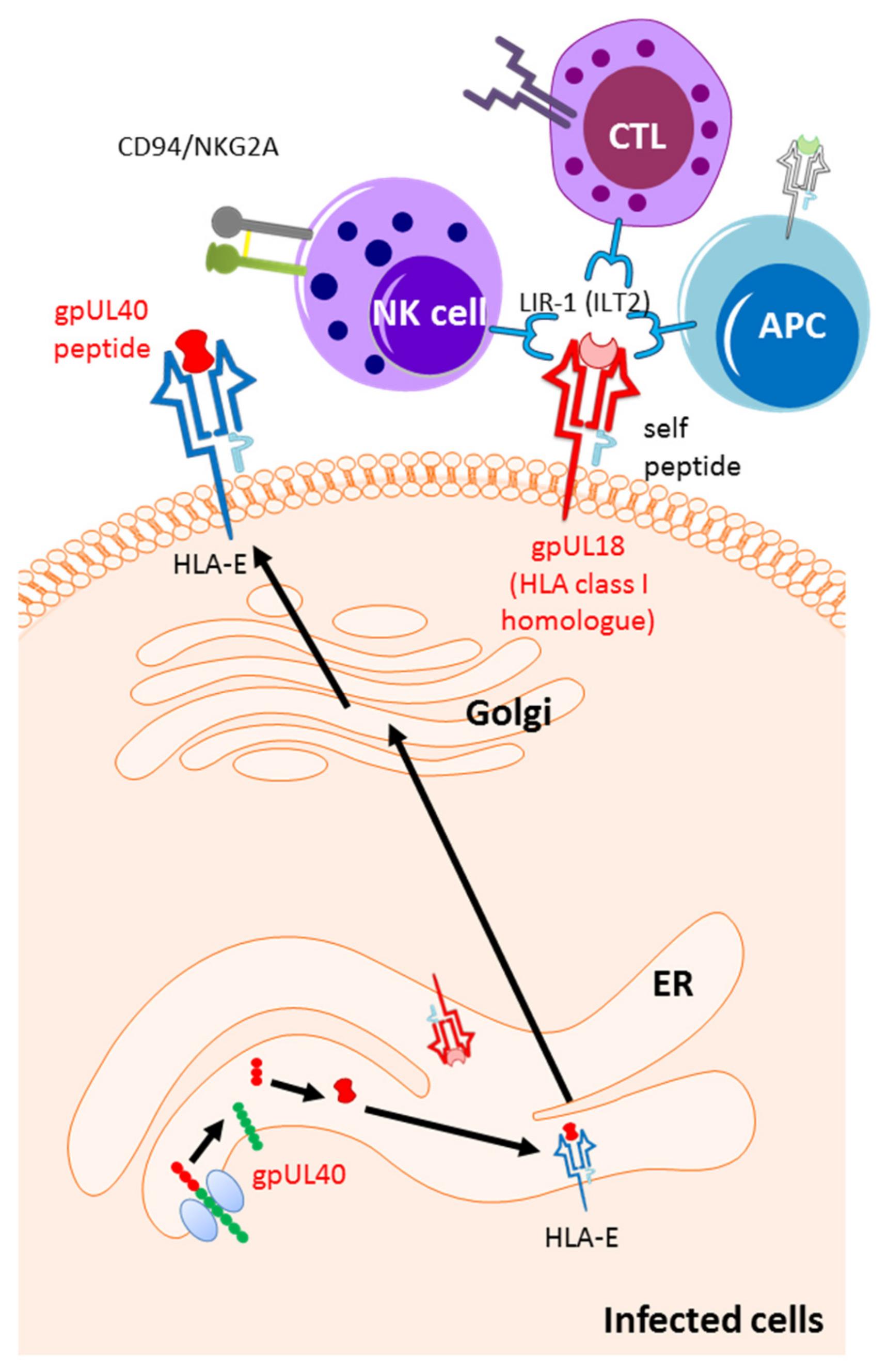
8. Immune Evasions in Regulation of Effector Cell Activation
9. Recent Advances in HCMV Therapeutics and Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ribbert, D. Uber protozoenartige zellen in der niere eines syphilitischen neugoborenen und in der parotis von kindern. Zentralbl. Allg. Pathol. 1904, 15, 945–948. [Google Scholar]
- Goodpasture, E.W.; Talbot, F.B. Concerning the nature of “proteozoan-like” cells in certain lesions of infancy. Am. J. Dis. Child. 1921, 21, 415–421. [Google Scholar]
- Smith, M.G. Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc. Soc. Exp. Biol. Med. 1956, 92, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.P.; Hartley, J.W.; Waterman, S.; Turner, H.C.; Huebner, R.J. Cytopathogenic agent resembling human salivary gland virus recovered from tissue cultures of human adenoids. Proc. Soc. Exp. Biol. Med. 1956, 92, 418–424. [Google Scholar] [PubMed]
- Craig, J.M.; Macauley, J.C.; Weller, T.H.; Wirth, P. Isolation of intranuclear inclusion producing agents from infants with illnesses resembling cytomegalic inclusion disease. Proc. Soc. Exp. Biol. Med. 1957, 94, 4–12. [Google Scholar] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Staras, S.A.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.F.; Cannon, M.J. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2006, 43, 1143–1151. [Google Scholar] [CrossRef]
- Ljungman, P.; Hakki, M.; Boeckh, M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol. Oncol. Clin. N. Am. 2011, 25, 151–169. [Google Scholar] [CrossRef]
- Kano, Y.; Shiohara, T. Current understanding of cytomegalovirus infection in immunocompetent individuals. J. Dermatol. Sci. 2000, 22, 196–204. [Google Scholar] [CrossRef]
- Slyker, J.A.; Lohman-Payne, B.L.; John-Stewart, G.C.; Maleche-Obimbo, E.; Emery, S.; Richardson, B.; Dong, T.; Iversen, A.K.; Mbori-Ngacha, D.; Overbaugh, J.; et al. Acute cytomegalovirus infection in Kenyan HIV-infected infants. Aids 2009, 23, 2173–2181. [Google Scholar] [CrossRef]
- Steininger, C.; Puchhammer-Stockl, E.; Popow-Kraupp, T. Cytomegalovirus disease in the era of highly active antiretroviral therapy (HAART). J. Clin. Virol. 2006, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gerard, L.; Leport, C.; Flandre, P.; Houhou, N.; Salmon-Ceron, D.; Pepin, J.M.; Mandet, C.; Brun-Vezinet, F.; Vilde, J.L. Cytomegalovirus (CMV) viremia and the CD4+ lymphocyte count as predictors of CMV disease in patients infected with human immunodeficiency virus. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1997, 24, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Fowler, K.B.; Stagno, S.; Pass, R.F.; Britt, W.J.; Boll, T.J.; Alford, C.A. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N. Engl. J. Med. 1992, 326, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Ross, S.A.; Fowler, K.B. Congenital Cytomegalovirus Infection: Clinical Outcome. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2013, 57 (Suppl. 4), S178–S181. [Google Scholar] [CrossRef] [PubMed]
- Townsend, C.L.; Forsgren, M.; Ahlfors, K.; Ivarsson, S.A.; Tookey, P.A.; Peckham, C.S. Long-term outcomes of congenital cytomegalovirus infection in Sweden and the United Kingdom. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2013, 56, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P.; Boeckh, M.; Hirsch, H.H.; Josephson, F.; Lundgren, J.; Nichols, G.; Pikis, A.; Razonable, R.R.; Miller, V.; Griffiths, P.D.; et al. Definitions of Cytomegalovirus Infection and Disease in Transplant Patients for Use in Clinical Trials. Clin. Infect. Dis. 2017, 64, 87–91. [Google Scholar] [PubMed]
- Adland, E.; Klenerman, P.; Goulder, P.; Matthews, P.C. Ongoing burden of disease and mortality from HIV/CMV coinfection in Africa in the antiretroviral therapy era. Front. Microbiol. 2015, 6, 1016. [Google Scholar] [CrossRef]
- Boeckh, M.; Geballe, A.P. Cytomegalovirus: Pathogen, paradigm, and puzzle. J. Clin. Investig. 2011, 121, 1673–1680. [Google Scholar] [CrossRef]
- Goncalves, C.; Cipriano, A.; Videira Santos, F.; Abreu, M.; Mendez, J.; Sarmento, E.C.R. Cytomegalovirus acute infection with pulmonary involvement in an immunocompetent patient. IDCases 2018, 14, e00445. [Google Scholar] [CrossRef]
- Cunha, B.A. Cytomegalovirus pneumonia: Community-acquired pneumonia in immunocompetent hosts. Infect. Dis. Clin. N. Am. 2010, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, N.; Hirsch, H.H.; Khanna, N.; Gerull, S.; Buser, A.; Bucher, C.; Halter, J.; Heim, D.; Tichelli, A.; Gratwohl, A.; et al. Evidence for a bidirectional relationship between cytomegalovirus replication and acute graft-versus-host disease. Biol. Blood Marrow Transplant. 2010, 16, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Appleton, A.L.; Sviland, L. Pathogenesis of GVHD: Role of herpes viruses. Bone Marrow Transplant. 1993, 11, 349–355. [Google Scholar] [PubMed]
- McCarthy, A.L.; Malik Peiris, J.S.; Taylor, C.E.; Green, M.A.; Sviland, L.; Pearson, A.D.; Malcolm, A.J. Increase in severity of graft versus host disease by cytomegalovirus. J. Clin. Pathol. 1992, 45, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Boeckh, M.; Nichols, W.G. The impact of cytomegalovirus serostatus of donor and recipient before hematopoietic stem cell transplantation in the era of antiviral prophylaxis and preemptive therapy. Blood 2004, 103, 2003–2008. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P. The role of cytomegalovirus serostatus on outcome of hematopoietic stem cell transplantation. Curr. Opin. Hematol. 2014, 21, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P.; Brand, R.; Einsele, H.; Frassoni, F.; Niederwieser, D.; Cordonnier, C. Donor CMV serologic status and outcome of CMV-seropositive recipients after unrelated donor stem cell transplantation: An EBMT megafile analysis. Blood 2003, 102, 4255–4260. [Google Scholar] [CrossRef]
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Van Endert, P.M.; Saveanu, L.; Hewitt, E.W.; Lehner, P.J. Powering the peptide pump: TAP crosstalk with energetic nucleotides. Trends Biochem. Sci. 2002, 27, 454–461. [Google Scholar] [CrossRef]
- Cresswell, P.; Bangia, N.; Dick, T.; Diedrich, G. The nature of the MHC class I peptide loading complex. Immunol. Rev. 1999, 172, 21–28. [Google Scholar] [CrossRef]
- Pamer, E.; Cresswell, P. Mechanisms of MHC class I restricted antigen processing. Annu. Rev. Immunol. 1998, 16, 323–358. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.N.; Powis, S.J.; Elliott, T. Assembly and export of MHC class I peptide ligands. Curr. Opin. Immunol. 2003, 15, 75–81. [Google Scholar] [CrossRef]
- Spiliotis, E.T.; Osorio, M.; Zúñiga, M.C.; Edidin, M. Selective export of MHC class I molecules from the ER after their dissociation from TAP. Immunity 2000, 13, 841–851. [Google Scholar] [CrossRef]
- Lautscham, G.; Rickinson, A.; Blake, N. TAP-independent antigen presentation on MHC class I molecules: Lessons from Epstein-Barr virus. Microbes Infect. 2003, 5, 291–299. [Google Scholar] [CrossRef]
- Fromm, S.V.; Duady-Ben Yaakov, S.; Schechter, C.; Ehrlich, R. Assembly and cell surface expression of TAP-independent, chloroquine-sensitive and interferon-gamma-inducible class I MHC complexes in transformed fibroblast cell lines are regulated by tapasin. Cell. Immunol. 2002, 215, 207–218. [Google Scholar] [CrossRef]
- Boyle, L.H.; Hermann, C.; Boname, J.M.; Porter, K.M.; Patel, P.A.; Burr, M.L.; Duncan, L.M.; Harbour, M.E.; Rhodes, D.A.; Skjødt, K.; et al. Tapasin-related protein TAPBPR is an additional component of the MHC class I presentation pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 3465–3470. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Rigoutsos, I.; Shibuya, T.; Shenk, T.E. Reevaluation of human cytomegalovirus coding potential. Proc. Natl. Acad. Sci. USA 2003, 100, 13585–13590. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Gatherer, D.; Hilfrich, B.; Baluchova, K.; Dargan, D.J.; Thomson, M.; Griffiths, P.D.; Wilkinson, G.W.; Schulz, T.F.; Davison, A.J. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J. Gen. Virol. 2010, 91, 605–615. [Google Scholar] [CrossRef]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus Cell Tropism. In Human Cytomegalovirus; Shenk, T., Stinski, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 325, pp. 63–83. [Google Scholar]
- Mach, M.; Kropff, B.; Kryzaniak, M.; Britt, W. Complex Formation by Glycoproteins M and N of Human Cytomegalovirus: Structural and Functional Aspects. J. Virol. 2005, 79, 2160–2170. [Google Scholar] [CrossRef]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef]
- Revello, M.G.; Gerna, G. Human cytomegalovirus tropism for endothelial/epithelial cells: Scientific background and clinical implications. Rev. Med. Virol. 2010, 20, 136–155. [Google Scholar] [CrossRef] [PubMed]
- Lopper, M.; Compton, T. Coiled-Coil Domains in Glycoproteins B and H Are Involved in Human Cytomegalovirus Membrane Fusion. J. Virol. 2004, 78, 8333–8341. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Feire, A. Early events in human cytomegalovirus infection. In Human Herpesviruses: Biology, Therapy and Immunoprophylaxis; 25 February 2011; Arvin, A.M., Mocarski, E.S., Moore, P., Whitley, R., Yamanishi, K., Campadelli-Fiume, G., Roizman, B., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 231–240. [Google Scholar]
- Demarchi, J.M. Human cytomegalovirus DNA: Restriction enzyme cleavage maps and map locations for immediate-early, early, and late RNAs. Virology 1981, 114, 23–38. [Google Scholar] [CrossRef]
- Wathen, M.W.; Stinski, M.F. Temporal patterns of human cytomegalovirus transcription: Mapping the viral RNAs synthesized at immediate early, early, and late times after infection. J. Virol. 1982, 41, 462–477. [Google Scholar] [PubMed]
- Stinski, M.F. Sequence of protein synthesis in cells infected by human cytomegalovirus: Early and late virus-induced polypeptides. J. Virol. 1978, 26, 686–701. [Google Scholar] [PubMed]
- Stenberg, R.M.; Thomsen, D.R.; Stinski, M.F. Structural analysis of the major immediate early gene of human cytomegalovirus. J. Virol. 1984, 49, 190–199. [Google Scholar]
- Stenberg, R.M.; Witte, P.R.; Stinski, M.F. Multiple spliced and unspliced transcripts from human cytomegalovirus immediate-early region 2 and evidence for a common initiation site within immediate-early region 1. J. Virol. 1985, 56, 665–675. [Google Scholar]
- White, E.A.; Spector, D.H. Early viral gene expression and function. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; 25 February 2011; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Malone, C.L.; Vesole, D.H.; Stinski, M.F. Transactivation of a human cytomegalovirus early promoter by gene products from the immediate-early gene IE2 and augmentation by IE1: Mutational analysis of the viral proteins. J. Virol. 1990, 64, 1498–1506. [Google Scholar]
- Klucher, K.M.; Sommer, M.; Kadonaga, J.T.; Spector, D.H. In vivo and in vitro analysis of transcriptional activation mediated by the human cytomegalovirus major immediate-early proteins. Mol. Cell. Biol. 1993, 13, 1238–1250. [Google Scholar] [CrossRef]
- Schwartz, R.; Sommer, M.H.; Scully, A.; Spector, D.H. Site-specific binding of the human cytomegalovirus IE2 86-kilodalton protein to an early gene promoter. J. Virol. 1994, 68, 5613–5622. [Google Scholar]
- Cherrington, J.M.; Khoury, E.L.; Mocarski, E.S. Human cytomegalovirus IE2 negatively regulates alpha gene expression via a short target sequence near the transcription start site. J. Virol. 1991, 65, 887–896. [Google Scholar] [PubMed]
- Huang, L.; Stinski, M.F. Binding of cellular repressor protein or the IE2 protein to a cis-acting negative regulatory element upstream of a human cytomegalovirus early promoter. J. Virol. 1995, 69, 7612–7621. [Google Scholar] [PubMed]
- Liu, B.; Hermiston, T.W.; Stinski, M.F. A cis-acting element in the major immediate-early (IE) promoter of human cytomegalovirus is required for negative regulation by IE2. J. Virol. 1991, 65, 897–903. [Google Scholar] [PubMed]
- Taylor, R.T.; Bresnahan, W.A. Human Cytomegalovirus Immediate-Early 2 Protein IE86 Blocks Virus-Induced Chemokine Expression. J. Virol. 2006, 80, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Tenney, D.J.; Colberg-Poley, A.M. Human cytomegalovirus UL36-38 and US3 immediate-early genes: Temporally regulated expression of nuclear, cytoplasmic, and polysome-associated transcripts during infection. J. Virol. 1991, 65, 6724–6734. [Google Scholar] [PubMed]
- Jones, T.R.; Wiertz, E.J.; Sun, L.; Fish, K.N.; Nelson, J.A.; Ploegh, H.L. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. USA 1996, 93, 11327–11333. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.; Chu, D.T.; O’Guin, A.K.; Yokoyama, W.M.; Virgin, H.W. Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J. Virol. 2005, 79, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Madera, S.; Rapp, M.; Firth, M.A.; Beilke, J.N.; Lanier, L.L.; Sun, J.C. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med. 2016, 213, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, J.F.; Warner, J.F.; Dennert, G.; Welsh, R.M. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J. Exp. Med. 1985, 161, 40–52. [Google Scholar] [CrossRef]
- Bukowski, J.F.; Woda, B.A.; Welsh, R.M. Pathogenesis of murine cytomegalovirus infection in natural killer cell-depleted mice. J. Virol. 1984, 52, 119–128. [Google Scholar]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe Herpesvirus Infections in an Adolescent without Natural Killer Cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Iversen, A.C.; Norris, P.S.; Ware, C.F.; Benedict, C.A. Human NK cells inhibit cytomegalovirus replication through a noncytolytic mechanism involving lymphotoxin-dependent induction of IFN-beta. J. Immunol. 2005, 175, 7568–7574. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sinzger, C.; Reichel, J.J.; Just, M.; Mertens, T. Natural Killer Cells Can Inhibit the Transmission of Human Cytomegalovirus in Cell Culture by Using Mechanisms from Innate and Adaptive Immune Responses. J. Virol. 2015, 89, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Zaia, J.A.; Sun, J.Y.; Gallez-Hawkins, G.M.; Thao, L.; Oki, A.; Lacey, S.F.; Dagis, A.; Palmer, J.; Diamond, D.J.; Forman, S.J.; et al. The effect of single and combined activating KIR genotypes on CMV infection and immunity after hematopoietic cell transplantation. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2009, 15, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Busson, M.; Rocha, V.; Appert, M.L.; Lepage, V.; Dulphy, N.; Haas, P.; Socie, G.; Toubert, A.; Charron, D.; et al. Activating KIR genes are associated with CMV reactivation and survival after non-T-cell depleted HLA-identical sibling bone marrow transplantation for malignant disorders. Bone Marrow Transplant. 2006, 38, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef]
- Limaye, A.P.; Bakthavatsalam, R.; Kim, H.W.; Randolph, S.E.; Halldorson, J.B.; Healey, P.J.; Kuhr, C.S.; Levy, A.E.; Perkins, J.D.; Reyes, J.D.; et al. Impact of cytomegalovirus in organ transplant recipients in the era of antiviral prophylaxis. Transplantation 2006, 81, 1645–1652. [Google Scholar] [CrossRef]
- Riddell, S.R.; Watanabe, K.S.; Goodrich, J.M.; Li, C.R.; Agha, M.E.; Greenberg, P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 257, 238–241. [Google Scholar] [CrossRef]
- Ramirez, N.; Olavarria, E. Viral-specific adoptive immunotherapy after allo-SCT: The role of multimer-based selection strategies. Bone Marrow Transplant. 2013, 48, 1265–1270. [Google Scholar] [CrossRef][Green Version]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mohty, M.; Or, R.; Maschan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef] [PubMed]
- Sellar, R.S.; Peggs, K.S. Therapeutic strategies for cytomegalovirus infection in haematopoietic transplant recipients: A focused update. Expert Opin. Biol. Ther. 2014, 14, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Solana, R.; Tarazona, R.; Aiello, A.E.; Akbar, A.N.; Appay, V.; Beswick, M.; Bosch, J.A.; Campos, C.; Cantisán, S.; Cicin-Sain, L.; et al. CMV and Immunosenescence: From basics to clinics. Immun. Ageing 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, C.; Diamond, D.J. The immune response to human CMV. Future Virol. 2012, 7, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. NK cells in autoimmune diseases: Linking innate and adaptive immune responses. Autoimmun. Rev. 2018, 17, 142–154. [Google Scholar] [CrossRef]
- Paust, S.; Blish, C.A.; Reeves, R.K. Redefining Memory: Building the Case for Adaptive NK Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Adams, N.M.; Geary, C.D.; Santosa, E.K.; Lumaquin, D.; Le Luduec, J.B.; Sottile, R.; van der Ploeg, K.; Hsu, J.; Whitlock, B.M.; Jackson, B.T.; et al. Cytomegalovirus Infection Drives Avidity Selection of Natural Killer Cells. Immunity 2019, 50, 1381–1390.e5. [Google Scholar] [CrossRef]
- Gerna, G.; Sarasini, A.; Patrone, M.; Percivalle, E.; Fiorina, L.; Campanini, G.; Gallina, A.; Baldanti, F.; Revello, M.G. Human cytomegalovirus serum neutralizing antibodies block virus infection of endothelial/epithelial cells, but not fibroblasts, early during primary infection. J. Gen. Virol. 2008, 89, 853–865. [Google Scholar] [CrossRef]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef]
- Revello, M.G.; Gerna, G. Diagnosis and management of human cytomegalovirus infection in the mother, fetus, and newborn Infant. Clin. Microbiol. Rev. 2002, 15, 680–715. [Google Scholar] [CrossRef]
- Chidrawar, S.; Khan, N.; Wei, W.; McLarnon, A.; Smith, N.; Nayak, L.; Moss, P. Cytomegalovirus-seropositivity has a profound influence on the magnitude of major lymphoid subsets within healthy individuals. Clin. Exp. Immunol. 2009, 155, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Akbar, A.; Caruso, C.; Solana, R.; Grubeck-Loebenstein, B.; Wikby, A. Human immunosenescence: Is it infectious? Immunol. Rev. 2005, 205, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.; Faulhaber, N.; Frommel, C.; Khatamzas, E.; Prosch, S.; Schonemann, C.; Kretzschmar, I.; Volkmer-Engert, R.; Volk, H.D.; Reinke, P. Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur. J. Immunol. 2000, 30, 1676–1682. [Google Scholar] [CrossRef]
- Elkington, R.; Walker, S.; Crough, T.; Menzies, M.; Tellam, J.; Bharadwaj, M.; Khanna, R. Ex Vivo Profiling of CD8+-T-Cell Responses to Human Cytomegalovirus Reveals Broad and Multispecific Reactivities in Healthy Virus Carriers. J. Virol. 2003, 77, 5226–5240. [Google Scholar] [CrossRef] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4(+) and CD8(+) T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.M.; Jackson, S.; Okecha, G.; Poole, E.; Sissons, J.G.P.; Sinclair, J.; Wills, M.R. Human cytomegalovirus latency-associated proteins elicit Immune-suppressive IL-10 producing CD4(+) T cells. PLoS Pathog. 2013, 9, e1003635. [Google Scholar] [CrossRef] [PubMed]
- Tey, S.-K.; Goodrum, F.; Khanna, R. CD8+ T-cell recognition of human cytomegalovirus latency-associated determinant pUL138. J. Gen. Virol. 2010, 91, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H.; Roosnek, E.; Rufer, N.; Sinzger, C.; Riegler, S.; Löffler, J.; Grigoleit, U.; Moris, A.; Rammensee, H.-G.; Kanz, L.; et al. Infusion of cytomegalovirus (CMV)–specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; La Gruta, N.L.; Davenport, M.P.; Turner, S.J.; Doherty, P.C. Contribution of T cell receptor affinity to overall avidity for virus-specific CD8+ T cell responses. Proc. Natl. Acad. Sci. USA 2005, 102, 11432–11437. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.C.; Dash, P.; McCullers, J.A.; Doherty, P.C.; Thomas, P.G. T-cell receptor αβ diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci. Transl. Med. 2012, 4, 128ra42. [Google Scholar] [CrossRef]
- Gavin, M.A.; Gilbert, M.J.; Riddell, S.R.; Greenberg, P.D.; Bevan, M.J. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J. Immunol. 1993, 151, 3971–3980. [Google Scholar]
- Ghanekar, S.A.; Nomura, L.E.; Suni, M.A.; Picker, L.J.; Maecker, H.T.; Maino, V.C. Gamma Interferon Expression in CD8(+) T Cells Is a Marker for Circulating Cytotoxic T Lymphocytes That Recognize an HLA A2-Restricted Epitope of Human Cytomegalovirus Phosphoprotein pp65. Clin. Diagn. Lab. Immunol. 2001, 8, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Chen, S.; Sharp, M.; Dekker, C.; Manganello, A.M.; Tongson, E.C.; Maecker, H.T.; Holmes, T.H.; Wang, Z.; Kemble, G.; et al. Persistent and Selective Deficiency of CD4+ T Cell Immunity to Cytomegalovirus in Immunocompetent Young Children. J. Immunol. 2004, 172, 3260–3267. [Google Scholar] [CrossRef] [PubMed]
- Sester, U.; Gartner, B.C.; Wilkens, H.; Schwaab, B.; Wossner, R.; Kindermann, I.; Girndt, M.; Meyerhans, A.; Mueller-Lantzsch, N.; Schafers, H.J.; et al. Differences in CMV-specific T-cell levels and long-term susceptibility to CMV infection after kidney, heart and lung transplantation. Am. J. Transplant. 2005, 5, 1483–1489. [Google Scholar] [CrossRef]
- Jeitziner, S.M.; Walton, S.M.; Torti, N.; Oxenius, A. Adoptive transfer of cytomegalovirus-specific effector CD4+ T cells provides antiviral protection from murine CMV infection. Eur. J. Immunol. 2013, 43, 2886–2895. [Google Scholar] [CrossRef]
- Reuter, J.D.; Wilson, J.H.; Idoko, K.E.; van den Pol, A.N. CD4+ T-cell reconstitution reduces cytomegalovirus in the immunocompromised brain. J. Virol. 2005, 79, 9527–9539. [Google Scholar] [CrossRef] [PubMed]
- Costa-Garcia, M.; Ataya, M.; Moraru, M.; Vilches, C.; Lopez-Botet, M.; Muntasell, A. Human Cytomegalovirus Antigen Presentation by HLA-DR+ NKG2C+ Adaptive NK Cells Specifically Activates Polyfunctional Effector Memory CD4+ T Lymphocytes. Front. Immunol. 2019, 10, 687. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Gratama, J.W.; Brooimans, R.A.; van der Holt, B.; Sintnicolaas, K.; van Doornum, G.; Niesters, H.G.; Löwenberg, B.; Cornelissen, J.J. Monitoring cytomegalovirus IE-1 and pp65-specific CD4+ and CD8+ T-cell responses after allogeneic stem cell transplantation may identify patients at risk for recurrent CMV reactivations. Cytom. Part B Clin. Cytom. 2008, 74, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.; Liang, X.; Wiesner, M.; Mautner, J.; Moosmann, A. A Diverse Repertoire of CD4 T Cells Targets the Immediate-Early 1 Protein of Human Cytomegalovirus. Front. Immunol. 2015, 6, 598. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Bilate, A.M.; Lafaille, J.J. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu. Rev. Immunol. 2012, 30, 733–758. [Google Scholar] [CrossRef] [PubMed]
- Terrazzini, N.; Bajwa, M.; Vita, S.; Cheek, E.; Thomas, D.; Seddiki, N.; Smith, H.; Kern, F. A novel cytomegalovirus-induced regulatory-type T-cell subset increases in size during older life and links virus-specific immunity to vascular pathology. J. Infect. Dis. 2014, 209, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Aandahl, E.M.; Michaëlsson, J.; Moretto, W.J.; Hecht, F.M.; Nixon, D.F. Human CD4+ CD25+ Regulatory T Cells Control T-Cell Responses to Human Immunodeficiency Virus and Cytomegalovirus Antigens. J. Virol. 2004, 78, 2454–2459. [Google Scholar] [CrossRef]
- Sinclair, J. Human cytomegalovirus: Latency and reactivation in the myeloid lineage. J. Clin. Virol. 2008, 41, 180–185. [Google Scholar] [CrossRef]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef]
- Patel, M.; Vlahava, V.M.; Forbes, S.K.; Fielding, C.A.; Stanton, R.J.; Wang, E.C.Y. HCMV-Encoded NK Modulators: Lessons From in vitro and in vivo Genetic Variation. Front. Immunol. 2018, 9, 2214. [Google Scholar] [CrossRef] [PubMed]
- McSharry, B.P.; Avdic, S.; Slobedman, B. Human Cytomegalovirus Encoded Homologs of Cytokines, Chemokines and their Receptors: Roles in Immunomodulation. Viruses 2012, 4, 2448–2470. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 2003, 3, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.; Beisser, P.S.; Bruggeman, C.A. Molecular Mimicry by Cytomegaloviruses. Intervirology 1999, 42, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Banks, T.A.; Rouse, B.T. Herpesviruses-immune escape artists? Clin. Infect. Dis. 1992, 14, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Vider-Shalit, T.; Fishbain, V.; Raffaeli, S.; Louzoun, Y. Phase-Dependent Immune Evasion of Herpesviruses. J. Virol. 2007, 81, 9536–9545. [Google Scholar] [CrossRef] [PubMed]
- Bresnahan, W.A.; Shenk, T.E. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14506–14511. [Google Scholar] [CrossRef] [PubMed]
- Schierling, K.; Stamminger, T.; Mertens, T.; Winkler, M. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J. Virol. 2004, 78, 9512–9523. [Google Scholar] [CrossRef] [PubMed]
- Trgovcich, J.; Cebulla, C.; Zimmerman, P.; Sedmak, D.D. Human Cytomegalovirus Protein pp71 Disrupts Major Histocompatibility Complex Class I Cell Surface Expression. J. Virol. 2006, 80, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Hesse, J.; Reyda, S.; Tenzer, S.; Besold, K.; Reuter, N.; Krauter, S.; Büscher, N.; Stamminger, T.; Plachter, B. Human Cytomegalovirus pp71 Stimulates Major Histocompatibility Complex Class I Presentation of IE1-Derived Peptides at Immediate Early Times of Infection. J. Virol. 2013, 87, 5229–5238. [Google Scholar] [CrossRef]
- Abate, D.A.; Watanabe, S.; Mocarski, E.S. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 2004, 78, 10995–11006. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Shenk, T. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11439–11444. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; Verschuuren, E.A.; Dekkers, C.A.; Adriaanse, H.M.; van der Bij, W.; The, T.H.; Middeldorp, J.M. Expression dynamics of human cytomegalovirus immune evasion genes US3, US6, and US11 in the blood of lung transplant recipients. J. Infect. Dis. 2001, 184, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Humar, A.; Kumar, D.; Gray, M.; Moussa, G.; Venkataraman, S.; Kumar, R.; Tipples, G.A. A prospective assessment of cytomegalovirus immune evasion gene transcription profiles in transplant patients with cytomegalovirus infection. Transplantation 2007, 83, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.R.; Hanson, L.K.; Sun, L.; Slater, J.S.; Stenberg, R.M.; Campbell, A.E. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J. Virol. 1995, 69, 4830–4841. [Google Scholar] [PubMed]
- Ahn, K.; Angulo, A.; Ghazal, P.; Peterson, P.A.; Yang, Y.; Früh, K. Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Proc. Natl. Acad. Sci. USA 1996, 93, 10990–10995. [Google Scholar] [CrossRef] [PubMed]
- Gruhler, A.; Peterson, P.A.; Früh, K. Human cytomegalovirus immediate early glycoprotein US3 retains MHC class I molecules by transient association. Traffic 2000, 1, 318–325. [Google Scholar] [CrossRef]
- Park, B.; Kim, Y.; Shin, J.; Lee, S.; Cho, K.; Früh, K.; Lee, S.; Ahn, K. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity 2004, 20, 71–85. [Google Scholar] [CrossRef]
- Park, B.; Lee, S.; Kim, E.; Cho, K.; Riddell, S.R.; Cho, S.; Ahn, K. Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell 2006, 127, 369–382. [Google Scholar] [CrossRef]
- Noriega, V.M.; Hesse, J.; Gardner, T.J.; Besold, K.; Plachter, B.; Tortorella, D. Human cytomegalovirus US3 modulates destruction of MHC class I molecules. Mol. Immunol. 2012, 51, 245–253. [Google Scholar] [CrossRef]
- Weston, K.; Barrell, B.G. Sequence of the short unique region, short repeats, and part of the long repeats of human cytomegalovirus. J. Mol. Biol. 1986, 192, 177–208. [Google Scholar] [CrossRef]
- Tenney, D.J.; Santomenna, L.D.; Goudie, K.B.; Colberg-Poley, A.M. The human cytomegalovirus US3 immediate-early protein lacking the putative transmembrane domain regulates gene expression. Nucleic Acids Res. 1993, 21, 2931–2937. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yoon, J.; Park, B.; Jun, Y.; Jin, M.; Sung, H.C.; Kim, I.-H.; Kang, S.; Choi, E.-J.; Ahn, B.Y.; et al. Structural and functional dissection of human cytomegalovirus US3 in binding major histocompatibility complex class I molecules. J. Virol. 2000, 74, 11262–11269. [Google Scholar] [CrossRef]
- Lee, S.; Park, B.; Ahn, K. Determinant for endoplasmic reticulum retention in the luminal domain of the human cytomegalovirus US3 glycoprotein. J. Virol. 2003, 77, 2147–2156. [Google Scholar] [CrossRef]
- Misaghi, S.; Sun, Z.-Y.J.; Stern, P.; Gaudet, R.; Wagner, G.; Ploegh, H. Structural and functional analysis of human cytomegalovirus US3 protein. J. Virol. 2004, 78, 413–423. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Z.; Winkler, M.; Biegalke, B. Human cytomegalovirus: Host immune modulation by the viral US3 gene. Int. J. Biochem. Cell Biol. 2009, 41, 503–506. [Google Scholar] [CrossRef]
- Liu, W.; Zhao, Y.; Biegalke, B. Analysis of human cytomegalovirus US3 gene products. Virology 2002, 301, 32–42. [Google Scholar] [CrossRef][Green Version]
- Shin, J.; Park, B.; Lee, S.; Kim, Y.; Biegalke, B.J.; Kang, S.; Ahn, K. A short isoform of human cytomegalovirus US3 functions as a dominant negative inhibitor of the full-length form. J. Virol. 2006, 80, 5397–5404. [Google Scholar] [CrossRef]
- Jones, T.R.; Sun, L. Human cytomegalovirus US2 destabilizes major histocompatibility complex class I heavy chains. J. Virol. 1997, 71, 2970–2979. [Google Scholar]
- Wiertz, E.J.H.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec6l-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar] [CrossRef]
- Chevalier, M.S.; Daniels, G.M.; Johnson, D.C. Binding of Human Cytomegalovirus US2 to Major Histocompatibility Complex Class I and II Proteins Is Not Sufficient for Their Degradation. J. Virol. 2002, 76, 8265–8275. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Gaudet, R.; Tortorella, D.; Wang, E.W.; Ploegh, H.L.; Wiley, D.C. Antigen presentation subverted: Structure of the human cytomegalovirus protein US2 bound to the class I molecule HLA-A2. Proc. Natl. Acad. Sci. USA 2001, 98, 6794–6799. [Google Scholar] [CrossRef] [PubMed]
- Halenius, A.; Gerke, C.; Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: So many targets[mdash]but how many arrows in the quiver? Cell. Mol. Immunol. 2015, 12, 139–153. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Wang, E.W.; Tortorella, D.; Schust, D.J.; Ploegh, H.L. Human cytomegalovirus US2 endoplasmic reticulum-lumenal domain dictates association with major histocompatibility complex class I in a locus-specific manner. J. Virol. 2001, 75, 5197–5204. [Google Scholar] [CrossRef] [PubMed]
- Barel, M.T.; Ressing, M.; Pizzato, N.; van Leeuwen, D.; Le Bouteiller, P.; Lenfant, F.; Wiertz, E.J. Human cytomegalovirus-encoded US2 differentially affects surface expression of MHC class I locus products and targets membrane-bound, but not soluble HLA-G1 for degradation. J. Immunol. 2003, 171, 6757–6765. [Google Scholar] [CrossRef] [PubMed]
- Stagg, H.R.; Thomas, M.; van den Boomen, D.; Wiertz, E.J.H.J.; Drabkin, H.A.; Gemmill, R.M.; Lehner, P.J. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 2009, 186, 685–692. [Google Scholar] [CrossRef]
- Van den Boomen, D.J.H.; Lehner, P.J. Identifying the ERAD ubiquitin E3 ligases for viral and cellular targeting of MHC class I. Mol. Immunol. 2015, 68, 106–111. [Google Scholar] [CrossRef]
- Van de Weijer, M.L.; Schuren, A.B.C.; van den Boomen, D.J.H.; Mulder, A.; Claas, F.H.J.; Lehner, P.J.; Lebbink, R.J.; Wiertz, E. Multiple E2 ubiquitin-conjugating enzymes regulate human cytomegalovirus US2-mediated immunoreceptor downregulation. J. Cell Sci. 2017, 130, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Rehm, A.; Stern, P.; Ploegh, H.L.; Tortorella, D. Signal peptide cleavage of a type I membrane protein, HCMV US11, is dependent on its membrane anchor. EMBO J. 2001, 20, 1573–1582. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Ploegh, H.L.; Tortorella, D. US2, a Human Cytomegalovirus-encoded Type I Membrane Protein, Contains a Non-cleavable Amino-terminal Signal Peptide. J. Biol. Chem. 2002, 277, 11306–11313. [Google Scholar] [CrossRef]
- Loureiro, J.; Lilley, B.N.; Spooner, E.; Noriega, V.; Tortorella, D.; Ploegh, H.L. Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature 2006, 441, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Boname, J.M.; Bloor, S.; Wandel, M.P.; Nathan, J.A.; Antrobus, R.; Dingwell, K.S.; Thurston, T.L.; Smith, D.L.; Smith, J.C.; Randow, F.; et al. Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J. Cell Biol. 2014, 205, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Cho, K.; Cho, S.; Kim, I.; Oh, C.; Ahn, K. Protein disulphide isomerase is required for signal peptide peptidase-mediated protein degradation. EMBO J. 2010, 29, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Furman, M.H.; Ploegh, H.L.; Tortorella, D. Membrane-specific, Host-derived Factors Are Required for US2- and US11-mediated Degradation of Major Histocompatibility Complex Class I Molecules. J. Biol. Chem. 2002, 277, 3258–3267. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; van den Boomen, D.J.H.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J.; et al. Plasma Membrane Profiling Defines an Expanded Class of Cell Surface Proteins Selectively Targeted for Degradation by HCMV US2 in Cooperation with UL141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef]
- Soetandyo, N.; Ye, Y. The p97 ATPase Dislocates MHC Class I Heavy Chain in US2-expressing Cells via a Ufd1-Npl4-independent Mechanism. J. Biol. Chem. 2010, 285, 32352–32359. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef]
- Richly, H.; Rape, M.; Braun, S.; Rumpf, S.; Hoege, C.; Jentsch, S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 2005, 120, 73–84. [Google Scholar] [CrossRef]
- Wang, Q.; Li, L.; Ye, Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 2006, 174, 963–971. [Google Scholar] [CrossRef]
- Wiertz, E.J.H.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Van der Wal, F.J.; Kikkert, M.; Wiertz, E. The HCMV Gene Products US2 and US11 Target MHC Class I Molecules for Degradation in the Cytosol. In Viral Proteins Counteracting Host Defenses; Koszinowski, U., Hengel, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 269, pp. 37–55. [Google Scholar]
- Jones, T.R.; Muzithras, V.P. Fine mapping of transcripts expressed from the US6 gene family of human cytomegalovirus strain AD169. J. Virol. 1991, 65, 2024–2036. [Google Scholar] [PubMed]
- Barel, M.T.; Hassink, G.C.; Voorden, S.V.; Wiertz, E.J.H.J. Human cytomegalovirus-encoded US2 and US11 target unassembled MHC class I heavy chains for degradation. Mol. Immunol. 2006, 43, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Barel, M.T.; Pizzato, N.; van Leeuwen, D.; Bouteiller, P.L.; Wiertz, E.J.; Lenfant, F. Amino acid composition of alpha1/alpha2 domains and cytoplasmic tail of MHC class I molecules determine their susceptibility to human cytomegalovirus US11-mediated down-regulation. Eur. J. Immunol. 2003, 33, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-O.; Hwang, S.; Park, J.; Park, B.; Jin, B.-S.; Lee, S.; Kim, E.; Cho, S.; Kim, Y.; Cho, K.; et al. Functional dissection of HCMV US11 in mediating the degradation of MHC class I molecules. Biochem. Biophys. Res. Commun. 2005, 330, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Tortorella, D.; Ploegh, H.L. Dislocation of a Type I Membrane Protein Requires Interactions between Membrane-spanning Segments within the Lipid Bilayer. Mol. Biol. Cell. 2003, 14, 3690–3698. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van den Boomen, D.J.H.; Timms, R.T.; Grice, G.L.; Stagg, H.R.; Skødt, K.; Dougan, G.; Nathan, J.A.; Lehner, P.J. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. USA 2014, 111, 11425–11430. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004, 429, 834–840. [Google Scholar] [CrossRef]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef]
- Cho, S.; Kim, B.Y.; Ahn, K.; Jun, Y. The C-Terminal Amino Acid of the MHC-I Heavy Chain Is Critical for Binding to Derlin-1 in Human Cytomegalovirus US11-Induced MHC-I Degradation. PLoS ONE 2013, 8, e72356. [Google Scholar] [CrossRef]
- Van de Weijer, M.L.; Bassik, M.C.; Luteijn, R.D.; Voorburg, C.M.; Lohuis, M.A.M.; Kremmer, E.; Hoeben, R.C.; LeProust, E.M.; Chen, S.; Hoelen, H.; et al. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat. Commun. 2014, 5, 3832. [Google Scholar] [CrossRef]
- Flierman, D.; Coleman, C.S.; Pickart, C.M.; Rapoport, T.A.; Chau, V. E2-25K mediates US11-triggered retro-translocation of MHC class I heavy chains in a permeabilized cell system. Proc. Natl. Acad. Sci. USA 2006, 103, 11589–11594. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.; Lilley, B.N.; Ploegh, H.L. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 2006, 175, 261–270. [Google Scholar] [CrossRef]
- Shamu, C.E.; Flierman, D.; Ploegh, H.L.; Rapoport, T.A.; Chau, V. Polyubiquitination Is Required for US11-dependent Movement of MHC Class I Heavy Chain from Endoplasmic Reticulum into Cytosol. Mol. Biol. Cell 2001, 12, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M.; Hassink, G.; Barel, M.; Hirsch, C.; van der Wal, F.J.; Wiertz, E. Ubiquitination is essential for human cytomegalovirus US11-mediated dislocation of MHC class I molecules from the endoplasmic reticulum to the cytosol. Biochem. J. 2001, 358, 369–377. [Google Scholar] [CrossRef]
- Shamu, C.E.; Story, C.M.; Rapoport, T.A.; Ploegh, H.L. The Pathway of Us11-Dependent Degradation of Mhc Class I Heavy Chains Involves a Ubiquitin-Conjugated Intermediate. J. Cell Biol. 1999, 147, 45–58. [Google Scholar] [CrossRef]
- Hassink, G.C.; Barel, M.T.; Van Voorden, S.B.; Kikkert, M.; Wiertz, E.J. Ubiquitination of MHC Class I Heavy Chains Is Essential for Dislocation by Human Cytomegalovirus-encoded US2 but Not US11. J. Biol. Chem. 2006, 281, 30063–30071. [Google Scholar] [CrossRef] [PubMed]
- Lehner, P.J.; Karttunen, J.T.; Wilkinson, G.W.G.; Cresswell, P. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc. Natl. Acad. Sci. USA 1997, 94, 6904–6909. [Google Scholar] [CrossRef] [PubMed]
- Hengel, H.; Koopmann, J.-O.; Flohr, T.; Muranyi, W.; Goulmy, E.; Hämmerling, G.J.; Koszinowski, U.H.; Momburg, F. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 1997, 6, 623–632. [Google Scholar] [CrossRef]
- Schölz, C.; Tampé, R. The peptide-loading complex—Antigen translocation and MHC class I loading. Biol. Chem. 2009, 390, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Neumann, L.; Tampe, R. Kinetic analysis of peptide binding to the TAP transport complex: Evidence for structural rearrangements induced by substrate binding. J. Mol. Biol. 1999, 294, 1203–1213. [Google Scholar] [CrossRef]
- Hewitt, E.W.; Gupta, S.S.; Lehner, P.J. The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J. 2001, 20, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Meyer, T.H.; Uebel, S.; Sempé, P.; Djaballah, H.; Yang, Y.; Peterson, P.A.; Früh, K.; Tampé, R. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 1996, 15, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, C.; Gorbulev, S.; Hutschenreiter, S.; Pawlitschko, K.; Abele, R.; Tampé, R. Molecular Mechanism and Structural Aspects of Transporter Associated with Antigen Processing Inhibition by the Cytomegalovirus Protein US6. J. Biol. Chem. 2001, 276, 48031–48039. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Gruhler, A.; Galocha, B.; Jones, T.R.; Wiertz, E.J.H.J.; Ploegh, H.L.; Peterson, P.A.; Yang, Y.; Früh, K. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997, 6, 613–621. [Google Scholar] [CrossRef]
- Halenius, A.; Momburg, F.; Reinhard, H.; Bauer, D.; Lobigs, M.; Hengel, H. Physical and functional interactions of the cytomegalovirus US6 glycoprotein with the transporter associated with antigen processing. J. Biol. Chem. 2006, 281, 5383–5390. [Google Scholar] [CrossRef] [PubMed]
- Dugan, G.E.; Hewitt, E.W. Structural and Functional Dissection of the Human Cytomegalovirus Immune Evasion Protein US6. J. Virol. 2008, 82, 3271–3282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Furman, M.H.; Dey, N.; Tortorella, D.; Ploegh, H.L. The human cytomegalovirus US10 gene product delays trafficking of major histocompatibility complex class I molecules. J. Virol. 2002, 76, 11753–11756. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Spooner, E.; Houser, B.L.; Strominger, J.L.; Ploegh, H.L. The HCMV membrane glycoprotein US10 selectively targets HLA-G for degradation. J. Exp. Med. 2010, 207, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Ben-Arieh, S.V.; Zimerman, B.; Smorodinsky, N.I.; Yaacubovicz, M.; Schechter, C.; Bacik, I.; Gibbs, J.; Bennink, J.R.; Yewdell, J.W.; Coligan, J.E.; et al. Human Cytomegalovirus Protein US2 Interferes with the Expression of Human HFE, a Nonclassical Class I Major Histocompatibility Complex Molecule That Regulates Iron Homeostasis. J. Virol. 2001, 75, 10557–10562. [Google Scholar] [CrossRef]
- Arieh, S.V.-B.; Laham, N.; Schechter, C.; Yewdell, J.W.; Coligan, J.E.; Ehrlich, R. A single viral protein HCMV US2 affects antigen presentation and intracellular iron homeostasis by degradation of classical HLA class I and HFE molecules. Blood 2003, 101, 2858–2864. [Google Scholar] [CrossRef]
- Fielding, C.A.; Aicheler, R.; Stanton, R.J.; Wang, E.C.Y.; Han, S.; Seirafian, S.; Davies, J.; McSharry, B.P.; Weekes, M.P.; Antrobus, P.R.; et al. Two Novel Human Cytomegalovirus NK Cell Evasion Functions Target MICA for Lysosomal Degradation. PLoS Pathog. 2014, 10, e1004058. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.W.; Tomasec, P.; Stanton, R.J.; Armstrong, M.; Prod’homme, V.; Aicheler, R.; McSharry, B.P.; Rickards, C.R.; Cochrane, D.; Llewellyn-Lacey, S.; et al. Modulation of natural killer cells by human cytomegalovirus. J. Clin. Virol. 2008, 41, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Kubin, M.; Cassiano, L.; Chalupny, J.; Chin, W.; Cosman, D.; Fanslow, W.; Mullberg, J.; Rousseau, A.M.; Ulrich, D.; Armitage, R. ULBP1, 2, 3: Novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur. J. Immunol. 2001, 31, 1428–1437. [Google Scholar] [CrossRef]
- Welte, S.A.; Sinzger, C.; Lutz, S.Z.; Singh-Jasuja, H.; Sampaio, K.L.; Eknigk, U.; Rammensee, H.G.; Steinle, A. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur. J. Immunol. 2003, 33, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host immune system gene targeting by a viral miRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Arnon, T.I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E.; et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 2005, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Ulbrecht, M.; Martinozzi, S.; Grzeschik, M.; Hengel, H.; Ellwart, J.W.; Pla, M.; Weiss, E.H. Cutting edge: The human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J. Immunol. 2000, 164, 5019–5022. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.; McSharry, B.; Retiere, C.; Tomasec, P.; Williams, S.; Borysiewicz, L.K.; Braud, V.M.; Wilkinson, G.W. UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2002, 99, 7570–7575. [Google Scholar] [CrossRef]
- Lee, N.; Goodlett, D.R.; Ishitani, A.; Marquardt, H.; Geraghty, D.E. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J. Immunol. 1998, 160, 4951–4960. [Google Scholar]
- Tomasec, P.; Braud, V.M.; Rickards, C.; Powell, M.B.; McSharry, B.P.; Gadola, S.; Cerundolo, V.; Borysiewicz, L.K.; McMichael, A.J.; Wilkinson, G.W. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000, 287, 1031. [Google Scholar] [CrossRef]
- Prod’homme, V.; Griffin, C.; Aicheler, R.J.; Wang, E.C.; McSharry, B.P.; Rickards, C.R.; Stanton, R.J.; Borysiewicz, L.K.; Lopez-Botet, M.; Wilkinson, G.W.; et al. The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1- NK cells. J. Immunol. 2007, 178, 4473–4481. [Google Scholar] [CrossRef] [PubMed]
- Reyburn, H.T.; Mandelboim, O.; Vales-Gomez, M.; Davis, D.M.; Pazmany, L.; Strominger, J.L. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature 1997, 386, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, B.; Cho, S.; Shin, J.; Cho, K.; Jun, Y.; Ahn, K. Human cytomegalovirus UL18 utilizes US6 for evading the NK and T-cell responses. PLoS Pathog. 2008, 4, e1000123. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Mousavi-Jazi, M.; Linde, A.; Soderstrom, K.; Brytting, M.; Wahren, B.; Karre, K.; Carbone, E. Human cytomegalovirus strain-dependent changes in NK cell recognition of infected fibroblasts. J. Immunol. 2000, 164, 4775–4782. [Google Scholar] [CrossRef] [PubMed]
- Tomasec, P.; Wang, E.C.; Davison, A.J.; Vojtesek, B.; Armstrong, M.; Griffin, C.; McSharry, B.P.; Morris, R.J.; Llewellyn-Lacey, S.; Rickards, C.; et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat. Immunol. 2005, 6, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Chalupny, N.J.; Rein-Weston, A.; Dosch, S.; Cosman, D. Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem. Biophys. Res. Commun. 2006, 346, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Dassa, L.; Seidel, E.; Oiknine-Djian, E.; Yamin, R.; Wolf, D.G.; Le-Trilling, V.T.K.; Mandelboim, O. The Human Cytomegalovirus Protein UL148A Downregulates the NK Cell-Activating Ligand MICA To Avoid NK Cell Attack. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.J.; Prod’homme, V.; Purbhoo, M.A.; Moore, M.; Aicheler, R.J.; Heinzmann, M.; Bailer, S.M.; Haas, J.; Antrobus, R.; Weekes, M.P.; et al. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 2014, 16, 201–214. [Google Scholar] [CrossRef]
- Wang, E.C.Y.; Pjechova, M.; Nightingale, K.; Vlahava, V.M.; Patel, M.; Ruckova, E.; Forbes, S.K.; Nobre, L.; Antrobus, R.; Roberts, D.; et al. Suppression of costimulation by human cytomegalovirus promotes evasion of cellular immune defenses. Proc. Natl. Acad. Sci. USA 2018, 115, 4998–5003. [Google Scholar] [CrossRef]
- Afessa, B.; Peters, S.G. Major complications following hematopoietic stem cell transplantation. Semin Respir. Crit. Care Med. 2006, 27, 297–309. [Google Scholar] [CrossRef]
- Amina, A. Antiviral Treatment of Cytomegalovirus Infection. Infect. Disord. Drug Targets 2011, 11, 475–503. [Google Scholar]
- Biron, K.K. Antiviral drugs for cytomegalovirus diseases. Antivir. Res. 2006, 71, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, R.F.; Ullmann, A.J.; Stoelben, S.; Richard, M.P.; Bornhauser, M.; Groth, C.; Einsele, H.; Silverman, M.; Mullane, K.M.; Brown, J.; et al. Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N. Engl. J. Med. 2014, 370, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Verfuerth, S.; Pizzey, A.; Khan, N.; Guiver, M.; Moss, P.A.; Mackinnon, S. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet 2003, 362, 1375–1377. [Google Scholar] [CrossRef]
- Bollard, C.M.; Heslop, H.E. T cells for viral infections after allogeneic hematopoietic stem cell transplant. Blood 2016, 127, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Koehne, G.; Hasan, A.; Doubrovina, E.; Prockop, S.; Tyler, E.; Wasilewski, G.; O’Reilly, R.J. Immunotherapy with donor T cells sensitized with overlapping pentadecapeptides for treatment of persistent cytomegalovirus infection or viremia. Biol. Blood Marrow Transplant. 2015, 21, 1663–1678. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Thomson, K.; Samuel, E.; Dyer, G.; Armoogum, J.; Chakraverty, R.; Pang, K.; Mackinnon, S.; Lowdell, M.W. Directly Selected Cytomegalovirus-Reactive Donor T Cells Confer Rapid and Safe Systemic Reconstitution of Virus-Specific Immunity Following Stem Cell Transplantation. Clin. Infect. Dis. 2011, 52, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Micklethwaite, K.; Hansen, A.; Foster, A.; Snape, E.; Antonenas, V.; Sartor, M.; Shaw, P.; Bradstock, K.; Gottlieb, D. Ex Vivo Expansion and Prophylactic Infusion of CMV-pp65 Peptide-Specific Cytotoxic T-Lymphocytes following Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2007, 13, 707–714. [Google Scholar] [CrossRef]
- Hyun, S.J.; Sohn, H.J.; Lee, H.J.; Lee, S.D.; Kim, S.; Sohn, D.H.; Hong, C.H.; Choi, H.; Cho, H.I.; Kim, T.G. Comprehensive Analysis of Cytomegalovirus pp65 Antigen-Specific CD8(+) T Cell Responses According to Human Leukocyte Antigen Class I Allotypes and Intraindividual Dominance. Front. Immunol. 2017, 8, 1591. [Google Scholar] [CrossRef]
- Gerdemann, U.; Keirnan, J.M.; Katari, U.L.; Yanagisawa, R.; Christin, A.S.; Huye, L.E.; Perna, S.K.; Ennamuri, S.; Gottschalk, S.; Brenner, M.K.; et al. Rapidly Generated Multivirus-specific Cytotoxic T Lymphocytes for the Prophylaxis and Treatment of Viral Infections. Mol. Ther. 2012, 20, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.; Williams, R.Y.; O’Reilly, R.J.; Koehne, G. Generation of CMV-specific T lymphocytes using protein-spanning pools of pp65-derived overlapping pentadecapeptides for adoptive immunotherapy. Blood 2005, 105, 2793–2801. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Gerdemann, U.; Katari, U.L.; Tzannou, I.; Liu, H.; Martinez, C.; Leung, K.; Carrum, G.; Gee, A.P.; Vera, J.F.; et al. Activity of Broad-Spectrum T Cells as Treatment for AdV, EBV, CMV, BKV, and HHV6 Infections after HSCT. Sci. Transl. Med. 2014, 6, 242ra83. [Google Scholar] [CrossRef] [PubMed]
- Pump, W.C.; Schulz, R.; Huyton, T.; Kunze-Schumacher, H.; Martens, J.; Ho, G.T.; Blasczyk, R.; Bade-Doeding, C. Releasing the concept of HLA-allele specific peptide anchors in viral infections: A non-canonical naturally presented human cytomegalovirus-derived HLA-A*24:02 restricted peptide drives exquisite immunogenicity. Hla 2019, 94, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Schleiss, M.R. Update on the current status of cytomegalovirus vaccines. Expert Rev. Vaccines 2010, 9, 1303–1314. [Google Scholar] [CrossRef]
- Schleiss, M.R. Current Status of Cytomegalovirus Vaccines; John Wiley & Sons Ltd.: Chichester, UK, 2016; Available online: http://www.els.net (accessed on 24 July 2019).
- Bernstein, D.I.; Munoz, F.M.; Callahan, S.T.; Rupp, R.; Wootton, S.H.; Edwards, K.M.; Turley, C.B.; Stanberry, L.R.; Patel, S.M.; McNeal, M.M.; et al. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: A randomized clinical trial. Vaccine 2016, 34, 313–319. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O’Beirne, J.; et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef]
- Paston, S.J.; Dodi, I.A.; Madrigal, J.A. Progress made towards the development of a CMV peptide vaccine. Hum. Immunol. 2004, 65, 544–549. [Google Scholar] [CrossRef]
- Chiurchiu, S.; Calo Carducci, F.I.; Rocchi, F.; Simonetti, A.; Bonatti, G.; Salmaso, S.; Melchiorri, D.; Pani, L.; Rossi, P. Is HCMV vaccine an unmet need? The state of art of vaccine development. Int. J. Immunopathol. Pharmacol. 2013, 26, 15–26. [Google Scholar] [CrossRef]
- Finnefrock, A.C.; Freed, D.C.; Tang, A.; Li, F.; He, X.; Wu, C.; Nahas, D.; Wang, D.; Fu, T.M. Preclinical evaluations of peptide-conjugate vaccines targeting the antigenic domain-2 of glycoprotein B of human cytomegalovirus. Hum. Vaccines Immunother. 2016, 12, 2106–2112. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manandhar, T.; Hò, G.-G.T.; Pump, W.C.; Blasczyk, R.; Bade-Doeding, C. Battle between Host Immune Cellular Responses and HCMV Immune Evasion. Int. J. Mol. Sci. 2019, 20, 3626. https://doi.org/10.3390/ijms20153626
Manandhar T, Hò G-GT, Pump WC, Blasczyk R, Bade-Doeding C. Battle between Host Immune Cellular Responses and HCMV Immune Evasion. International Journal of Molecular Sciences. 2019; 20(15):3626. https://doi.org/10.3390/ijms20153626
Chicago/Turabian StyleManandhar, Trishna, Gia-Gia T. Hò, Wiebke C. Pump, Rainer Blasczyk, and Christina Bade-Doeding. 2019. "Battle between Host Immune Cellular Responses and HCMV Immune Evasion" International Journal of Molecular Sciences 20, no. 15: 3626. https://doi.org/10.3390/ijms20153626
APA StyleManandhar, T., Hò, G.-G. T., Pump, W. C., Blasczyk, R., & Bade-Doeding, C. (2019). Battle between Host Immune Cellular Responses and HCMV Immune Evasion. International Journal of Molecular Sciences, 20(15), 3626. https://doi.org/10.3390/ijms20153626