The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance



 ,
, 
Abstract
1. Introduction
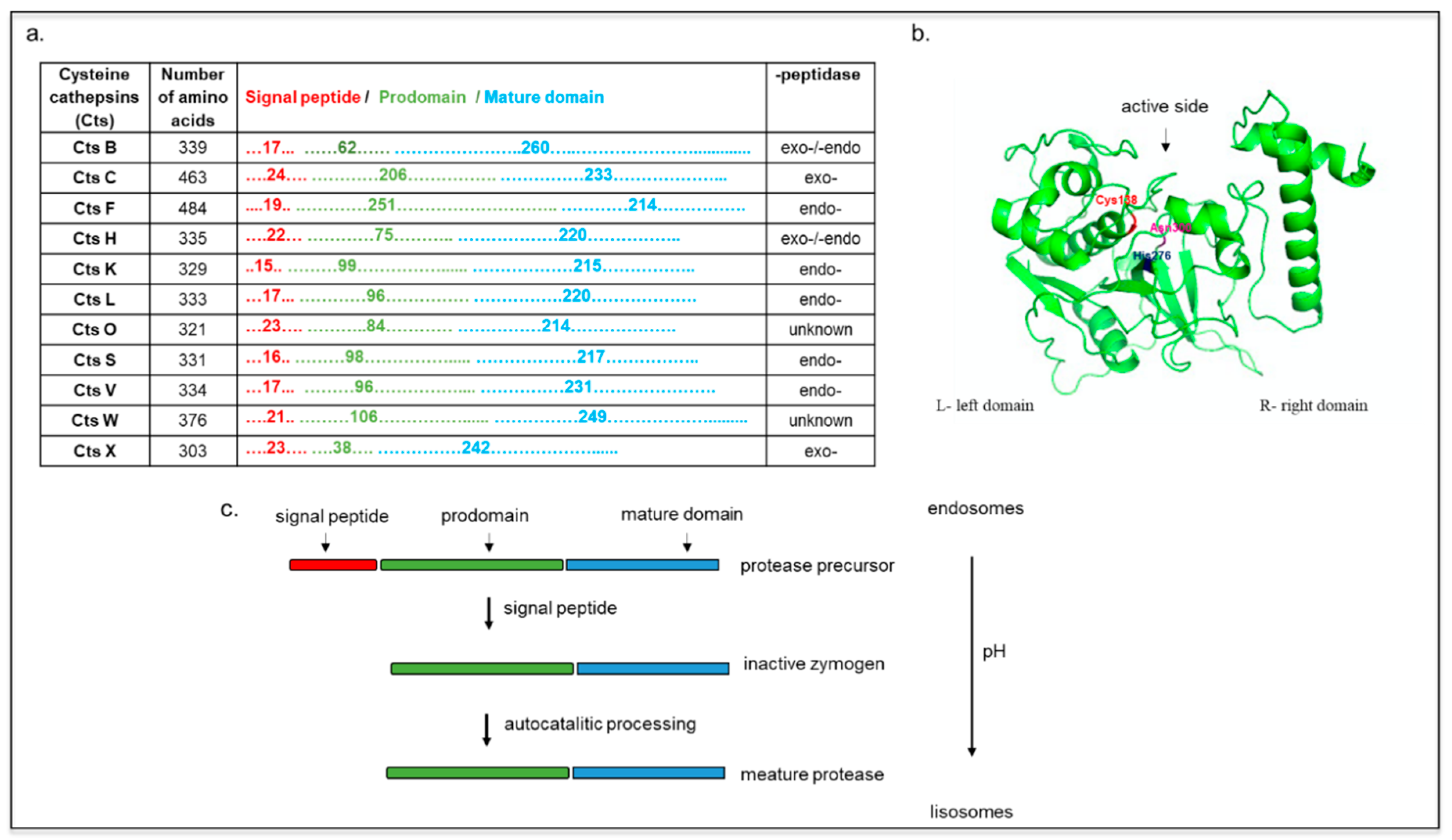
2. Cts Synthesis, Structure, and Localization
Inhibitors of Cysteine Cathepsins
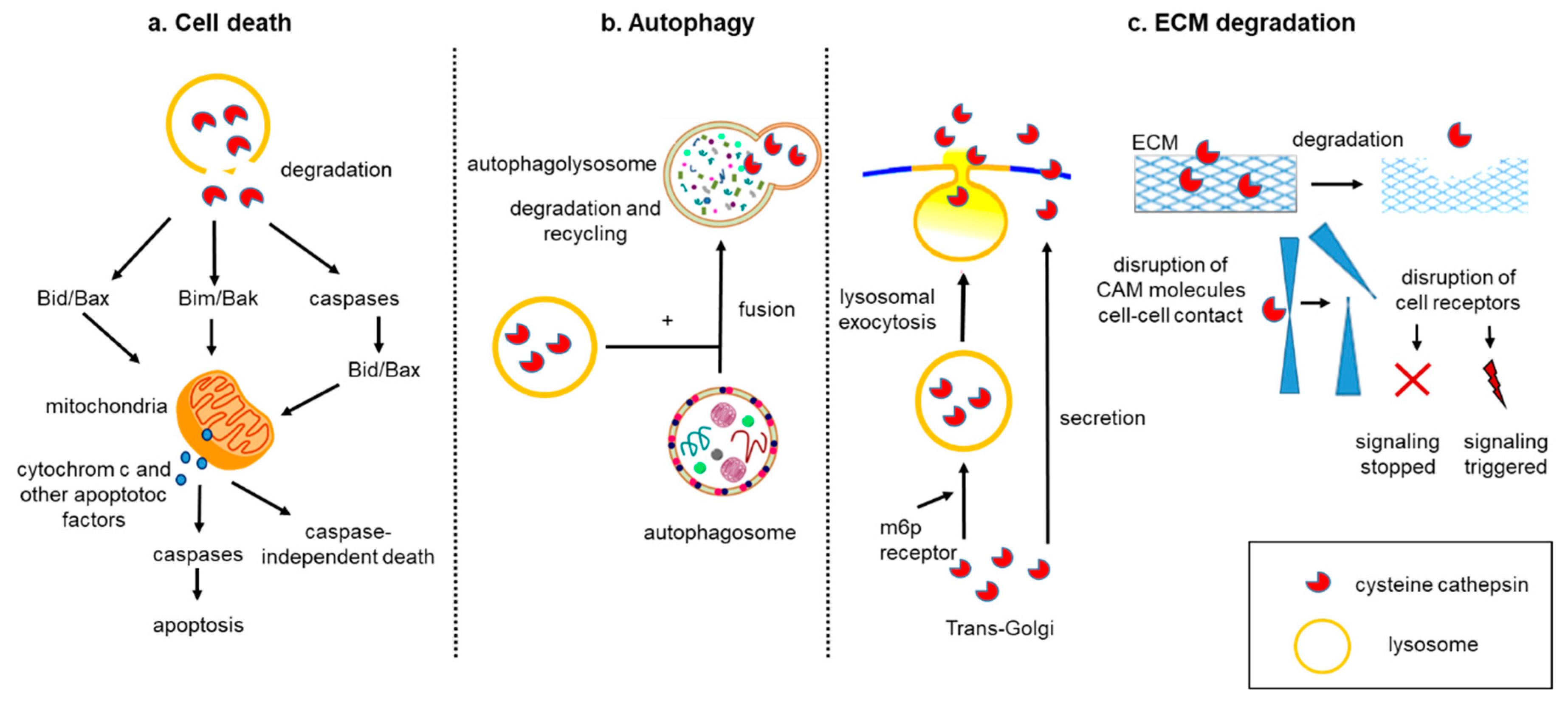
3. Cell Death
4. Autophagy
5. Tumor Matrix Cellular Degradation
6. Crosstalk between Cell Death, Autophagy, and Tumor Matrix Degradation
7. Drug Resistance
8. Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CTS | Cysteine Cathepsins |
| LOH | Loss of Heterozygosity |
| BID | BH3 Interacting Domain Death Agonist |
| BAK | BCL2 Antagonist/Killer |
| BAX | BCL2 Associated X, Apoptosis Regulator |
| XIAP | X-Chromosome-Linked Inhibitor of Apoptosis |
| ROS | Reactive Oxygen Species |
| TNF | Tumor Necrosis Factor |
| LAMP-1 | Lysosome-Associated Membrane Protein-1 |
| HRPC | Hormone-Refractory Prostate Cancer |
| NSCLC | Non-Small Cell Lung Cancer |
| E-64-d | Protease Inhibitor |
| EMT | Epithelial–Mesenchymal Transition |
| MET | Mesenchymal-Epithelial Transition |
| SNAIL1 | Snail Family Transcriptional Repressor 1 |
| SLUG | Snail Family Transcriptional Repressor 2 |
| ZEB | Zinc Finger E-Box Binding Homeobox |
| NF-κB | Nuclear Factor Kappa B |
| PI3K | Phosphatidylinositol 3-Kinase |
| MMP | Matrix Metalloproteinase |
| ECM | Extracellular Matrix |
| MV | Microvesicle |
| KGP94 | Cathpesin L Inhibitor |
References
- Brix, K. Lysosomal Proteases: Revival of the Sleeping Beauty. In Madame Curie Bioscience Database [Internet]; Paul Saftig; Landes Bioscience: Austin, TX, USA, 2005. [Google Scholar]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Fonović, M.; Turk, B. Cysteine cathepsins and extracellular matrix degradation. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Kukor, Z.; Mayerle, J.; Kruger, B.; Toth, M.; Steed, P.M.; Halangk, W.; Lerch, M.M.; Sahin-Toth, M. Presence of cathepsin B in the human pancreatic secretory pathway and its role in trypsinogen activation during hereditary pancreatitis. J. Biol. Chem. 2002, 277, 21389–21396. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar] [CrossRef]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin. Exp. Metastasis 2013, 30, 891–902. [Google Scholar] [CrossRef]
- Chen, S.; Dong, H.; Yang, S.; Guo, H. Cathepsins in digestive cancers. Oncotarget 2017, 8, 41690. [Google Scholar] [CrossRef]
- Sever, S.; Altintas, M.M.; Nankoe, S.R.; Möller, C.C.; Ko, D.; Wei, C.; Henderson, J.; del Re, E.C.; Hsing, L.; Erickson, A. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J. Clin. Investig. 2007, 117, 2095–2104. [Google Scholar] [CrossRef]
- Goulet, B.; Baruch, A.; Moon, N.-S.; Poirier, M.; Sansregret, L.L.; Erickson, A.; Bogyo, M.; Nepveu, A. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol. Cell 2004, 14, 207–219. [Google Scholar] [CrossRef]
- Cheng, X.W.; Shi, G.-P.; Kuzuya, M.; Sasaki, T.; Okumura, K.; Murohara, T. Role for cysteine protease cathepsins in heart disease: Focus on biology and mechanisms with clinical implication. Circulation 2012, 125, 1551–1562. [Google Scholar] [CrossRef]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta. 2000, 291, 113–135. [Google Scholar] [CrossRef]
- Vidak, E.; Javorsek, U.; Vizovisek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chou, P.M.; Mirkin, B.L.; Rebbaa, A. Senescence-initiated reversal of drug resistance: Specific role of cathepsin L. Cancer Res. 2004, 64, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Shi, C.; Yan, Z.; Wu, M. Overexpression of Cathepsin L is associated with chemoresistance and invasion of epithelial ovarian cancer. Oncotarget 2016, 7, 45995. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Turk, B.; Turk, D. Lysosomal cysteine proteases: Facts and opportunities. EMBO J. 2001, 20, 4629–4633. [Google Scholar] [CrossRef] [PubMed]
- Brömme, D.; Wilson, S. Role of cysteine cathepsins in extracellular proteolysis; Springer: Berlin/Heidelberg, Germany, 2011; pp. 23–51. [Google Scholar]
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: Essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des. 2002, 8, 1623–1637. [Google Scholar] [CrossRef]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine proteases: Modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef]
- Lecaille, F.; Kaleta, J.; Brömme, D. Human and parasitic papain-like cysteine proteases: Their role in physiology and pathology and recent developments in inhibitor design. Chem. Rev. 2002, 102, 4459–4488. [Google Scholar] [CrossRef]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef]
- Grzonka, Z.; Jankowska, E.; Kasprzykowski, F.; Kasprzykowska, R.; Lankiewicz, L.; Wiczk, W.; Wieczerzak, E.; Ciarkowski, J.; Drabik, P.; Janowski, R.; et al. Structural studies of cysteine proteases and their inhibitors. Acta Biochim. Pol. 2001, 48, 1–20. [Google Scholar] [PubMed]
- Stoka, V.; Turk, B.; Turk, V. Lysosomal cysteine proteases: Structural features and their role in apoptosis. IUBMB Life 2005, 57, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Wiederanders, B. The function of propeptide domains of cysteine proteinases; Springer: Berlin/Heidelberg, Germany, 2002; pp. 261–270. [Google Scholar]
- Canuel, M.; Korkidakis, A.; Konnyu, K.; Morales, C.R. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem. Biophys. Res. Commun. 2008, 373, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Dahl, S.W.; Halkier, T.; Lauritzen, C.; Dolenc, I.; Pedersen, J.; Turk, V.; Turk, B. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 2001, 40, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Yamaza, T.; Goto, T.; Kamiya, T.; Kobayashi, Y.; Sakai, H.; Tanaka, T. Study of immunoelectron microscopic localization of cathepsin K in osteoclasts and other bone cells in the mouse femur. Bone 1998, 23, 499–509. [Google Scholar] [CrossRef]
- Perišić Nanut, M.; Sabotič, J.; Jewett, A.; Kos, J. Cysteine cathepsins as regulators of the cytotoxicity of NK and T cells. Front. Immunol. 2014, 5, 616. [Google Scholar] [CrossRef]
- Magister, Š.; Obermajer, N.; Mirković, B.; Švajger, U.; Renko, M.; Softić, A.; Colbert, J.D.; Watts, C.; Kos, J. Regulation of cathepsins S and L by cystatin F during maturation of dendritic cells. Eur. J. Cell Biol. 2012, 91, 391–401. [Google Scholar] [CrossRef]
- Wendt, W.; Schulten, R.; Stichel, C.C.; Lübbert, H. Intra-versus extracellular effects of microglia-derived cysteine proteases in a conditioned medium transfer model. J. Neurochem. 2009, 110, 1931–1941. [Google Scholar] [CrossRef]
- Sullivan, S.; Tosetto, M.; Kevans, D.; Coss, A.; Wang, L.; O’Donoghue, D.; Hyland, J.; Sheahan, K.; Mulcahy, H.; O’Sullivan, J. Localization of nuclear cathepsin L and its association with disease progression and poor outcome in colorectal cancer. Int. J. Cancer 2009, 125, 54–61. [Google Scholar] [CrossRef]
- Sloane, B.F.; Rozhin, J.; Johnson, K.; Taylor, H.; Crissman, J.D.; Honn, K.V. Cathepsin B: Association with plasma membrane in metastatic tumors. Proc. Natl. Acad. Sci. 1986, 83, 2483–2487. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Tamhane, T.; Lu, S.; Maelandsmo, G.M.; Haugen, M.H.; Brix, K. Nuclear cathepsin L activity is required for cell cycle progression of colorectal carcinoma cells. Biochimie 2016, 122, 208–218. [Google Scholar] [CrossRef] [PubMed]
- MAGNY, M.-C. Cathepsin B: An alternative protease for the generation of an aggrecan ‘metalloproteinase’cleavage neoepitope. Biochem. J. 1998, 335, 491–494. [Google Scholar]
- Maciewicz, R.A.; Wotton, S.F.; Etherington, D.J.; Duance, V.C. Susceptibility of the cartilage collagens types II, IX and XI to degradation by the cysteine proteinases, cathepsins B and L. FEBS Lett. 1990, 269, 189–193. [Google Scholar] [CrossRef]
- Roughley, P.J.; Barrett, A.J. The degradation of cartilage proteoglycans by tissue proteinases. Proteoglycan structure and its susceptibility to proteolysis. Biochem. J. 1977, 167, 629–637. [Google Scholar] [CrossRef]
- Buck, M.; Karustis, D.G.; Day, N.; Honn, K.; Sloane, B.F. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. Biochem. J. 1992, 282, 273–278. [Google Scholar] [CrossRef]
- Sires, U.I.; Schmid, T.M.; Fliszar, C.J.; Wang, Z.-Q.; Gluck, S.L.; Welgus, H.G. Complete degradation of type X collagen requires the combined action of interstitial collagenase and osteoclast-derived cathepsin-B. J. Clin. Investig. 1995, 95, 2089–2095. [Google Scholar] [CrossRef]
- Baumgrass, R.; Williamson, M.K.; Price, P.A. Identification of peptide fragments generated by digestion of bovine and human osteocalcin with the lysosomal proteinases cathepsin B, D, L, H, and S. J. Bone Mineral. Res. 1997, 12, 447–455. [Google Scholar] [CrossRef]
- Page, A.; Hayman, A.; Andersson, L.; Chambers, T.; Warburton, M. Degradation of bone matrix proteins by osteoclast cathepsins. Int. J. Biochem. 1993, 25, 545–550. [Google Scholar] [CrossRef]
- Öörni, K.; Sneck, M.; Brömme, D.; Pentikäinen, M.O.; Lindstedt, K.A.; Mäyränpää, M.; Aitio, H.; Kovanen, P.T. Cysteine protease cathepsin F is expressed in human atherosclerotic lesions, is secreted by cultured macrophages, and modifies low density lipoprotein particles in vitro. J. Biol. Chem. 2004, 279, 34776–34784. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.-S.; Li, Z.; BüTTNER, F.H.; Bartnik, E.; Brömme, D. Cleavage site specificity of cathepsin K toward cartilage proteoglycans and protease complex formation. Biol. Chem. 2003, 384, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Brömme, D.; Okamoto, K.; Wang, B.B.; Biroc, S. Human cathepsin O2, a matrix protein-degrading cysteine protease expressed in osteoclasts functional expression of human cathepsin O2 in Spodoptera frugiperda and characterization of the enzyme. J. Biol. Chem. 1996, 271, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Bossard, M.J.; Tomaszek, T.A.; Thompson, S.K.; Amegadzie, B.Y.; Hanning, C.R.; Jones, C.; Kurdyla, J.T.; McNulty, D.E.; Drake, F.H.; Gowen, M. Proteolytic activity of human osteoclast cathepsin K expression, purification, activation, and substrate identification. J. Biol. Chem. 1996, 271, 12517–12524. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, A.Y.; Kanaori, K.; Teno, N.; Togame, H.; Inaoka, T.; Takai, M.; Kokubo, T. Conformational studies on the specific cleavage site of type I collagen (α-1) fragment (157–192) by cathepsins K and L by proton NMR spectroscopy. Bioorganic Med. Chem. 1999, 7, 375–379. [Google Scholar] [CrossRef]
- Ishidoh, K.; Kominami, E. Procathepsin L degrades extracellular matrix proteins in the presence of glycosaminoglycans in vitro. Biochem. Biophys. Res. Commun. 1995, 217, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Li, Z.; Greenbaum, D.; Bogyo, M.; Weber, E.; Brömme, D. Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J. Biol. Chem. 2004, 279, 36761–36770. [Google Scholar] [CrossRef] [PubMed]
- Staudt, N.D.; Aicher, W.K.; Kalbacher, H.; Stevanovic, S.; Carmona, A.K.; Bogyo, M.; Klein, G. Cathepsin X is secreted by human osteoblasts, digests CXCL-12 and impairs adhesion of hematopoietic stem and progenitor cells to osteoblasts. haematologica 2010, 95, 1452–1460. [Google Scholar] [CrossRef]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease inhibitors: Current status and future prospects. J. Med. Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, D.P. Cysteine Peptidases of Mammals: Their Biological Roles and Potential Effects in the Oral Cavity and Other Tissues in Health and Disease. Crit. Rev. Oral. Biol. Med. 2002, 13, 238–275. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed]
- Pulukuri, S.; Gorantla, B.; Knost, J.A.; Rao, J.S. Frequent loss of cystatin E/M expression implicated in the progression of prostate cancer. Oncogene 2009, 28, 2829. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, R.; Bailey, R.D.; Kusmartseva, I.; Mane, S.; Epling-Burnette, P.; Loughran, T.P. Constitutive expression of cytotoxic proteases and down-regulation of protease inhibitors in LGL leukemia. Int. J. Oncol. 2003, 22, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Jiborn, T.; Abrahamson, M.; Helczynski, L.; Otterbein, L.; Persson, J.L.; Bjartell, A. Cystatin C is downregulated in prostate cancer and modulates invasion of prostate cancer cells via MAPK/Erk and androgen receptor pathways. PLoS ONE 2009, 4, e7953. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Mitrović, A.; Lah, T.T.; Kos, J. Cystatins in cancer progression: More than just cathepsin inhibitors. Biochimie 2019. [Google Scholar] [CrossRef] [PubMed]
- Duivenvoorden, H.M.; Rautela, J.; Edgington-Mitchell, L.E.; Spurling, A.; Greening, D.W.; Nowell, C.J.; Molloy, T.J.; Robbins, E.; Brockwell, N.K.; Lee, C.S.; et al. Myoepithelial cell-specific expression of stefin A as a suppressor of early breast cancer invasion. J. Pathol. 2017, 243, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Kokalj-Kunovar, M.; Štrukelj, B.; Pungerčar, J.; Barlič-Maganja, D.; Drobnič-Košorok, M.; Kastelic, L.; Babnik, J.; Golouh, R.; Turk, V. Stefins and lysosomal cathepsins B, L and D in human breast carcinoma. Int. J. Cancer 1992, 50, 36–44. [Google Scholar] [CrossRef]
- Strojnik, T.; Zajc, I.; Bervar, A.; Zidanik, B.; Golouh, R.; Kos, J.; Dolenc, V.; Lah, T. Cathepsin B and its inhibitor stefin A in brain tumors. Pflug. Arch. 2000, 439, R122–R123. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ding, F.; Zhang, L.; Liu, Z.; Wu, Y.; Luo, A.; Wu, M.; Wang, M.; Zhan, Q. Overexpression of stefin A in human esophageal squamous cell carcinoma cells inhibits tumor cell growth, angiogenesis, invasion, and metastasis. Clin. Cancer Res. 2005, 11, 8753–8762. [Google Scholar] [CrossRef]
- Strojan, P.; Budihna, M.; Smid, L.; Svetic, B.; Vrhovec, I.; Kos, J.; Skrk, J. Prognostic significance of cysteine proteinases cathepsins B and L and their endogenous inhibitors stefins A and B in patients with squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2000, 6, 1052–1062. [Google Scholar]
- Ma, Y.; Chen, Y.; Li, Y.; Grun, K.; Berndt, A.; Zhou, Z.; Petersen, I. Cystatin A suppresses tumor cell growth through inhibiting epithelial to mesenchymal transition in human lung cancer. Oncotarget 2018, 9, 14084–14098. [Google Scholar] [CrossRef] [PubMed]
- Mirtti, T.; Alanen, K.; Kallajoki, M.; Rinne, A.; Söderström, K.O. Expression of cystatins, high molecular weight cytokeratin, and proliferation markers in prostatic adenocarcinoma and hyperplasia. Prostate 2003, 54, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Kuopio, T.; Kankaanranta, A.; Jalava, P.; Kronqvist, P.; Kotkansalo, T.; Weber, E.; Collan, Y. Cysteine proteinase inhibitor cystatin A in breast cancer. Cancer Res. 1998, 58, 432–436. [Google Scholar] [PubMed]
- Lin, Y.Y.; Chen, Z.W.; Lin, Z.P.; Lin, L.B.; Yang, X.M.; Xu, L.Y.; Xie, Q. Tissue Levels of Stefin A and Stefin B in Hepatocellular Carcinoma. Anat. Rec. (Hoboken) 2016, 299, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Gole, B.; Huszthy, P.C.; Popović, M.; Jeruc, J.; Ardebili, Y.S.; Bjerkvig, R.; Lah, T.T. The regulation of cysteine cathepsins and cystatins in human gliomas. Int. J. Cancer 2012, 131, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Levičar, N.; Kos, J.; Blejec, A.; Golouh, R.; Vrhovec, I.; Frkovič-Grazio, S.; Lah, T.T. Comparison of potential biological markers cathepsin B, cathepsin L, stefin A and stefin B with urokinase and plasminogen activator inhibitor-1 and clinicopathological data of breast carcinoma patients. Cancer Detect. Prev. 2002, 26, 42–49. [Google Scholar] [CrossRef]
- Wang, X.; Gui, L.; Zhang, Y.; Zhang, J.; Shi, J.; Xu, G. Cystatin B is a progression marker of human epithelial ovarian tumors mediated by the TGF-β signaling pathway. Int. J. Oncol. 2014, 44, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.S.; Banyard, J.; Wu, C.L.; McDougal, W.S.; Zetter, B.R. Cystatin B as a tissue and urinary biomarker of bladder cancer recurrence and disease progression. Clin. Cancer Res. 2009, 15, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, K.; Iida, A.; Kasumi, F.; Sakamoto, G.; Akimoto, M.; Nakamura, Y.; Emi, M. Mapping of a new target region of allelic loss to a 6-cM interval at 21q21 in primary breast cancers. Genes Chromosomes Cancer 1998, 23, 244–247. [Google Scholar] [CrossRef]
- Widschwendter, M.; Jones, P.A. DNA methylation and breast carcinogenesis. Oncogene 2002, 21, 5462. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; Coleman, W.B. Epigenetic regulation of cystatins in cancer. Front. Biosci. 2009, 14, 453–462. [Google Scholar] [CrossRef]
- Keppler, D. Towards novel anti-cancer strategies based on cystatin function. Cancer Lett. 2006, 235, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, M.; Olafsson, I.; Palsdottir, A.; Ulvsbäck, M.; Lundwall, Å.; Jensson, O.; Grubb, A. Structure and expression of the human cystatin C gene. Biochem. J. 1990, 268, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Lignelid, H.; Collins, V.P.; Jacobsson, B. Cystatin C and transthyretin expression in normal and neoplastic tissues of the human brain and pituitary. Acta Neuropathol. 1997, 93, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Jacobsson, B.; Lignelid, H.; Bergerheim, U. Transthyretin and cystatin C are catabolized in proximal tubular epithelial cells and the proteins are not useful as markers for renal cell carcinomas. Histopathology 1995, 26, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Vigneswaran, N.; Wu, J.; Muller, S.; Zacharias, W.; Narendran, S.; Middleton, L. Expression analysis of cystatin C and M in laser-capture microdissectioned human breast cancer cells—A preliminary study. Pathol. Res. Pract. 2005, 200, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Yokoyama, M.; Asano, G.; Tanaka, S. Expression of cathepsin B and cystatin C in human colorectal cancer. Hum. Pathol. 1999, 30, 680–686. [Google Scholar] [CrossRef]
- Kos, J.; Lah, T.T. Cysteine proteinases and their endogenous inhibitors: Target proteins for prognosis, diagnosis and therapy in cancer. Oncol. Rep. 1998, 5, 1349–1410. [Google Scholar] [CrossRef]
- Zore, I.; Krasovec, M.; Cimerman, N.; Kuhelj, R.; Werle, B.; Nielsen, H.J.; Brünner, N.; Kos, J. Cathepsin B/cystatin C complex levels in sera from patients with lung and colorectal cancer. Biol. Chem. 2001, 382, 805–810. [Google Scholar] [CrossRef]
- Werle, B.; Schanzenbächer, U.; Lah, T.T.; Ebert, E.; Jülke, B.; Ebert, W.; Fiehn, W.; Kayser, K.; Spiess, E.; Abrahamson, M. Cystatins in non-small cell lung cancer: Tissue levels, localization and relation to prognosis. Oncol. Rep. 2006, 16, 647–655. [Google Scholar] [CrossRef]
- Strojan, P.; Oblak, I.; Svetic, B.; Šmid, L.; Kos, J. Cysteine proteinase inhibitor cystatin C in squamous cell carcinoma of the head and neck: Relation to prognosis. Br. J. Cancer 2004, 90, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Hara, M.; Shimuzu, K. Clinicopathologic significance of cystatin C expression in gliomas. Hum. Pathol. 2005, 36, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.; Håkansson, K.; Nathanson, C.-M.; Thorgeirsson, U.; Jonsson, N.; Grubb, A.; Abrahamson, M.; Karlsson, S. Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. Mol. Pathol. 1999, 52, 332. [Google Scholar] [CrossRef] [PubMed]
- Završnik, J.; Butinar, M.; Prebanda, M.T.; Krajnc, A.; Vidmar, R.; Fonović, M.; Grubb, A.; Turk, V.; Turk, B.; Vasiljeva, O. Cystatin C deficiency suppresses tumor growth in a breast cancer model through decreased proliferation of tumor cells. Oncotarget 2017, 8, 73793. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cao, X.; Dong, W.; Xia, M.; Luo, S.; Fan, Q.; Xie, J. Cystatin M expression is reduced in gastric carcinoma and is associated with promoter hypermethylation. Biochem. Biophys. Res. Commun. 2010, 391, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; Jones, W.D.; Coleman, W.B. DNA methylation-dependent silencing of CST6 in human breast cancer cell lines. Lab. Investig. 2006, 86, 1233. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; Jones, W.D.; Risher, J.D.; Coleman, W.B. DNA methylation-dependent epigenetic regulation of gene expression in MCF-7 breast cancer cells. Epigenetics 2006, 1, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; Livasy, C.A.; Boyd, C.E.; Keppler, D.; Coleman, W.B. Methylation-dependent Silencing of CST6 in Primary Human Breast Tumors and Metastatic Lesions. Exp. Mol. Pathol. 2007, 83, 188–197. [Google Scholar] [CrossRef][Green Version]
- Vigneswaran, N.; Wu, J.; Zacharias, W. Upregulation of cystatin M during the progression of oropharyngeal squamous cell carcinoma from primary tumor to metastasis. Oral Oncol. 2003, 39, 559–568. [Google Scholar] [CrossRef]
- Langerholc, T.; Zavasnik-Bergant, V.; Turk, B.; Turk, V.; Abrahamson, M.; Kos, J. Inhibitory properties of cystatin F and its localization in U937 promonocyte cells. Febs. J. 2005, 272, 1535–1545. [Google Scholar] [CrossRef]
- Morita, M.; Yoshiuchi, N.; Arakawa, H.; Nishimura, S. CMAP: A novel cystatin-like gene involved in liver metastasis. Cancer Res. 1999, 59, 151–158. [Google Scholar] [PubMed]
- Yoneda, K.; Iida, H.; Endo, H.; Hosono, K.; Akiyama, T.; Takahashi, H.; Inamori, M.; Abe, Y.; Yoneda, M.; Fujita, K.; et al. Identification of Cystatin SN as a novel tumor marker for colorectal cancer. Int. J. Oncol. 2009, 35, 33–40. [Google Scholar] [PubMed]
- Utsunomiya, T.; Hara, Y.; Kataoka, A.; Morita, M.; Arakawa, H.; Mori, M.; Nishimura, S. Cystatin-like metastasis-associated protein mRNA expression in human colorectal cancer is associated with both liver metastasis and patient survival. Clin. Cancer Res. 2002, 8, 2591–2594. [Google Scholar] [PubMed]
- Alvarez-Diaz, S.; Valle, N.; Garcia, J.M.; Pena, C.; Freije, J.M.; Quesada, V.; Astudillo, A.; Bonilla, F.; Lopez-Otin, C.; Munoz, A. Cystatin D is a candidate tumor suppressor gene induced by vitamin D in human colon cancer cells. J. Clin Invest. 2009, 119, 2343–2358. [Google Scholar] [CrossRef] [PubMed]
- Balbin, M.; Hall, A.; Grubb, A.; Mason, R.W.; Lopez-Otin, C.; Abrahamson, M. Structural and functional characterization of two allelic variants of human cystatin D sharing a characteristic inhibition spectrum against mammalian cysteine proteinases. J. Biol. Chem. 1994, 269, 23156–23162. [Google Scholar] [PubMed]
- Hunten, S.; Hermeking, H. p53 directly activates cystatin D/CST5 to mediate mesenchymal-epithelial transition: A possible link to tumor suppression by vitamin D3. Oncotarget 2015, 6, 15842–15856. [Google Scholar] [CrossRef]
- Magister, S.; Kos, J. Cystatins in immune system. J. Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef]
- Choi, E.H.; Kim, J.T.; Kim, J.H.; Kim, S.Y.; Song, E.Y.; Kim, J.W.; Yeom, Y.I.; Kim, I.H.; Lee, H.G. Upregulation of the cysteine protease inhibitor, cystatin SN, contributes to cell proliferation and cathepsin inhibition in gastric cancer. Clin. Chim. Acta 2009, 406, 45–51. [Google Scholar] [CrossRef]
- Dai, D.; Li, Y.; Chen, B.; Du, Y.; Li, S.; Lu, S.; Zhao, Z.; Zhou, A.; Xue, N.; Xia, T.; et al. Elevated expression of CST1 promotes breast cancer progression and predicts a poor prognosis. J. Mol. Med. (Berl) 2017, 95, 873–886. [Google Scholar] [CrossRef]
- Blanco, M.A.; LeRoy, G.; Khan, Z.; Aleckovic, M.; Zee, B.M.; Garcia, B.A.; Kang, Y. Global secretome analysis identifies novel mediators of bone metastasis. Cell Res. 2012, 22, 1339–1355. [Google Scholar] [CrossRef]
- Oh, S.S.; Park, S.; Lee, K.W.; Madhi, H.; Park, S.G.; Lee, H.G.; Cho, Y.Y.; Yoo, J.; Dong Kim, K. Extracellular cystatin SN and cathepsin B prevent cellular senescence by inhibiting abnormal glycogen accumulation. Cell Death Dis. 2017, 8, e2729. [Google Scholar] [CrossRef] [PubMed]
- Chwieralski, C.; Welte, T.; Bühling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Andreau, K.; Poncet, D.; Zamzami, N.; Perfettini, J.-L.; Metivier, D.; Ojcius, D.M.; Jäättelä, M.; Kroemer, G. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J. Exp. Med. 2003, 197, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.-C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Öllinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Droga-Mazovec, G.; Bojič, L.; Petelin, A.; Ivanova, S.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D. Noncaspase proteases in apoptosis. Leukemia 2000, 14, 1695. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Jäättelä, M. Triggering of apoptosis by cathepsins. Cell Death Differ. 2001, 8, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Antunes, F.; Eaton, J.W.; Brunk, U.T. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur. J. Biochem. 2003, 270, 3778–3786. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-α–mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137. [Google Scholar] [CrossRef]
- Cirman, T.; Orešić, K.; Mazovec, G.D.; Turk, V.; Reed, J.C.; Myers, R.M.; Salvesen, G.S.; Turk, B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 2004, 279, 3578–3587. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, B.; Schendel, S.L.; Kim, T.-H.; Cirman, T.; Snipas, S.J.; Ellerby, L.M.; Bredesen, D.; Freeze, H.; Abrahamson, M. Lysosomal protease pathways to apoptosis cleavage of Bid, not pro-caspases, is the most likely route. J. Biol. Chem. 2001, 276, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Billen, L.; Shamas-Din, A.; Andrews, D. Bid: A Bax-like BH3 protein. Oncogene 2009, 27, S93. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Zdolsek, J.; Zhang, H.; Roberg, K.; Brunk, U.; Sies, H. H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic. Res. Commun. 1993, 18, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Persson, H.L.; Eaton, J.W.; Brunk, U.T. Intralysosomal iron: A major determinant of oxidant-induced cell death. Free Radic. Biol. Med. 2003, 34, 1243–1252. [Google Scholar] [CrossRef]
- Appelqvist, H.; Waster, P.; Eriksson, I.; Rosdahl, I.; Ollinger, K. Lysosomal exocytosis and caspase-8-mediated apoptosis in UVA-irradiated keratinocytes. J. Cell Sci. 2013, 126, 5578–5584. [Google Scholar] [CrossRef] [PubMed]
- Groth-Pedersen, L.; Jäättelä, M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013, 332, 265–274. [Google Scholar] [CrossRef]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Pacheco, F.J.; Almaguel, F.; Perez, J.; Sahakian, E.; Daniels, T.R.; Leoh, L.S.; Padilla, A.; Wall, N.R.; Lilly, M.B. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol. Cancer 2009, 8, 68. [Google Scholar] [CrossRef]
- Bröker, L.E.; Huisman, C.; Span, S.W.; Rodriguez, J.A.; Kruyt, F.A.; Giaccone, G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004, 64, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Klionsky, D.J. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu. Rev. Biochem. 2000, 69, 303–342. [Google Scholar] [CrossRef] [PubMed]
- Dielschneider, R.F.; Henson, E.S.; Gibson, S.B. Lysosomes as oxidative targets for cancer therapy. Oxidative Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wen, X.; Cheng, Y. Survival or death: Disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013, 4, e892. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, W.; Lin, J.; Wei, P.; Zhang, Y.; Jin, P.; Chen, M.; Man, N.; Wen, L. Autophagic lysosomal reformation depends on mTOR reactivation in H2O2-induced autophagy. Int. J. Biochem. Cell Biol. 2016, 70, 76–81. [Google Scholar] [CrossRef]
- Hasegawa, J.; Maejima, I.; Iwamoto, R.; Yoshimori, T. Selective autophagy: Lysophagy. Methods 2015, 75, 128–132. [Google Scholar] [CrossRef]
- Lamparska-Przybysz, M.; Gajkowska, B.; Motyl, T. Cathepsins and BID are involved in the molecular switch between apoptosis and autophagy in breast cancer MCF-7 cells exposed to camptothecin. J. Physiol. Pharmacol. 2005, 56, 159. [Google Scholar]
- Cartledge, D.M.; Colella, R.; Glazewski, L.; Lu, G.; Mason, R.W. Inhibitors of cathepsins B and L induce autophagy and cell death in neuroblastoma cells. Investig. New Drugs 2013, 31, 20–29. [Google Scholar] [CrossRef]
- Colella, R.; Lu, G.; Glazewski, L.; Korant, B.; Matlapudi, A.; England, M.R.; Craft, C.; Frantz, C.N.; Mason, R.W. Induction of cell death in neuroblastoma by inhibition of cathepsins B and L. Cancer Lett. 2010, 294, 195–203. [Google Scholar] [CrossRef]
- Stahl, S.; Reinders, Y.; Asan, E.; Mothes, W.; Conzelmann, E.; Sickmann, A.; Felbor, U. Proteomic analysis of cathepsin B and L-deficient mouse brain lysosomes. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2007, 1774, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Soori, M.; Lu, G.; Mason, R.W. Cathepsin Inhibition Prevents Autophagic Protein Turnover and Downregulates Insulin Growth Factor-1 Receptor–Mediated Signaling in Neuroblastoma. J. Pharmacol. Exp. Ther. 2016, 356, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Racoma, I.O.; Meisen, W.H.; Wang, Q.-E.; Kaur, B.; Wani, A.A. Thymoquinone inhibits autophagy and induces cathepsin-mediated, caspase-independent cell death in glioblastoma cells. PLoS ONE 2013, 8, e72882. [Google Scholar] [CrossRef] [PubMed]
- Gos12, M.; Joanna Miłoszewska, M.P. Epithelial-mesenchymal transition in cancer progression. POSTĘPY Biochem. 2009, 55, 121–128. [Google Scholar]
- Han, M.-l.; Zhao, Y.-f.; Tan, C.-h.; Xiong, Y.-j.; Wang, W.-j.; Wu, F.; Fei, Y.; Wang, L.; Liang, Z.-q. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol. Sin. 2016, 37, 1606. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Xiong, Y.; Shen, X.; Zhao, Y.; Zhu, Y.; Wang, L.; Liang, Z. Cathepsin L promotes ionizing radiation-induced U251 glioma cell migration and invasion through regulating the GSK-3β/CUX1 pathway. Cell. Signal. 2018, 44, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Han, M.; Wang, W.; Song, Y.; Chen, G.; Wang, Z.; Liang, Z. Downregulation of cathepsin L suppresses cancer invasion and migration by inhibiting transforming growth factor-β-mediated epithelial-mesenchymal transition. Oncol. Rep. 2015, 33, 1851–1859. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.M.; Assfalg-Machleidt, I.; Zahler, S.; Stoeckelhuber, M.; Machleidt, W.; Jochum, M.; Nägler, D.K. RGD-dependent binding of procathepsin X to integrin αvβ3 mediates cell-adhesive properties. J. Biol. Chem. 2006, 281, 39588–39597. [Google Scholar] [CrossRef]
- Sevenich, L.; Schurigt, U.; Sachse, K.; Gajda, M.; Werner, F.; Müller, S.; Vasiljeva, O.; Schwinde, A.; Klemm, N.; Deussing, J. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 2497–2502. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Li, Y.; Guan, X.-Y. Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS ONE 2011, 6, e24967. [Google Scholar] [CrossRef] [PubMed]
- Cavallo-Medved, D.; Mai, J.; Dosescu, J.; Sameni, M.; Sloane, B.F. Caveolin-1 mediates the expression and localization of cathepsin B, pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J. Cell Sci. 2005, 118, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Affara, N.I.; Andreu, P.; Coussens, L.M. Delineating protease functions during cancer development; Proteases and Cancer, Humana Press: New York, NY, USA, 2009; pp. 1–32. [Google Scholar]
- Kirschke, H.; Eerola, R.; Hopsu-Havu, V.; Brömme, D.; Vuorio, E. Antisense RNA inhibition of cathepsin L expression reduces tumorigenicity of malignant cells. Eur. J. Cancer 2000, 36, 787–795. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Bailey, K.M.; Wojtkowiak, J.W.; Ben-Nun, Y.; Bogyo, M.; Weber, E.; Moin, K.; Blum, G.; Mattingly, R.R.; Gillies, R.J. Acid-mediated tumor proteolysis: Contribution of cysteine cathepsins. Neoplasia 2013, 15, 1125–IN1129. [Google Scholar] [CrossRef] [PubMed]
- Kasabova, M.; Joulin-Giet, A.; Lecaille, F.; Gilmore, B.F.; Marchand-Adam, S.; Saidi, A.; Lalmanach, G. Regulation of TGF-β1-driven differentiation of human lung fibroblasts: Emerging roles of cathepsin B and cystatin C. J. Biol. Chem. 2014, 289, 16239–16251. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef] [PubMed]
- Munson, P.; Shukla, A. Exosomes: Potential in cancer diagnosis and therapy. Medicines 2015, 2, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W.; Engelse, M.A.; Quax, P.H. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 716–728. [Google Scholar] [CrossRef]
- Rath, B.; Klameth, L.; Plangger, A.; Hochmair, M.; Ulsperger, E.; Huk, I.; Zeillinger, R.; Hamilton, G. Expression of Proteolytic Enzymes by Small Cell Lung Cancer Circulating Tumor Cell Lines. Cancers 2019, 11, 114. [Google Scholar] [CrossRef]
- Lopes-Bastos, B.M.; Jiang, W.G.; Cai, J. Tumour–Endothelial Cell Communications: Important and Indispensable Mediators of Tumour Angiogenesis. Anticancer Res. 2016, 36, 1119–1126. [Google Scholar]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Waguri, S.; Sato, N.; Watanabe, T.; Ishido, K.; Kominami, E. Cell and tissue distribution of lysosomal cysteine proteinases, cathepsins B, H, and L, and their biological roles. Acta Histochem. Et Cytochem. 1994, 27, 287–308. [Google Scholar] [CrossRef]
- Gocheva, V.; Chen, X.; Peters, C.; Reinheckel, T.; Joyce, J.A. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol. Chem. 2010, 391, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, Y.; Zhu, K. Inhibition of glioma cell lysosome exocytosis inhibits glioma invasion. PLoS ONE 2012, 7, e45910. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kondo, C.; Kojima, T.; Nagata, H.; Moriyama, A.; Hayakawa, T.; Katunuma, N. Significance of 32-kDa cathepsin L secreted from cancer cells. Cancer Biother. Radiopharm. 2006, 21, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Gondi, C.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 2004, 23, 4681. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef]
- Giusti, I.; D’Ascenzo, S.; Millimaggi, D.; Taraboletti, G.; Carta, G.; Franceschini, N.; Pavan, A.; Dolo, V. Cathepsin B mediates the pH-dependent proinvasive activity of tumor-shed microvesicles. Neoplasia 2008, 10, 481–488. [Google Scholar] [CrossRef]
- Cavallo-Medved, D.; Rudy, D.; Blum, G.; Bogyo, M.; Caglic, D.; Sloane, B.F. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp. Cell Res. 2009, 315, 1234–1246. [Google Scholar] [CrossRef]
- Caglič, D.; Pungerčar, J.R.; Pejler, G.; Turk, V.; Turk, B. Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 2007, 282, 33076–33085. [Google Scholar] [CrossRef]
- Wang, B.; Sun, J.; Kitamoto, S.; Yang, M.; Grubb, A.; Chapman, H.A.; Kalluri, R.; Shi, G.-P. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J. Biol. Chem. 2006, 281, 6020–6029. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Kuehn, D.; Burden, R.E.; Gormley, J.A.; Jaquin, T.J.; Gazdoiu, M.; Small, D.; Bicknell, R.; Johnston, J.A.; Scott, C.J. Antibody targeting of cathepsin S inhibits angiogenesis and synergistically enhances anti-VEGF. PLoS ONE 2010, 5, e12543. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Heeschen, C.; Aicher, A.; Sasaki, K.-i.; Bruhl, T.; Farhadi, M.R.; Vajkoczy, P.; Hofmann, W.K.; Peters, C.; Pennacchio, L.A. Cathepsin L is required for endothelial progenitor cell–induced neovascularization. Nat. Med. 2005, 11, 206. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, T.; Nishida, S.; Nakano, T.; Funasaka, K.; Kimoto, S.; Murata, K.; Tomura, T. Study on cathepsin B activity in human thyroid tumors. Auris Nasus Larynx 1995, 22, 43–48. [Google Scholar] [CrossRef]
- Novinec, M.; Grass, R.N.; Stark, W.J.; Turk, V.; Baici, A.; Lenarčič, B. Interaction between Human Cathepsins K, L, and S and Elastins mechanism of elastinolysis and inhibition by macromolecular inhibitors. J. Biol. Chem. 2007, 282, 7893–7902. [Google Scholar] [CrossRef]
- Zhou, G.; Yu, W.; Li, X. The experimental study of specific inhibitor-CA-074Me of Cathepsin B suppressing retinal neovascularization. [Zhonghua Yan Ke Za Zhi] Chin. J. Ophthalmol. 2008, 44, 207–211. [Google Scholar]
- Jiang, H.; Cheng, X.W.; Shi, G.-P.; Hu, L.; Inoue, A.; Yamamura, Y.; Wu, H.; Takeshita, K.; Li, X.; Huang, Z. Cathepsin K-mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat. Commun. 2014, 5, 3838. [Google Scholar] [CrossRef]
- Krueger, S.; Kalinski, T.; Hundertmark, T.; Wex, T.; Küster, D.; Peitz, U.; Ebert, M.; Nägler, D.K.; Kellner, U.; Malfertheiner, P. Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2005, 207, 32–42. [Google Scholar]
- Nägler, D.K.; Krüger, S.; Kellner, A.; Ziomek, E.; Menard, R.; Buhtz, P.; Krams, M.; Roessner, A.; Kellner, U. Up-regulation of cathepsin X in prostate cancer and prostatic intraepithelial neoplasia. Prostate 2004, 60, 109–119. [Google Scholar] [CrossRef]
- Rumpler, G.; Becker, B.; Hafner, C.; McClelland, M.; Stolz, W.; Landthaler, M.; Schmitt, R.; Bosserhoff, A.; Vogt, T. Identification of differentially expressed genes in models of melanoma progression by cDNA array analysis: SPARC, MIF and a novel cathepsin protease characterize aggressive phenotypes. Exp. Dermatol. 2003, 12, 761–771. [Google Scholar] [CrossRef]
- Jechorek, D.; Votapek, J.; Meyer, F.; Kandulski, A.; Roessner, A.; Franke, S. Characterization of cathepsin X in colorectal cancer development and progression. Pathol. Res. Pract. 2014, 210, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Premzl, A.; Bergant, T.Z.; Turk, B.; Kos, J. Carboxypeptidase cathepsin X mediates β2-integrin-dependent adhesion of differentiated U-937 cells. Exp. Cell Res. 2006, 312, 2515–2527. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, T.; Thorburn, A. Autophagy and Cell Death. Essays Biochem. 2013, 55, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Conus, S.; Simon, H.U. Cathepsins: Key modulators of cell death and inflammatory responses. Biochem. Pharm. 2008, 76, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Nat. 2015, 7, 17–28. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Mendelson, D.S.; Kato, G. Tumor angiogenesis and novel antiangiogenic strategies. Int. J. Cancer 2010, 126, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143. [Google Scholar] [CrossRef]
- Guo, B.; Tam, A.; Santi, S.A.; Parissenti, A.M. Role of autophagy and lysosomal drug sequestration in acquired resistance to doxorubicin in MCF-7 cells. BMC Cancer 2016, 16, 762. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Grabski, S.; Hoff, E.; Simon, S.M. Defective pH regulation of acidic compartments in human breast cancer cells (MCF-7) is normalized in adriamycin-resistant cells (MCF-7adr). Biochemistry 1996, 35, 2811–2817. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.J.; Sykes, H.R.; Fox, M.E.; Furlong, I.J. Subcellular distribution of the anticancer drug mitoxantrone in human and drug-resistant murine cells analyzed by flow cytometry and confocal microscopy and its relationship to the induction of DNA damage. Cancer Res. 1992, 52, 4000–4008. [Google Scholar] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; De Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Duvvuri, M.; Duncan, M.B.; Liu, J.; Krise, J.P. Niemann-Pick C1 protein facilitates the efflux of the anticancer drug daunorubicin from cells according to a novel vesicle-mediated pathway. J. Pharmacol. Exp. Ther. 2006, 316, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Regev, R.; Yeheskely-Hayon, D.; Katzir, H.; Eytan, G.D. Transport of anthracyclines and mitoxantrone across membranes by a flip-flop mechanism. Biochem. Pharmacol. 2005, 70, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321. [Google Scholar] [CrossRef]
- Asgharzadeh, M.R.; Barar, J.; Pourseif, M.M.; Eskandani, M.; Niya, M.J.; Mashayekhi, M.R.; Omidi, Y. Molecular machineries of pH dysregulation in tumor microenvironment: Potential targets for cancer therapy. BioImpacts BI 2017, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Hämälistö, S.; Jäättelä, M. Lysosomes in cancer—Living on the edge (of the cell). Curr. Opin. Cell Biol. 2016, 39, 69–76. [Google Scholar] [CrossRef]
- Zheng, X.; Chu, F.; Chou, P.M.; Gallati, C.; Dier, E.; Mirkin, B.L.; Mousa, S.A.; Rebbaa, A. Cathepsin L inhibition suppresses drug resistance in vitro and in vivo: A putative mechanism. Am. J. Physiol. Cell Physiol. 2009, 296, C65–C74. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Transformation-dependent secretion of a low molecular weight protein by murine fibroblasts. Proc. Natl. Acad. Sci. USA 1978, 75, 2767–2771. [Google Scholar] [CrossRef] [PubMed]
- Burden, R.E.; Gormley, J.A.; Jaquin, T.J.; Small, D.M.; Quinn, D.J.; Hegarty, S.M.; Ward, C.; Walker, B.; Johnston, J.A.; Olwill, S.A.; et al. Antibody-mediated inhibition of cathepsin S blocks colorectal tumor invasion and angiogenesis. Clin. Cancer Res. 2009, 15, 6042–6051. [Google Scholar] [CrossRef] [PubMed]
- Burden, R.E.; Gormley, J.A.; Kuehn, D.; Ward, C.; Kwok, H.F.; Gazdoiu, M.; McClurg, A.; Jaquin, T.J.; Johnston, J.A.; Scott, C.J.; et al. Inhibition of Cathepsin S by Fsn0503 enhances the efficacy of chemotherapy in colorectal carcinomas. Biochimie 2012, 94, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhou, K.; Wang, L.; Wang, F.; Chen, X.; Fan, Q. Clinical significance of serum cathepsin B and cystatin C levels and their ratio in the prognosis of patients with esophageal cancer. Onco Targets Ther. 2017, 10, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.D.; Williams, R.; Scott, C.J.; Burden, R.E. Cathepsin S: Therapeutic, diagnostic, and prognostic potential. Biol. Chem. 2015, 396, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nun, Y.; Fichman, G.; Adler-Abramovich, L.; Turk, B.; Gazit, E.; Blum, G. Cathepsin nanofiber substrates as potential agents for targeted drug delivery. J. Control. Release 2017, 257, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.H.; Kang, S.W.; Seo, J.W.; Hyun, S.W.; Park, J.H.; et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J. Control. Release 2019, 294, 376–389. [Google Scholar] [CrossRef]
- Choi, K.Y.; Swierczewska, M.; Lee, S.; Chen, X. Protease-activated drug development. Theranostics 2012, 2, 156. [Google Scholar] [CrossRef]
- Gong, F.; Peng, X.; Luo, C.; Shen, G.; Zhao, C.; Zou, L.; Li, L.; Sang, Y.; Zhao, Y.; Zhao, X. Cathepsin B as a potential prognostic and therapeutic marker for human lung squamous cell carcinoma. Mol. Cancer 2013, 12, 125. [Google Scholar] [CrossRef]
- Chan, A.T.; Baba, Y.; Shima, K.; Nosho, K.; Chung, D.C.; Hung, K.E.; Mahmood, U.; Madden, K.; Poss, K.; Ranieri, A.; et al. Cathepsin B expression and survival in colon cancer: Implications for molecular detection of neoplasia. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2777–2785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, S.; Wang, Q.; Yang, Z.; Pan, Z.; Li, L. Overexpression of cysteine cathepsin L is a marker of invasion and metastasis in ovarian cancer. Oncol. Rep. 2014, 31, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Withana, N.P.; Blum, G.; Sameni, M.; Slaney, C.; Anbalagan, A.; Olive, M.B.; Bidwell, B.N.; Edgington, L.; Wang, L.; Moin, K.; et al. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res. 2012, 72, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Pranjol, M.Z.I.; Gutowski, N.; Hannemann, M.; Whatmore, J. The Potential Role of the Proteases Cathepsin D and Cathepsin L in the Progression and Metastasis of Epithelial Ovarian Cancer. Biomolecules 2015, 5, 3260–3279. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cysteine Aathepsins (Cts) | Extracellular Localization | ECM Proteins Degraded by Cts | References |
|---|---|---|---|
| CtsB | + | aggrecan, proteoglycan, collagen I, II, IV, IV, IX, X, XI, laminin fibronectin, osteocalcin, osteonectin | [37,38,39,40,41,42,43] |
| CtsC | N/A | N/A | - |
| CtsF | + | proteoglycan | [44] |
| CtsH | + | osteocalcin | [42] |
| CtsK | + | aggrecan, elastin, osteonectin, collagen I, II | [45,46,47] |
| CtsL | + | proteoglycan, aggrecan, collagen I, II, IX, XI, fibronectin, laminin, osteocalcin | [38,39,42,45,48,49] |
| CtsO | N/A | N/A | - |
| CtsS | + | aggrecan, proteoglycan, collagen, elastin, fibronectin, osteocalcin | [3,38,45] |
| CtsV | + | elastin | [50] |
| CtsW | N/A | N/A | - |
| CtsX | + | N/A | [51] |
| Stefin | Cancer Type | Function | References |
|---|---|---|---|
| Stefin A | Breast | The low expression level is associated with cancer development and aggressiveness | [59,60] |
| Brain | [61] | ||
| Esophageal squamous | [62,63] | ||
| Lung | [64] | ||
| Prostate | [65] | ||
| Stefin A | Breast | The low expression correlates with a better outcome of patients | [66] |
| Liver | [67] | ||
| Brain | [68] | ||
| Sfefin B | Colorectal | The low expression level is associated with cancer development and aggressiveness | [36] |
| Breast | [69] | ||
| Head and neck | [63] | ||
| Stefin B | Liver | The low expression correlates with a better outcome of patients | [67] |
| Ovarian | [70] | ||
| Brain | [68] | ||
| Bladder cancer | [71] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20, 3602. https://doi.org/10.3390/ijms20143602
Rudzińska M, Parodi A, Soond SM, Vinarov AZ, Korolev DO, Morozov AO, Daglioglu C, Tutar Y, Zamyatnin AA Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. International Journal of Molecular Sciences. 2019; 20(14):3602. https://doi.org/10.3390/ijms20143602
Chicago/Turabian StyleRudzińska, Magdalena, Alessandro Parodi, Surinder M. Soond, Andrey Z. Vinarov, Dmitry O. Korolev, Andrey O. Morozov, Cenk Daglioglu, Yusuf Tutar, and Andrey A. Zamyatnin, Jr. 2019. "The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance" International Journal of Molecular Sciences 20, no. 14: 3602. https://doi.org/10.3390/ijms20143602
APA StyleRudzińska, M., Parodi, A., Soond, S. M., Vinarov, A. Z., Korolev, D. O., Morozov, A. O., Daglioglu, C., Tutar, Y., & Zamyatnin, A. A., Jr. (2019). The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. International Journal of Molecular Sciences, 20(14), 3602. https://doi.org/10.3390/ijms20143602