Ureaplasma Species Modulate Cytokine and Chemokine Responses in Human Brain Microvascular Endothelial Cells

 , and
, and
Abstract
1. Introduction
2. Results
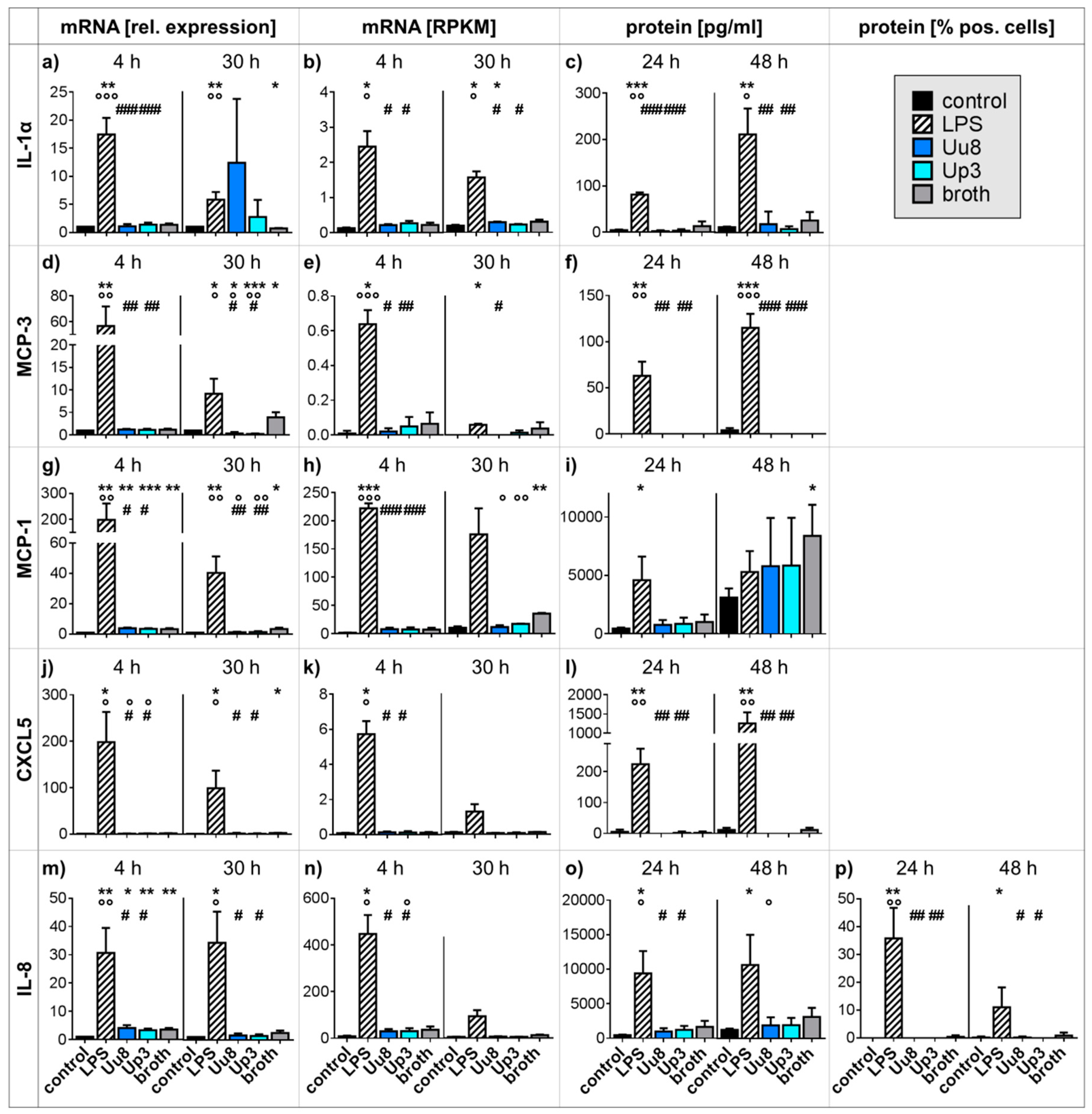
2.1. Pro-inflammatory Cytokine and Chemokine Responses in HBMEC upon Stimulation with Ureaplasma Isolates or LPS
2.2. Pro-inflammatory Cytokine and Chemokine Responses in Co-stimulated HBMEC
2.3. Anti-inflammatory Cytokine Responses in Mono- and Co-stimulated HBMEC
2.4. CXCR4 Responses in Mono- and Co-stimulated HBMEC
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
4.2. Cell Line and Culture Conditions
4.3. Stimulation Assays
4.4. RNA Extraction and Reverse Transcriptase PCR (RT-PCR)
4.5. Quantitative Real-Time RT-PCR (qRT-PCR)
4.6. RNA Sequencing
4.7. Multi-Analyte Immunoassay
4.8. Flow Cytometry
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATCC | American Tissue Culture Collection |
| BBB | blood–brain barrier |
| CCL | CC chemokine ligand |
| CCU | color-changing units |
| CD | cluster of differentiation |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| CXCL | C-X-C chemokine ligand |
| CXCR | C-X-C chemokine receptor |
| E. | Escherichia |
| HBMEC | human brain microvascular endothelial cells |
| HPRT | hypoxanthine phosphoribosyltransferase |
| IL | interleukin |
| IVH | intraventricular hemorrhage |
| LPS | lipopolysaccharide |
| MCP | monocyte chemoattractant protein |
| MMP | matrix metalloproteinase |
| qRT-PCR | quantitative real-time reverse transcriptase polymerase chain reaction |
| RA | receptor antagonist |
| RPKM | reads per kilo base per million mapped reads |
| SD | standard deviation |
| spp. | species |
| TJ | tight junction |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| U. | Ureaplasma |
| Up3 | Ureaplasma parvum serovar 3 |
| Uu8 | Ureaplasma urealyticum serovar 8 |
References
- Waites, K.B.; Katz, B.; Schelonka, R.L. Mycoplasmas and ureaplasmas as neonatal pathogens. Clin. Microbiol. Rev. 2005, 18, 757–789. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, R.L.; Andrews, W.W.; Goepfert, A.R.; Faye-Petersen, O.; Cliver, S.P.; Carlo, W.A.; Hauth, J.C. The Alabama Preterm Birth Study: Umbilical cord blood Ureaplasma urealyticum and Mycoplasma hominis cultures in very preterm newborn infants. Am. J. Obstet. Gynecol. 2008, 198, 43.e1–43.e5. [Google Scholar] [CrossRef] [PubMed]
- Rittenschober-Bohm, J.; Waldhoer, T.; Schulz, S.M.; Stihsen, B.; Pimpel, B.; Goeral, K.; Hafner, E.; Sliutz, G.; Kasper, D.C.; Witt, A.; et al. First Trimester Vaginal Ureaplasma Biovar Colonization and Preterm Birth: Results of a Prospective Multicenter Study. Neonatology 2018, 113, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, E.L.; Dando, S.J.; Kallapur, S.G.; Knox, C.L. The Human Ureaplasma Species as Causative Agents of Chorioamnionitis. Clin. Microbiol. Rev. 2017, 30, 349–379. [Google Scholar] [CrossRef] [PubMed]
- Silwedel, C.; Speer, C.P.; Glaser, K. Ureaplasma-associated prenatal, perinatal, and neonatal morbidities. Expert Rev. Clin. Immunol. 2017, 13, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.; Ratliff, A.; Crabb, D.; Waites, K.B.; Bharat, A. Ureaplasma Transmitted from Donor Lungs is Pathogenic after Lung Transplantation. Ann. Thorac. Surg. 2017, 103, 670–671. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Cunningham, S.A.; Scott Budinger, G.R.; Kreisel, D.; DeWet, C.J.; Gelman, A.E.; Waites, K.; Crabb, D.; Xiao, L.; Bhorade, S.; et al. Disseminated Ureaplasma infection as a cause of fatal hyperammonemia in humans. Sci. Transl. Med. 2015, 7, 284re3. [Google Scholar] [CrossRef]
- George, M.D.; Cardenas, A.M.; Birnbaum, B.K.; Gluckman, S.J. Ureaplasma septic arthritis in an immunosuppressed patient with juvenile idiopathic arthritis. J. Clin. Rheumatol. 2015, 21, 221–224. [Google Scholar] [CrossRef]
- Panero, A.; Pacifico, L.; Rossi, N.; Roggini, M.; Chiesa, C. Ureaplasma urealyticum as a cause of pneumonia in preterm infants: Analysis of the white cell response. Arch. Dis. Child. Fetal Neonatal Ed. 1995, 73, F37–F40. [Google Scholar] [CrossRef]
- Viscardi, R.M. Ureaplasma species: Role in neonatal morbidities and outcomes. Arch. Dis. Child. Fetal Neonatal. Ed. 2014, 99, F87–F92. [Google Scholar] [CrossRef]
- Glaser, K.; Speer, C.P. Neonatal CNS infection and inflammation caused by Ureaplasma species: Rare or relevant? Expert Rev. Anti-Infect. Ther. 2015, 13, 233–248. [Google Scholar] [CrossRef]
- Glaser, K.; Wohlleben, M.; Speer, C.P. An 8-month history of meningitis in an extremely low birth weight infant?—Long-lasting Infection with Ureaplasma parvum. Z. Geburtshilfe Neonatol. 2015, 219, 52–56. [Google Scholar] [CrossRef]
- Barichello, T.; Fagundes, G.D.; Generoso, J.S.; Elias, S.G.; Simoes, L.R.; Teixeira, A.L. Pathophysiology of neonatal acute bacterial meningitis. J. Med. Microbiol. 2013, 62, 1781–1789. [Google Scholar] [CrossRef]
- Kim, K.S. Pathogenesis of bacterial meningitis: From bacteraemia to neuronal injury. Nat. Rev. Neurosci. 2003, 4, 376–385. [Google Scholar] [CrossRef]
- Dammann, O.; Leviton, A. Intermittent or sustained systemic inflammation and the preterm brain. Pediatr. Res. 2014, 75, 376–380. [Google Scholar] [CrossRef]
- Moscuzza, F.; Belcari, F.; Nardini, V.; Bartoli, A.; Domenici, C.; Cuttano, A.; Ghirri, P.; Boldrini, A. Correlation between placental histopathology and fetal/neonatal outcome: Chorioamnionitis and funisitis are associated to intraventricular haemorrage and retinopathy of prematurity in preterm newborns. Gynecol. Endocrinol. 2011, 27, 319–323. [Google Scholar] [CrossRef]
- Gantert, M.; Been, J.V.; Gavilanes, A.W.; Garnier, Y.; Zimmermann, L.J.; Kramer, B.W. Chorioamnionitis: A multiorgan disease of the fetus? J. Perinatol. 2010, 30 (Suppl. 1), S21–S30. [Google Scholar] [CrossRef]
- Back, S.A.; Volpe, J.J. Cellular and molecular pathogenesis of periventricular white matter injury. Ment. Retard. Dev. Disabil. Res. Rev. 1997, 3, 96–107. [Google Scholar] [CrossRef]
- Kasper, D.C.; Mechtler, T.P.; Bohm, J.; Petricevic, L.; Gleiss, A.; Spergser, J.; Witt, A.; Herkner, K.R.; Berger, A. In utero exposure to Ureaplasma spp. is associated with increased rate of bronchopulmonary dysplasia and intraventricular hemorrhage in preterm infants. J. Perinat. Med. 2011, 39, 331–336. [Google Scholar] [CrossRef]
- Viscardi, R.M.; Hashmi, N.; Gross, G.W.; Sun, C.C.; Rodriguez, A.; Fairchild, K.D. Incidence of invasive ureaplasma in VLBW infants: Relationship to severe intraventricular hemorrhage. J. Perinatol. 2008, 28, 759–765. [Google Scholar] [CrossRef]
- Le Thuc, O.; Blondeau, N.; Nahon, J.L.; Rovere, C. The complex contribution of chemokines to neuroinflammation: Switching from beneficial to detrimental effects. Ann. N. Y. Acad. Sci. 2015, 1351, 127–140. [Google Scholar] [CrossRef]
- Atkinson, J.J.; Senior, R.M. Matrix metalloproteinase-9 in lung remodeling. Am. J. Respir. Cell Mol. Biol. 2003, 28, 12–24. [Google Scholar] [CrossRef]
- Risau, W.; Wolburg, H. Development of the blood-brain barrier. Trends Neurosci. 1990, 13, 174–178. [Google Scholar] [CrossRef]
- Williams, J.L.; Holman, D.W.; Klein, R.S. Chemokines in the balance: Maintenance of homeostasis and protection at CNS barriers. Front. Cell. Neurosci. 2014, 8, 154. [Google Scholar] [CrossRef]
- Silwedel, C.; Haarmann, A.; Fehrholz, M.; Claus, H.; Speer, C.P.; Glaser, K. More than just inflammation: Ureaplasma species induce apoptosis in human brain microvascular endothelial cells. J. Neuroinflamm. 2019, 16, 38. [Google Scholar] [CrossRef]
- Silwedel, C.; Speer, C.P.; Haarmann, A.; Fehrholz, M.; Claus, H.; Buttmann, M.; Glaser, K. Novel insights into neuroinflammation: Bacterial lipopolysaccharide, tumor necrosis factor α, and Ureaplasma species differentially modulate atypical chemokine receptor 3 responses in human brain microvascular endothelial cells. J. Neuroinflamm. 2018, 15, 156. [Google Scholar] [CrossRef]
- Chau, A.; Markley, J.C.; Juang, J.; Tsen, L.C. Cytokines in the perinatal period—Part I. Int. J. Obstet. Anesth. 2016, 26, 39–47. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.D. Function and regulation of IL-1alpha in inflammatory diseases and cancer. Immunol. Rev. 2018, 281, 124–137. [Google Scholar] [CrossRef]
- Bajetto, A.; Bonavia, R.; Barbero, S.; Florio, T.; Schettini, G. Chemokines and their receptors in the central nervous system. Front. Neuroendocrinol. 2001, 22, 147–184. [Google Scholar] [CrossRef]
- Kallapur, S.G.; Kramer, B.W.; Knox, C.L.; Berry, C.A.; Collins, J.J.; Kemp, M.W.; Nitsos, I.; Polglase, G.R.; Robinson, J.; Hillman, N.H.; et al. Chronic fetal exposure to Ureaplasma parvum suppresses innate immune responses in sheep. J. Immunol. 2011, 187, 2688–2695. [Google Scholar] [CrossRef]
- Glaser, K.; Gradzka-Luczewska, A.; Szymankiewicz-Breborowicz, M.; Kawczynska-Leda, N.; Henrich, B.; Waaga-Gasser, A.M.; Speer, C.P. Perinatal Ureaplasma exposure is associated with increased risk of late onset sepsis and imbalanced inflammation in preterm infants and may add to lung injury. Front. Cell. Infect. Microbiol. 2019. [Google Scholar] [CrossRef]
- Meng, S.Z.; Oka, A.; Takashima, S. Developmental expression of monocyte chemoattractant protein-1 in the human cerebellum and brainstem. Brain Dev. 1999, 21, 30–35. [Google Scholar] [CrossRef]
- Stowe, A.M.; Wacker, B.K.; Cravens, P.D.; Perfater, J.L.; Li, M.K.; Hu, R.; Freie, A.B.; Stuve, O.; Gidday, J.M. CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J. Neuroinflamm. 2012, 9, 33. [Google Scholar] [CrossRef]
- Loetscher, M.; Geiser, T.; O’Reilly, T.; Zwahlen, R.; Baggiolini, M.; Moser, B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J. Biol. Chem. 1994, 269, 232–237. [Google Scholar]
- Liu, K.K.; Dorovini-Zis, K. Regulation of CXCL12 and CXCR4 expression by human brain endothelial cells and their role in CD4+ and CD8+ T cell adhesion and transendothelial migration. J. Neuroimmunol. 2009, 215, 49–64. [Google Scholar] [CrossRef]
- Moll, N.M.; Cossoy, M.B.; Fisher, E.; Staugaitis, S.M.; Tucky, B.H.; Rietsch, A.M.; Chang, A.; Fox, R.J.; Trapp, B.D.; Ransohoff, R.M. Imaging correlates of leukocyte accumulation and CXCR4/CXCL12 in multiple sclerosis. Arch. Neurol. 2009, 66, 44–53. [Google Scholar] [CrossRef]
- Gupta, S.K.; Pillarisetti, K.; Aiyar, N. CXCR4 undergoes complex lineage and inducing agent-dependent dissociation of expression and functional responsiveness to SDF-1alpha during myeloid differentiation. J. Leukoc. Biol. 2001, 70, 431–438. [Google Scholar]
- Huang, J.; Li, Y.; Tang, Y.; Tang, G.; Yang, G.Y.; Wang, Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke 2013, 44, 190–197. [Google Scholar] [CrossRef]
- Forster, C.; Silwedel, C.; Golenhofen, N.; Burek, M.; Kietz, S.; Mankertz, J.; Drenckhahn, D. Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in a murine in vitro system. J. Physiol. 2005, 565, 475–486. [Google Scholar] [CrossRef]
- Berger, A.; Witt, A.; Haiden, N.; Kaider, A.; Klebermasz, K.; Fuiko, R.; Langgartner, M.; Pollak, A. Intrauterine infection with Ureaplasma species is associated with adverse neuromotor outcome at 1 and 2 years adjusted age in preterm infants. J. Perinat. Med. 2009, 37, 72–78. [Google Scholar] [CrossRef]
- Frigerio, S.; Gelati, M.; Ciusani, E.; Corsini, E.; Dufour, A.; Massa, G.; Salmaggi, A. Immunocompetence of human microvascular brain endothelial cells: Cytokine regulation of IL-1beta, MCP-1, IL-10, sICAM-1 and sVCAM-1. J. Neurol. 1998, 245, 727–730. [Google Scholar] [CrossRef]
- Londoño, D.; Carvajal, J.; Strle, K.; Kim, K.S.; Cadavid, D. IL-10 Prevents Apoptosis of Brain Endothelium during Bacteremia. J. Immunol. 2011, 186, 7176–7186. [Google Scholar] [CrossRef]
- Peltier, M.R.; Freeman, A.J.; Mu, H.H.; Cole, B.C. Characterization of the macrophage-stimulating activity from Ureaplasma urealyticum. Am. J. Reprod. Immunol. 2007, 57, 186–192. [Google Scholar] [CrossRef]
- Triantafilou, M.; De Glanville, B.; Aboklaish, A.F.; Spiller, O.B.; Kotecha, S.; Triantafilou, K. Synergic activation of toll-like receptor (TLR) 2/6 and 9 in response to Ureaplasma parvum & urealyticum in human amniotic epithelial cells. PLoS ONE 2013, 8, e61199. [Google Scholar] [CrossRef]
- Dammann, O.; O’Shea, T.M. Cytokines and perinatal brain damage. Clin. Perinatol. 2008, 35, 643–663. [Google Scholar] [CrossRef]
- Glaser, K.; Silwedel, C.; Fehrholz, M.; Henrich, B.; Waaga-Gasser, A.M.; Claus, H.; Speer, C.P. Ureaplasma isolates stimulate pro-inflammatory CC chemokines and matrix metalloproteinase-9 in neonatal and adult monocytes. PLoS ONE 2018, 13, e0194514. [Google Scholar] [CrossRef]
- Glaser, K.; Silwedel, C.; Fehrholz, M.; Waaga-Gasser, A.M.; Henrich, B.; Claus, H.; Speer, C.P. Ureaplasma Species Differentially Modulate Pro- and Anti-Inflammatory Cytokine Responses in Newborn and Adult Human Monocytes Pushing the State Toward Pro-Inflammation. Front. Cell. Infect. Microbiol. 2017, 7, 480. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 31 August 2017).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| LPS Effect | LPS + Ureaplasma spp. | Potential In Vivo Relevance | |
|---|---|---|---|
| CXCL5 | mRNA and protein ↑ | antagonistic (mRNA) | reduced immune responses |
| CXCR4 | mRNA ↑ | additive (mRNA) | compromised BBB integrity |
| IL-1α | mRNA and protein ↑ | antagonistic (protein) | reduced immune responses |
| IL-8 | mRNA and protein ↑ | antagonistic (mRNA, protein) | reduced immune responses |
| MCP-1 | mRNA and protein ↑ | antagonistic (mRNA) | reduced immune responses |
| MCP-3 | mRNA and protein ↑ | antagonistic (mRNA) | reduced immune responses |
| Name | Gene Symbol | Sequence Accession No. | Orientation | Sequence [5′ to 3′] |
|---|---|---|---|---|
| CXCL5 | CXCL5 | NM_002994.5 | forward | GTTGCGTTTGTTTACAGACC |
| reverse | TTTCCTTGTTTCCACCGT | |||
| CXCR4 | CXCR4 | NM_001008540.2 | forward | AAGACCACAGTCATCCTC |
| reverse | GTTCTCAAACTCACACCCT | |||
| HPRT1 | HPRT1 | NM_000194.2 | forward | CTGGCGTCGTGATTAGTG |
| reverse | AGTCCTGTCCATAATTAGTCC | |||
| IL-1α | IL1α | NM_000575.4 | forward | ATGGATCAATCTGTGTCTCTG |
| reverse | GCTTGATGATTTCTTCCTCTG | |||
| IL-1β | IL1β | NM_000576.2 | forward | TTCATTGCTCAAGTGTCTG |
| reverse | GCACTTCATCTGTTTAGGG | |||
| IL-8 | IL8 | NM_000584.4 | forward | CAGTGCATAAAGACATACTCC |
| reverse | TTTATGAATTCTCAGCCCTC | |||
| IL-10 | IL10 | NM_000572.3 | forward | GCTGTCATCGATTTCTTCC |
| reverse | GTCAAACTCACTCATGGCT | |||
| MCP-1 | CCL2 | NM_002982.4 | forward | GCTGTGATCTTCAAGACC |
| reverse | AAGTCTTCGGAGTTTGGG | |||
| MCP-3 | CCL7 | NM_006273.3 | forward | CTGAGACCAAACCAGAAACC |
| reverse | TATTAATCCCAACTGGCTGAG | |||
| MMP-9 | MMP9 | NM_004994.3 | forward | GGATGGGAAGTACTGGCGA |
| reverse | CTCCTCAAAGACCGAGTCC | |||
| TNF-α | TNFα | NM_000594.4 | forward | CAGCCTCTTCTCCTTCCT |
| reverse | GGGTTTGCTACAACATGG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silwedel, C.; Speer, C.P.; Haarmann, A.; Fehrholz, M.; Claus, H.; Schlegel, N.; Glaser, K. Ureaplasma Species Modulate Cytokine and Chemokine Responses in Human Brain Microvascular Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 3583. https://doi.org/10.3390/ijms20143583
Silwedel C, Speer CP, Haarmann A, Fehrholz M, Claus H, Schlegel N, Glaser K. Ureaplasma Species Modulate Cytokine and Chemokine Responses in Human Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences. 2019; 20(14):3583. https://doi.org/10.3390/ijms20143583
Chicago/Turabian StyleSilwedel, Christine, Christian P. Speer, Axel Haarmann, Markus Fehrholz, Heike Claus, Nicolas Schlegel, and Kirsten Glaser. 2019. "Ureaplasma Species Modulate Cytokine and Chemokine Responses in Human Brain Microvascular Endothelial Cells" International Journal of Molecular Sciences 20, no. 14: 3583. https://doi.org/10.3390/ijms20143583
APA StyleSilwedel, C., Speer, C. P., Haarmann, A., Fehrholz, M., Claus, H., Schlegel, N., & Glaser, K. (2019). Ureaplasma Species Modulate Cytokine and Chemokine Responses in Human Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences, 20(14), 3583. https://doi.org/10.3390/ijms20143583