In Silico Analysis of Gene Expression Change Associated with Copy Number of Enhancers in Pancreatic Adenocarcinoma



Abstract
1. Introduction
2. Results
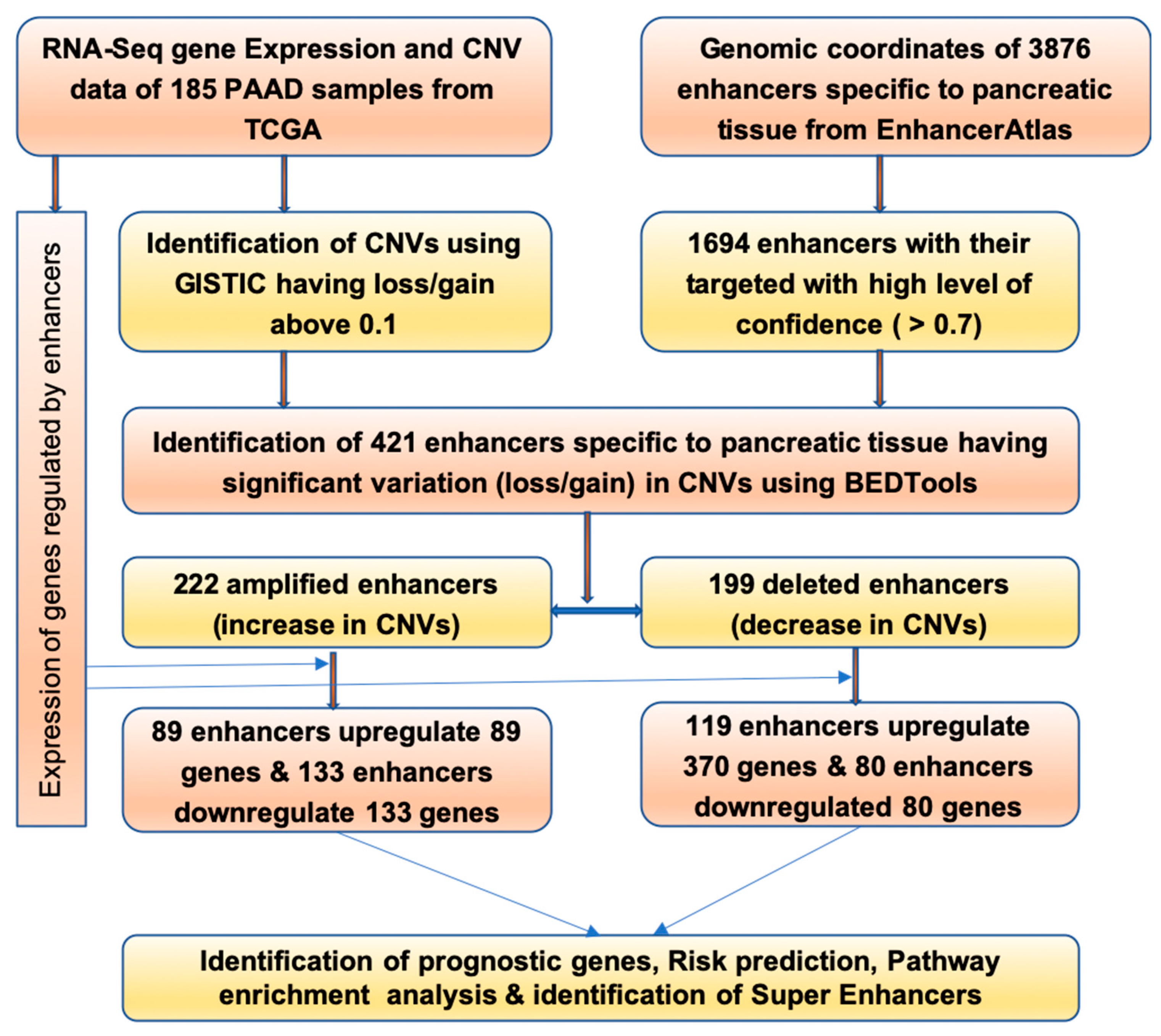
2.1. Identification of Enhancers Having Change in CNV
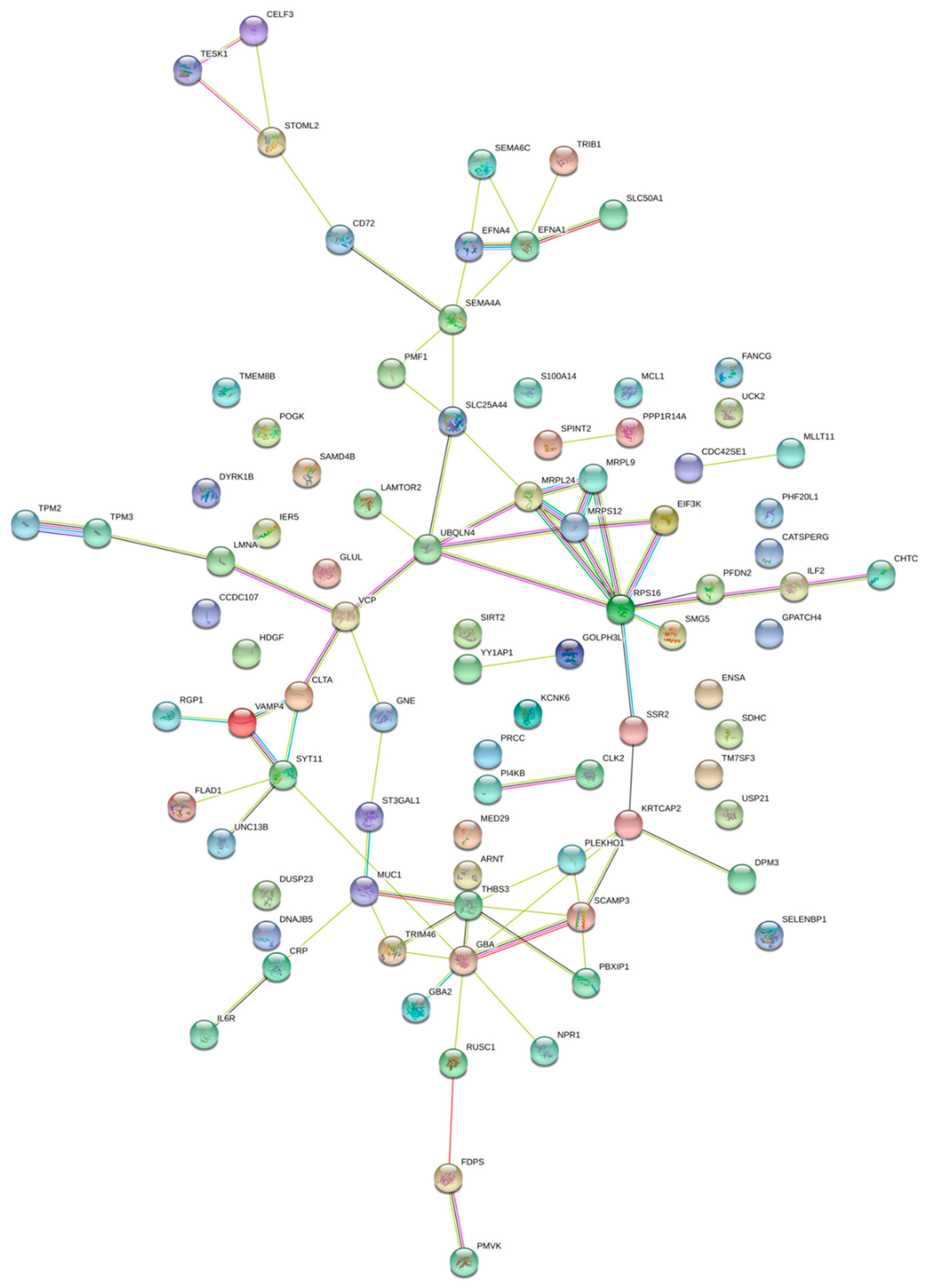
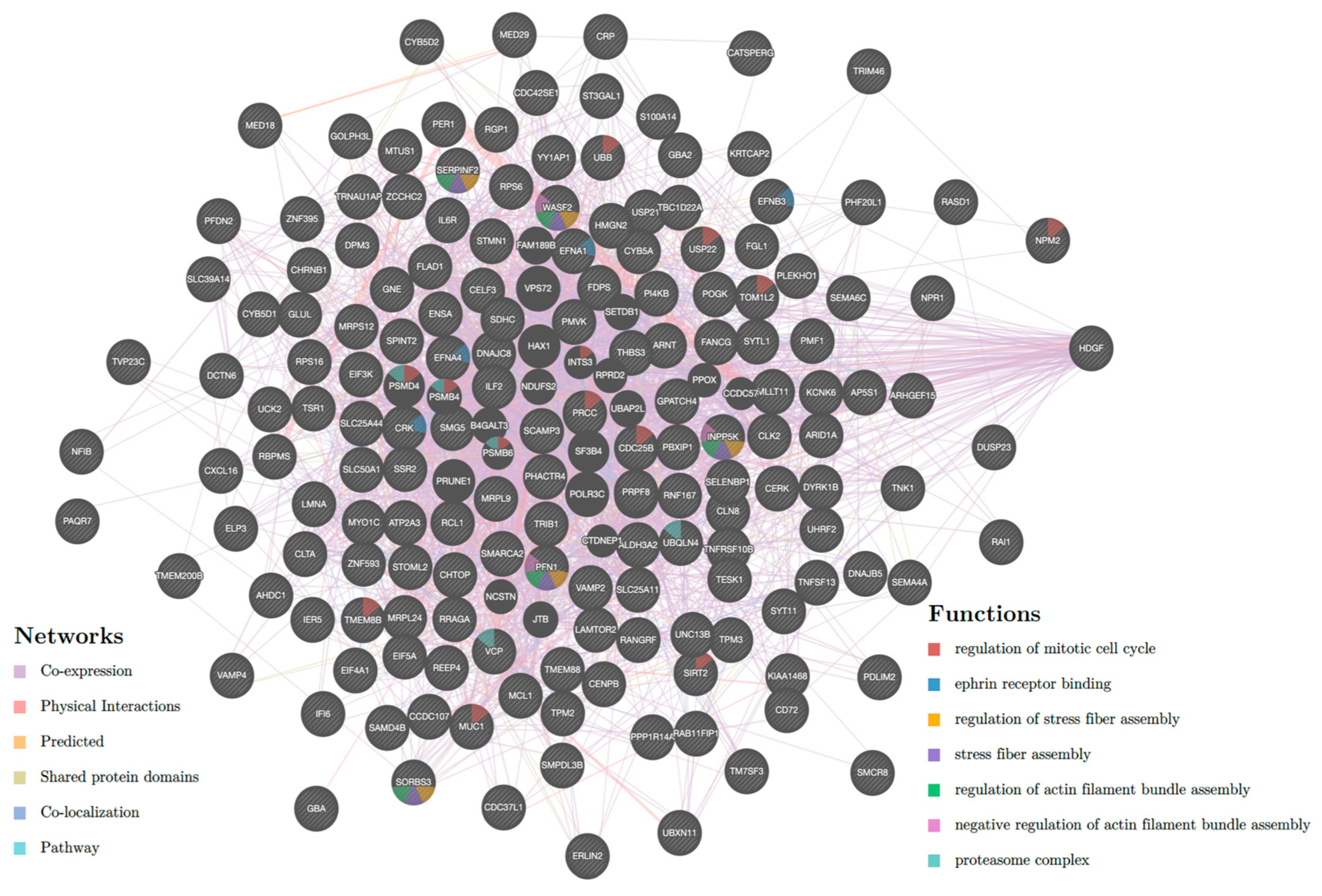
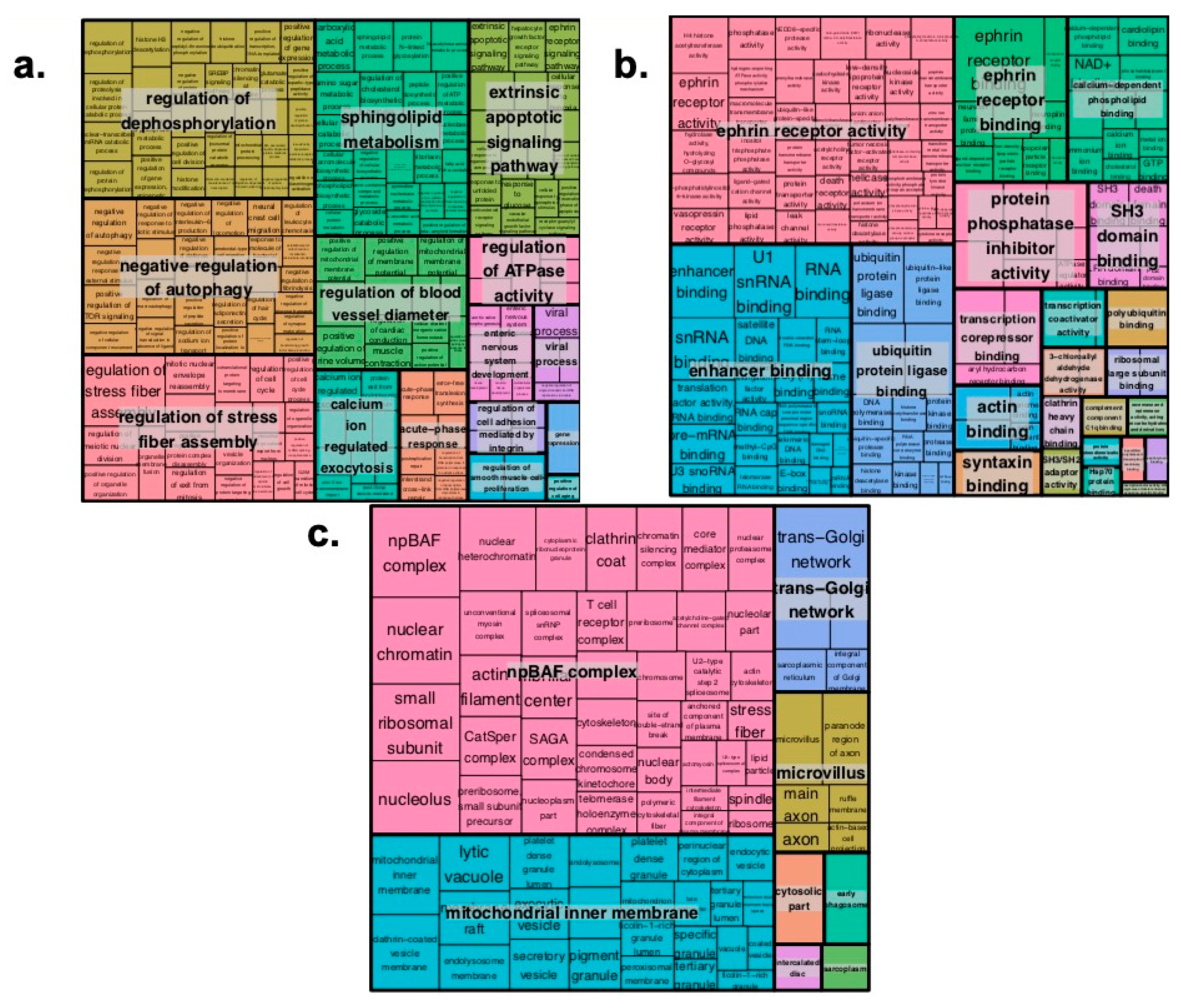
2.2. Pathway Enrichment Analysis of Genes Regulated by Enhancers
2.3. Cox Proportional Hazard Analysis of Enhancer Regulated Genes
2.4. Prognostic Potential of Regulatory Genes
2.5. Genes Regulated by Multiple Enhancers or Super Enhancers
2.6. Enhancer Expression Correlation with Patient Survival
2.7. Prognostic Potential of Negatively Correlated Genes
3. Conclusions and Discussion
4. Materials and Methods
4.1. Enhancers and Target Gene for Human Pancreatic Tissue
4.2. Pancreatic Cancer Dataset from TCGA
4.3. Copy Number Variations Identification
4.4. Screening for Differential Gene Expression
4.5. Combination of Gene Expression and Copy Number Variation
4.6. Gene Network Construction, GO Enrichment and Super Enhancer Based Regulation
4.7. Correlation Analysis of Enhancer Expression with Patient Survival
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TCGA | The Cancer Genome Atlas |
| ICGC | International Cancer Genome Consortium |
| DGE | Differential Gene Expression |
| CNV | Copy Number variation |
| PAAD | Pancreatic Adenocarcinoma |
| CAF | cancer-associated fibroblasts |
| COSMIC | Catalogue of Somatic Mutation in Cancer |
| GISTIC2.0 | Genomic Identification of Significant Target in Cancer |
References
- Wong, M.C.S.; Jiang, J.Y.; Liang, M.; Fang, Y.; Yeung, M.S.; Sung, J.J.Y. Global temporal patterns of pancreatic cancer and association with socioeconomic development. Sci. Rep. 2017, 7, 3165. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Chen, L.-T.; Shan, Y.-S.; Chu, P.-Y.; Tsai, C.-R.; Tsai, H.-J. The incidence and survival of pancreatic cancer by histology, including rare subtypes: A nation-wide cancer registry-based study from Taiwan. Cancer Med. 2018, 7, 5775–5788. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, C.; Peng, X.; Zhou, Z.; Weinstein, J.N.; Cancer Genome Atlas Research Network, H.; Liang, H.; Demchok, J.A.; Felau, I.; Kasapi, M.; et al. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173, 386–399.e12. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.L.; Cenik, C.; Reuter, J.A.; Kiss, G.; Pande, V.S.; Snyder, M.P.; Greenleaf, W.J. Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations. Nat. Genet. 2016, 48, 117–125. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Fröse, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef]
- Perera, D.; Chacon, D.; Thoms, J.A.I.; Poulos, R.C.; Shlien, A.; Beck, D.; Campbell, P.J.; Pimanda, J.E.; Wong, J.W.H. OncoCis: Annotation of cis-regulatory mutations in cancer. Genome Biol. 2014, 15, 485. [Google Scholar]
- Hornshøj, H.; Nielsen, M.M.; Sinnott-Armstrong, N.A.; Świtnicki, M.P.; Juul, M.; Madsen, T.; Sallari, R.; Kellis, M.; Ørntoft, T.; Hobolth, A.; et al. Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. NPJ Genomic Med. 2018, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Herz, H.-M. Enhancer deregulation in cancer and other diseases. BioEssays 2016, 38, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Herman-Izycka, J.; Wlasnowolski, M.; Wilczynski, B. Taking promoters out of enhancers in sequence based predictions of tissue-specific mammalian enhancers. BMC Med. Genomics 2017, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Symmons, O.; Spitz, F. From remote enhancers to gene regulation: Charting the genome’s regulatory landscapes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Shi, J.; Zhu, S.; Lan, Y.; Xu, L.; Yuan, H.; Liao, G.; Liu, X.; Zhang, Y.; Xiao, Y.; et al. DiseaseEnhancer: A resource of human disease-associated enhancer catalog. Nucleic Acids Res. 2018, 46, D78–D84. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Stranger, B.E. The impact of human copy number variation on gene expression. Brief. Funct. Genomics 2015, 14, 352–357. [Google Scholar] [CrossRef]
- Jia, Y.; Chen, L.; Jia, Q.; Dou, X.; Xu, N.; Liao, D.J. The well-accepted notion that gene amplification contributes to increased expression still remains, after all these years, a reasonable but unproven assumption. J. Carcinog. 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Gao, G.F.; Campbell, J.D.; Ramachandran, A.; Mitsuishi, Y.; Ha, G.; Shih, J.; Vazquez, F.; et al. Somatic Superenhancer Duplications and Hotspot Mutations Lead to Oncogenic Activation of the KLF5 Transcription Factor. Cancer Discov. 2018, 8, 108–125. [Google Scholar] [CrossRef]
- Gao, T.; He, B.; Liu, S.; Zhu, H.; Tan, K.; Qian, J. EnhancerAtlas: A resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics 2016, 32, 3543–3551. [Google Scholar] [CrossRef]
- He, X.; Fuller, C.K.; Song, Y.; Meng, Q.; Zhang, B.; Yang, X.; Li, H. Sherlock: Detecting Gene-Disease Associations by Matching Patterns of Expression QTL and GWAS. Am. J. Hum. Genet. 2013, 92, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Shi, C.; Jiang, H.-X.; Qin, S.-Y. Identification of novel therapeutic target genes and pathway in pancreatic cancer by integrative analysis. Medicine (Baltimore). 2017, 96, e8261. [Google Scholar] [CrossRef] [PubMed]
- Basso, D. Altered glucose metabolism and proteolysis in pancreatic cancer cell conditioned myoblasts: Searching for a gene expression pattern with a microarray analysis of 5000 skeletal muscle genes. Gut 2004, 53, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sheng, J.; Zhang, Y.; Tian, Y.; Zhu, J.; Luo, N.; Xiao, C.; Li, R. Overexpression of SCAMP3 is an indicator of poor prognosis in hepatocellular carcinoma. Oncotarget 2017, 8, 109247–109257. [Google Scholar] [CrossRef] [PubMed]
- Sahraei, M.; Roy, L.D.; Curry, J.M.; Teresa, T.L.; Nath, S.; Besmer, D.; Kidiyoor, A.; Dalia, R.; Gendler, S.J.; Mukherjee, P. MUC1 regulates PDGFA expression during pancreatic cancer progression. Oncogene 2012, 31, 4935–4945. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Park, S.; Lee, E.K.; Yoo, Y.J. Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci. Rep. 2013, 3, 2623. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Xu, X.-Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Messenger, S.W.; Jones, E.K.; Holthaus, C.L.; Thomas, D.D.H.; Cooley, M.M.; Byrne, J.A.; Mareninova, O.A.; Gukovskaya, A.S.; Groblewski, G.E. Acute acinar pancreatitis blocks vesicle-associated membrane protein 8 (VAMP8)-dependent secretion, resulting in intracellular trypsin accumulation. J. Biol. Chem. 2017, 292, 7828–7839. [Google Scholar] [CrossRef]
- Rivera, I.-G.; Ordoñez, M.; Presa, N.; Gangoiti, P.; Gomez-Larrauri, A.; Trueba, M.; Fox, T.; Kester, M.; Gomez-Muñoz, A. Ceramide 1-phosphate regulates cell migration and invasion of human pancreatic cancer cells. Biochem. Pharmacol. 2016, 102, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Wang, J.; Ma, W.; Wang, X.; Cheng, Y. HDGF: A novel jack-of-all-trades in cancer. Future Oncol. 2014, 10, 2675–2685. [Google Scholar] [CrossRef] [PubMed]
- Fisher, W.E.; Wu, Y.; Amaya, F.; Berger, D.H. Somatostatin receptor subtype 2 gene therapy inhibits pancreatic cancer in vitro. J. Surg. Res. 2002, 105, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Kugler, J.-M.; Loya, A.C.; Cohen, S.M. USP21 regulates Hippo pathway activity by mediating MARK protein turnover. Oncotarget 2017, 8, 64095–64105. [Google Scholar] [CrossRef] [PubMed]
- Muniyan, S.; Haridas, D.; Chugh, S.; Rachagani, S.; Lakshmanan, I.; Gupta, S.; Seshacharyulu, P.; Smith, L.M.; Ponnusamy, M.P.; Batra, S.K. MUC16 contributes to the metastasis of pancreatic ductal adenocarcinoma through focal adhesion mediated signaling mechanism. Genes Cancer 2016, 7, 110–124. [Google Scholar] [PubMed]
- Takadate, T.; Onogawa, T.; Fukuda, T.; Motoi, F.; Suzuki, T.; Fujii, K.; Kihara, M.; Mikami, S.; Bando, Y.; Maeda, S.; et al. Novel prognostic protein markers of resectable pancreatic cancer identified by coupled shotgun and targeted proteomics using formalin-fixed paraffin-embedded tissues. Int. J. Cancer 2013, 132, 1368–1382. [Google Scholar] [CrossRef]
- Shin, H.Y. Targeting Super-Enhancers for Disease Treatment and Diagnosis. Mol. Cells 2018, 41, 506–514. [Google Scholar]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Kaveh, F.; Baumbusch, L.O.; Nebdal, D.; Børresen-Dale, A.-L.; Lingjærde, O.C.; Edvardsen, H.; Kristensen, V.N.; Solvang, H.K. A systematic comparison of copy number alterations in four types of female cancer. BMC Cancer 2016, 16, 913. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Lu, J.; Zhao, M. Integrative analysis to identify oncogenic gene expression changes associated with copy number variations of enhancer in ovarian cancer. Oncotarget 2017, 8, 91558–91567. [Google Scholar] [CrossRef] [PubMed][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Hazard Ratio (Median Value) | p-Value |
|---|---|---|
| VAMP2 | 2.917023 | 0.000221728 |
| GBA2 | 2.641652475 | 0.000497427 |
| RANGRF | 2.512853346 | 0.000794802 |
| CERK | 2.483219821 | 0.001137485 |
| SORBS3 | 2.450014903 | 0.001099829 |
| AP5S1 | 2.375174975 | 0.001570686 |
| INPP5K | 2.317435264 | 0.00206786 |
| SLC25A44 | 2.291295263 | 0.002326752 |
| RNF167 | 2.239662092 | 0.003430129 |
| DPM3 | 2.196709622 | 0.004076415 |
| SEMA6C | 2.192865982 | 0.005221015 |
| NPR1 | 2.170189229 | 0.00497616 |
| SLC25A11 | 2.104895785 | 0.006106619 |
| USP22 | 2.093982578 | 0.006492927 |
| DNAJB5 | 2.058641673 | 0.008559552 |
| Gene | Name | Z-Score |
|---|---|---|
| CLK2 | CDC-like kinase 2 | 2.39128 |
| EIF4A1 | Eukaryotic translation initiation factor 4A1 | 2.24365 |
| FDPS | Farnesyl diphosphate synthase | 4.03062 |
| FLAD1 | FAD1 flavin adenine dinucleotide synthetase homolog | 2.95895 |
| HDGF | Hepatoma-derived growth factor | 2.16932 |
| ILF2 | Interleukin enhancer binding factor 2, 45kDa | 2.80112 |
| MLLT11 | Myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 11 | 2.43407 |
| PFN1 | Profilin 1 | 2.49119 |
| PRCC | Papillary renal cell carcinoma (translocation-associated) | 3.93837 |
| REEP4 | Receptor accessory protein 4 | 2.98392 |
| RUSC1 | RUN and SH3 domain containing 1 | 3.28846 |
| SCAMP3 | Secretory carrier membrane protein 3 | 2.32083 |
| UBB | In multiple clusters | 2.54009 |
| UBQLN4 | Ubiquilin 4 | 2.79194 |
| USP21 | Ubiquitin specific peptidase 21 | 2.40871 |
| Gene Ensembl ID | Enhancer Coordinates | Chromosome |
|---|---|---|
| ENSG00000010278 | 6337690-6341770;6387100-6388270;6449480-6450940;6579320-6579700 | Chromosome 12 |
| ENSG00000068745 | 49028850-49029210;49044960-49046260;49044960-49046260;49486410-49486940;49486410-49486940 | Chromosome 3 |
| ENSG00000099622 | 1248660-1249890;1248660-1249890;1248660-1249890;1250530-1251560;1259140-1263220;1275830-1277180;1275830-1277180;1275830-1277180;1275830-1277180 | Chromosome 19 |
| ENSG00000099875 | 1941420-1943940;2031500-2033130;2047440-2050790;2047440-2050790 | Chromosome 19 |
| ENSG00000111319 | 6444030-6449420;6449480-6450940;6449480-6450940;6662010-6662810 | Chromosome 12 |
| ENSG00000111674 | 6662010-6662810;6999750-7001150;6999750-7001150;7046720-7047010;7047230-7047600 | Chromosome 12 |
| ENSG00000114353 | 50126460-50126820;50328720-50329650;50358380-50358800;50359200-50359520;50359200-50359520 | Chromosome 3 |
| ENSG00000115524 | 198055790-198057570;198152000-198152560;198318390-198319050;198318390-198319050 | Chromosome 2 |
| ENSG00000116285 | 8181000-8181800;8181000-8181800;8193790-8194520;8318930-8320010 | Chromosome 1 |
| ENSG00000116473 | 112134680-112136260;112134680-112136260;112134680-112136260;112202750-112203810 | Chromosome 1 |
| ENSG00000117632 | 26221860-26223380;26221860-26223380;26323300-26324510;26452760-26454880 | Chromosome 1 |
| ENSG00000129968 | 1875340-1876950;1905440-1906150;1905440-1906150;2042030-2042430;2166660-2167720;2579200-2579650 | Chromosome 19 |
| ENSG00000130005 | 1040200-1040500;1040580-1040860;1383940-1385280;1407800-1410200 | Chromosome 19 |
| ENSG00000137154 | 19183280-19185190;19232860-19233120;19379620-19380190;19456550-19457860 | Chromosome 9 |
| ENSG00000142910 | 32013850-32015900;32109070-32109980;32109070-32109980;111308340-111308980;111948770-111949470 | Chromosome 1 |
| ENSG00000143294 | 156659170-156659710;156716040-156721140;156659170-156659710;156716040-156721140 | Chromosome 1 |
| Coordinates of Enhancer on Genome | Correlation (CNV vs. Survival Time) | |
|---|---|---|
| Cancer | Healthy | |
| chr19:2059605-2060167 | 0.49837818 | 0.2314589 |
| chr5:172380663-172381064 | 0.46715342 | 0.16699777 |
| chr9:136999790-136999893 | 0.46557032 | −0.0626969 |
| chr16:85496879-85497321 | 0.45373278 | −0.6866357 |
| chr14:102415545-102415736 | 0.44115981 | 0.2792461 |
| chr5:10352620-10352938 | 0.43075964 | 0.52245209 |
| chr1:234746093-234747674 | 0.42760743 | 0.98845399 |
| chr19:863983-864016 | 0.42492329 | −0.3898893 |
| chr10:31892273-31892723 | 0.42465511 | −0.5260193 |
| chr4:124621616-124621886 | 0.41837579 | 0.17175469 |
| chr5:964244-964536 | −0.1737289 | 0.52446177 |
| chr6:169573934-169574190 | −0.1747893 | −0.7388221 |
| chr22:50980817-50981280 | −0.1776918 | 0.05853257 |
| chr8:128306934-128307283 | −0.1778833 | −0.3632054 |
| chr14:105500629-105500990 | −0.1798565 | 0.11625874 |
| chr15:99992993-99993428 | −0.1823302 | 0.31824116 |
| chrX:100792680-100793554 | −0.1832455 | −0.6606904 |
| chr22:50979060-50979802 | −0.1916855 | 0.01347895 |
| chr18:12306995-12307375 | −0.1946683 | 0.65842313 |
| chr6:169569548-169569688 | −0.1974295 | 0.1498992 |
| chr8:70042378-70042779 | −0.2421095 | 0.22802202 |
| Negatively correlated genes (E+G−/E−G+) | ||
|---|---|---|
| Gene Symbol | Hazard Ratio | p-Value |
| MUM1 | 3.558401305 | 1.33 × 10−5 |
| MUM1.1 | 3.558401305 | 1.33 × 10−5 |
| MUM1.2 | 3.558401305 | 1.33 × 10−5 |
| RBM6 | 3.444153767 | 1.83 × 10−5 |
| MBD3 | 3.027911665 | 0.00010983 |
| TLE2 | 2.983689472 | 0.000127766 |
| TLE2.1 | 2.983689472 | 0.000127766 |
| PLD3 | 2.853819471 | 0.00022269 |
| PLD3.1 | 2.853819471 | 0.00022269 |
| LRP3 | 2.81420074 | 0.00030683 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Patiyal, S.; Kumar, V.; Nagpal, G.; Raghava, G.P.S. In Silico Analysis of Gene Expression Change Associated with Copy Number of Enhancers in Pancreatic Adenocarcinoma. Int. J. Mol. Sci. 2019, 20, 3582. https://doi.org/10.3390/ijms20143582
Kumar R, Patiyal S, Kumar V, Nagpal G, Raghava GPS. In Silico Analysis of Gene Expression Change Associated with Copy Number of Enhancers in Pancreatic Adenocarcinoma. International Journal of Molecular Sciences. 2019; 20(14):3582. https://doi.org/10.3390/ijms20143582
Chicago/Turabian StyleKumar, Rajesh, Sumeet Patiyal, Vinod Kumar, Gandharva Nagpal, and Gajendra P.S. Raghava. 2019. "In Silico Analysis of Gene Expression Change Associated with Copy Number of Enhancers in Pancreatic Adenocarcinoma" International Journal of Molecular Sciences 20, no. 14: 3582. https://doi.org/10.3390/ijms20143582
APA StyleKumar, R., Patiyal, S., Kumar, V., Nagpal, G., & Raghava, G. P. S. (2019). In Silico Analysis of Gene Expression Change Associated with Copy Number of Enhancers in Pancreatic Adenocarcinoma. International Journal of Molecular Sciences, 20(14), 3582. https://doi.org/10.3390/ijms20143582