Targeting HIV/HCV Coinfection Using a Machine Learning-Based Multiple Quantitative Structure-Activity Relationships (Multiple QSAR) Method



Abstract
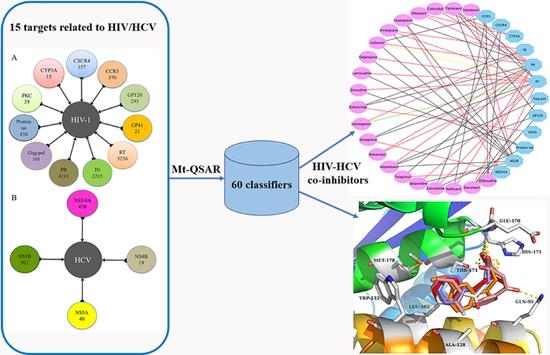
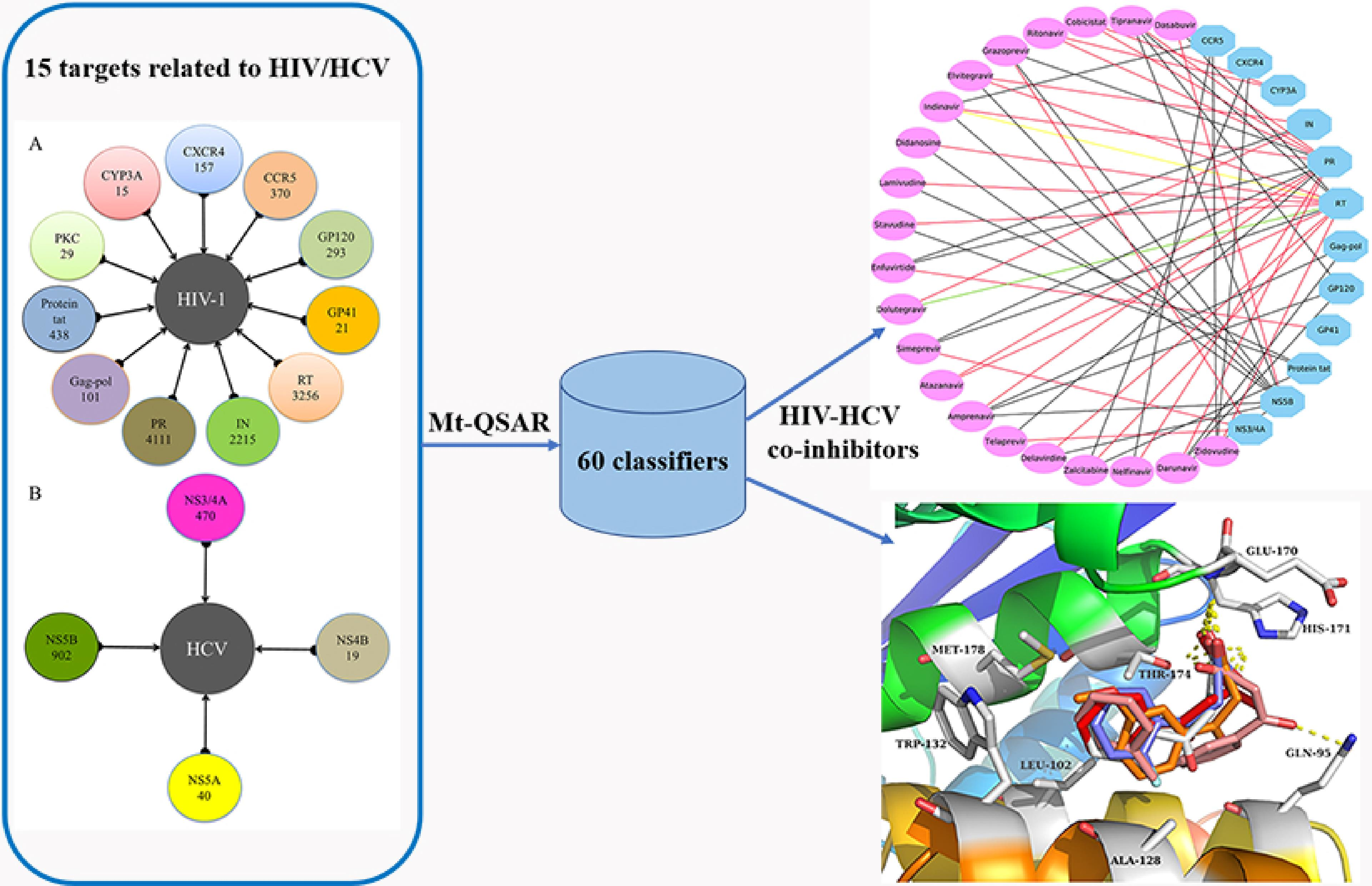
1. Introduction
2. Results
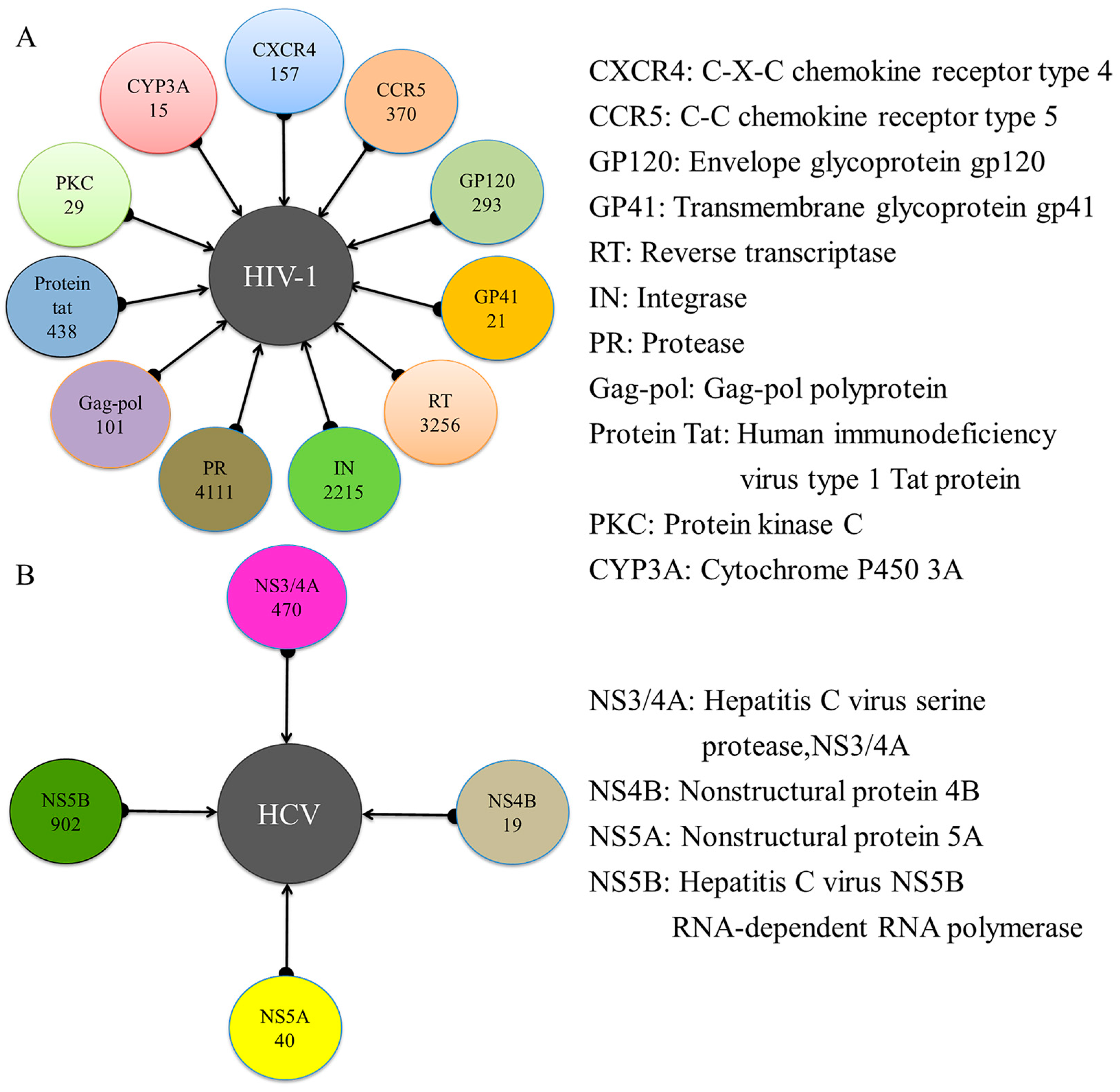
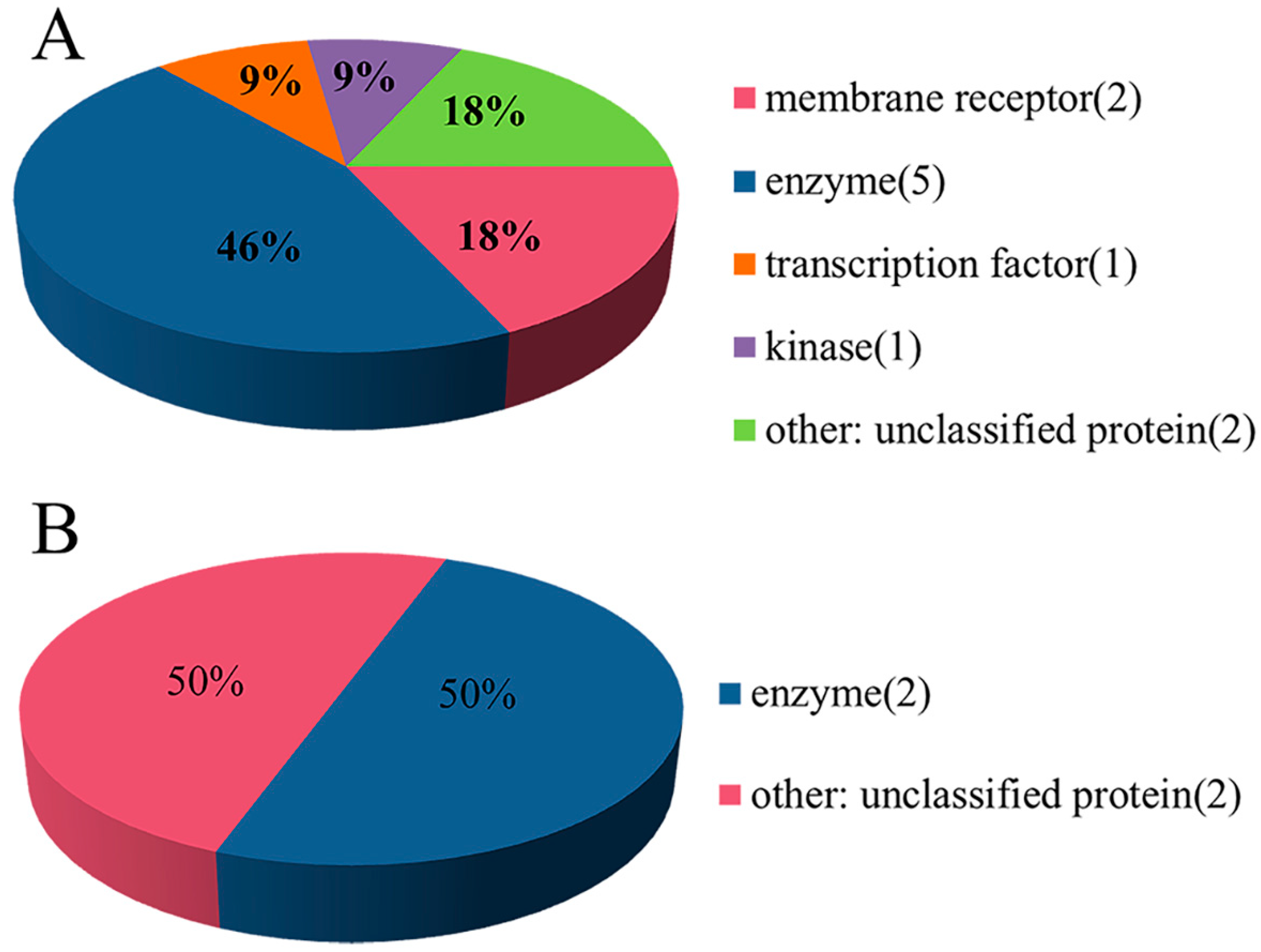
2.1. Experimental Dataset Analysis
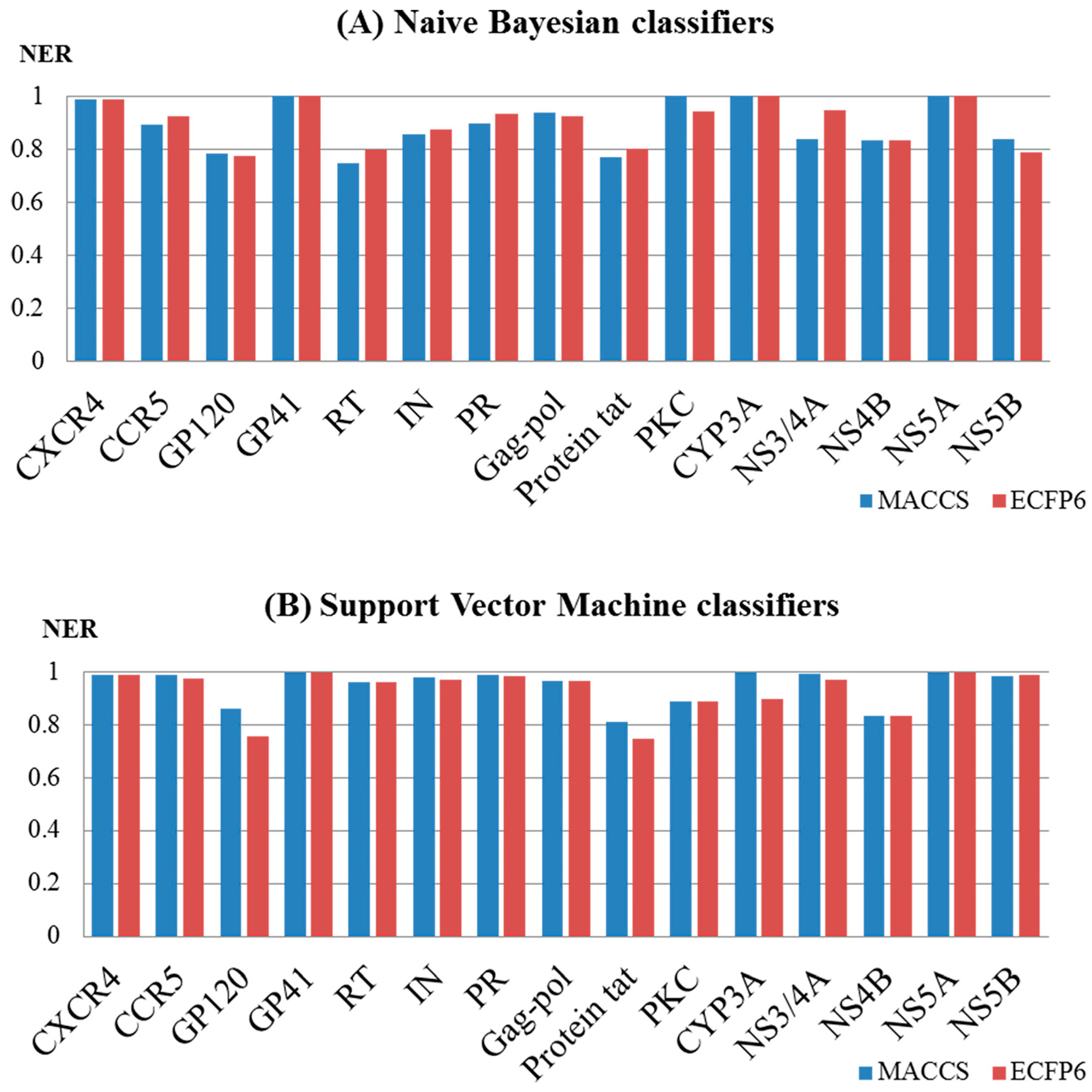
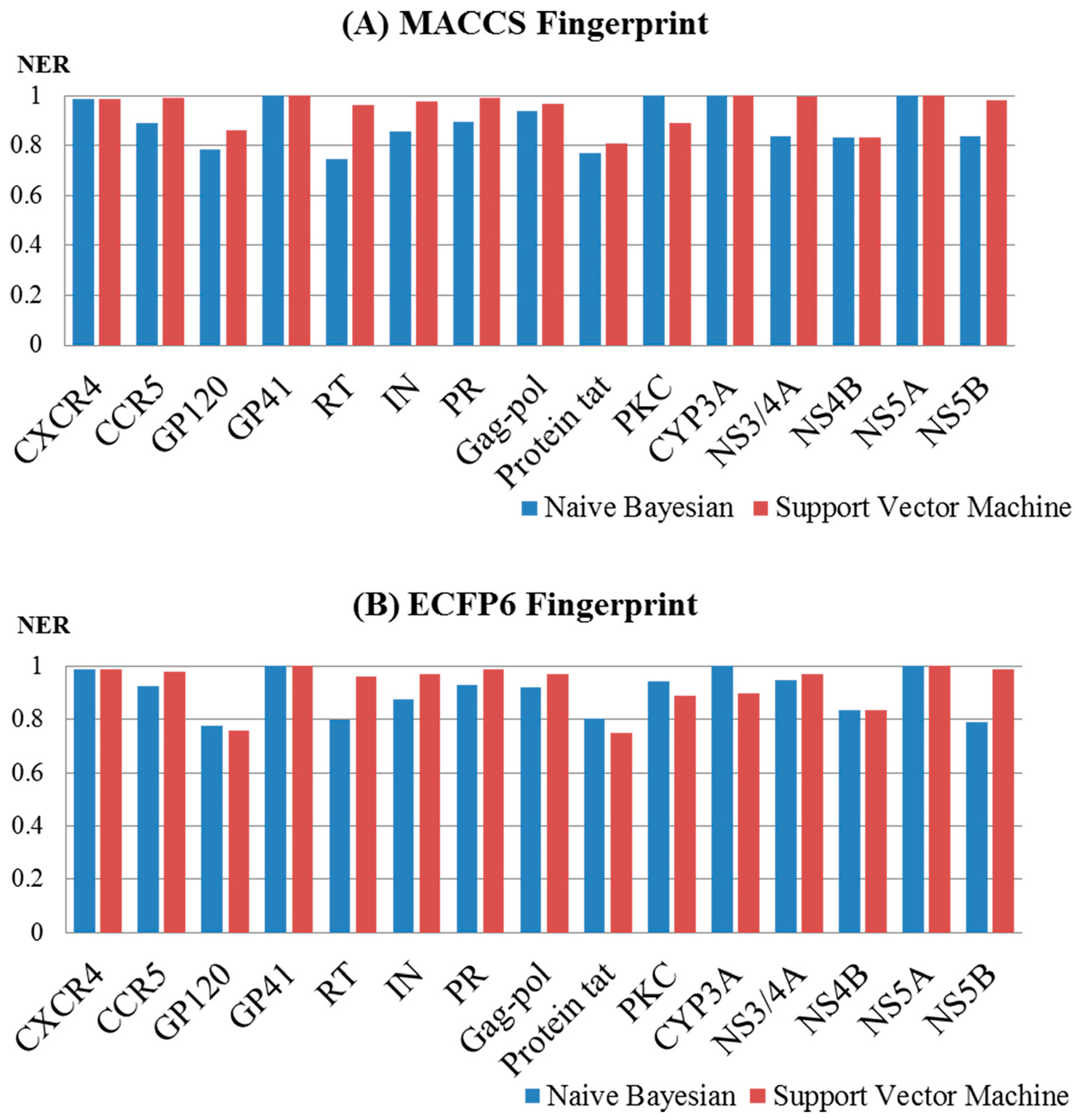
2.2. Performance Evaluation and the Comparison of the Models
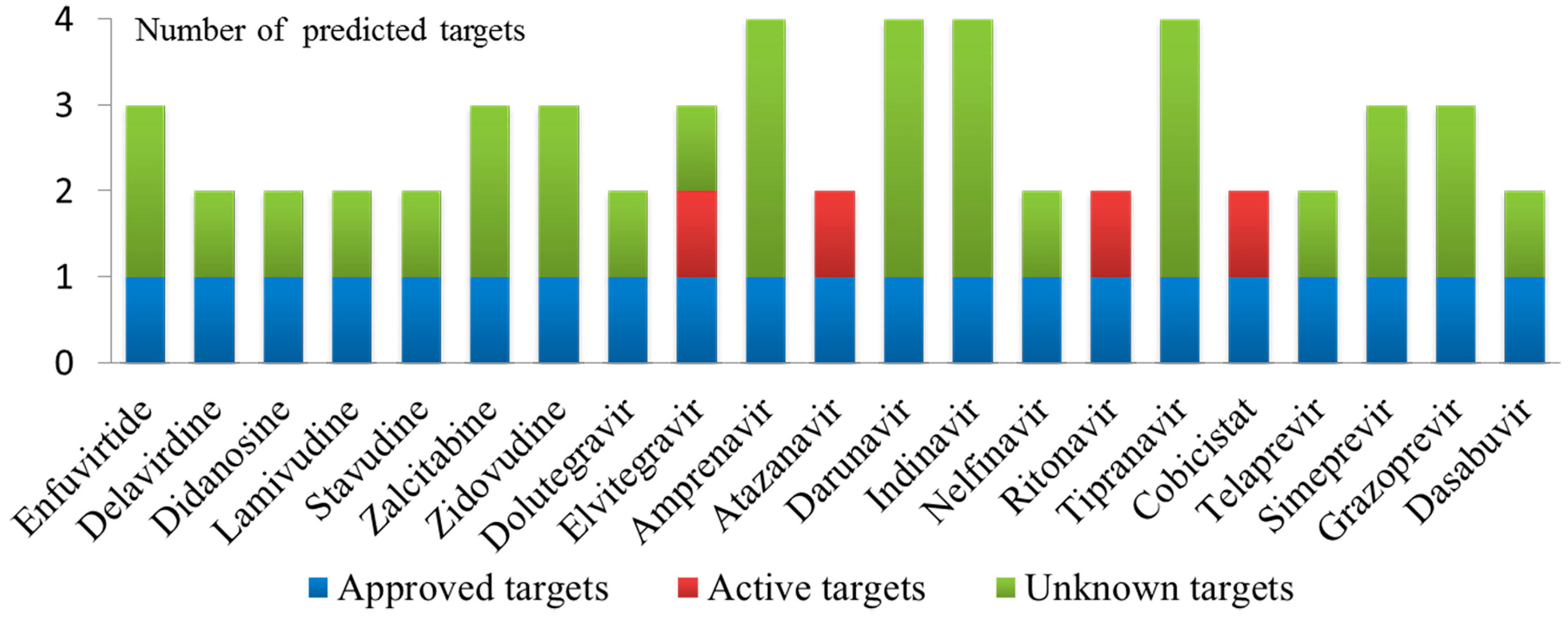
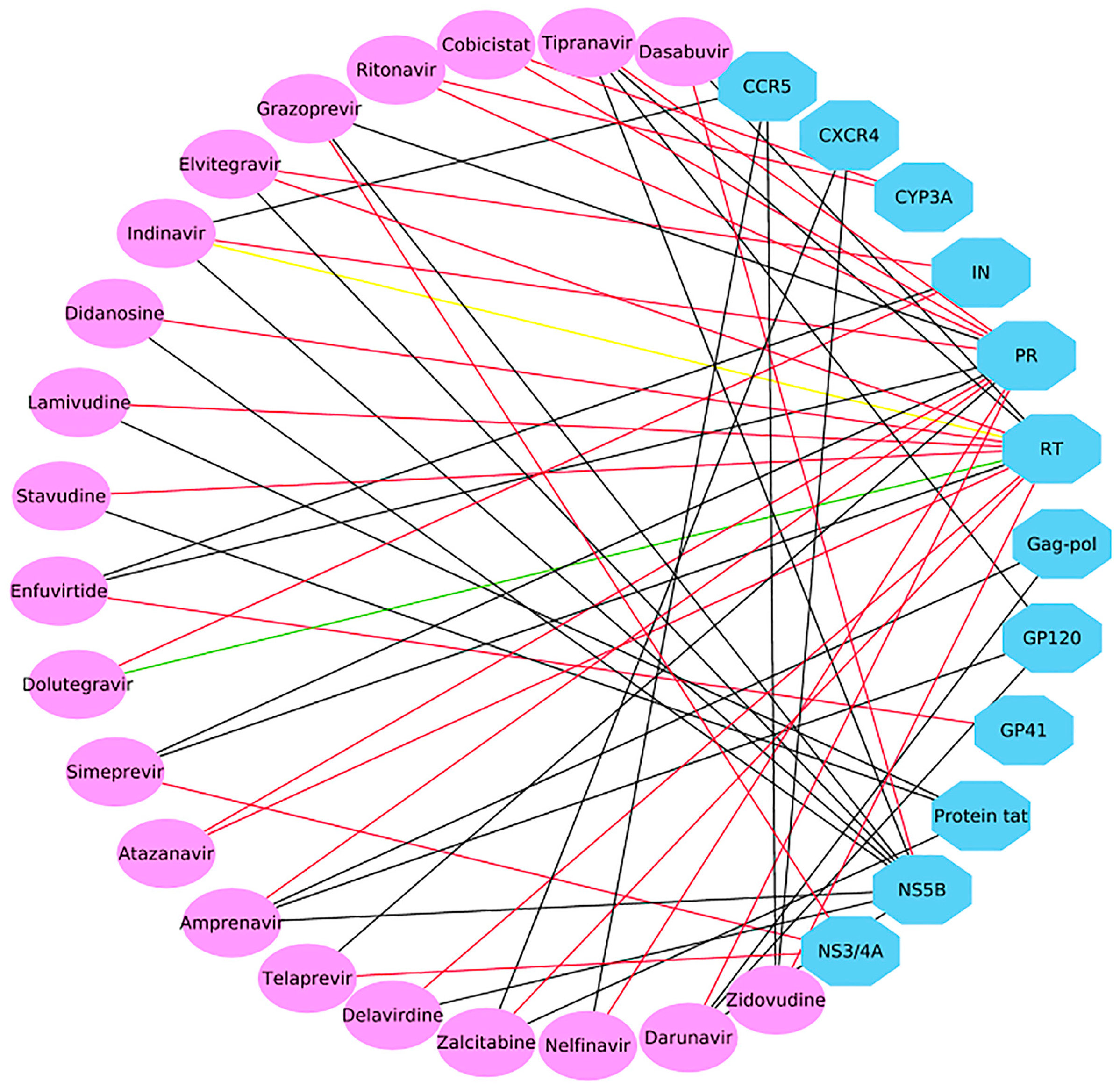
2.3. Case 1: Prediction and Analysis of the Polypharmacology of Known HIV-1 and HCV Drugs
2.4. Case 2: Target prediction and Analysis of Known Inhibitors
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Dataset Preparation
5.2. Molecular Representation
5.3. Multiple QSAR Models Generation
5.3.1. Naïve Bayes (NB)
5.3.2. Support Vector Machine (SVM)
5.4. Performance Evaluation of the Multiple QSAR Models
5.5. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, S.; Milne, G.W.A.; Yan, X.; Posey, I.J.; Nicklaus, M.C.; Graham, L.; Rice, W.G. Discovery of novel, non-peptide HIV-1 protease inhibitors by pharmacophore searching. J. Med. Chem. 1996, 39, 2047–2054. [Google Scholar] [CrossRef]
- Therese, P.J.; Manvar, D.; Kondepudi, S.; Battu, M.B.; Sriram, D.; Basu, A.; Yogeeswari, P.; Kaushik-Basu, N. Multiple e-pharmacophore modeling, 3D-QSAR, and high-throughput virtual screening of hepatitis C virus NS5B polymerase inhibitors. J. Chem. Inf. Model. 2014, 54, 539–552. [Google Scholar] [CrossRef]
- Laguno, M.; Murillas, J.; Blanco, J.L.; Martínez, E.; Miquel, R.; Sánchez-Tapias, J.M.; Bargallo, X.; García-Criado, A.; de Lazzari, E.; Larrousse, M.; et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for treatment of HIV/HCV co-infected patients. AIDS 2004, 18, F27–F36. [Google Scholar] [CrossRef]
- WHO Global Hepatitis Report. 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 24 March 2019).
- Wang, Z.; Bennett, E.M.; Wilson, D.J.; Salomon, C.; Vince, R. Rationally designed dual inhibitors of HIV reverse transcriptase and integrase. J. Med. Chem. 2007, 50, 3416–3419. [Google Scholar] [CrossRef]
- Cox, B.D.; Prosser, A.R.; Sun, Y.; Li, Z.; Lee, S.; Huang, M.B.; Bond, V.C.; Snyder, J.P.; Krystal, M.; Wilson, L.J.; et al. Pyrazolo-Piperidines Exhibit Dual Inhibition of CCR5/CXCR4 HIV Entry and Reverse Transcriptase. ACS Med. Chem. Lett. 2015, 6, 753–757. [Google Scholar] [CrossRef]
- Operskalski, E.A.; Kovacs, A. HIV/HCV co-infection: Pathogenesis, clinical complications, treatment, and new therapeutic technologies. Curr. HIV/AIDS Rep. 2011, 8, 12–22. [Google Scholar] [CrossRef]
- Bajorath, J. Computational analysis of ligand relationships within target families. Curr. Opin. Chem. Biol. 2008, 12, 352–358. [Google Scholar] [CrossRef]
- Rognan, D. Chemogenomic approaches to rational drug design. Br. J. Pharmacol. 2007, 152, 38–52. [Google Scholar] [CrossRef]
- Yang, L.; Wang, K.J.; Wang, L.S.; Jegga, A.G.; Qin, S.Y.; He, G.; Chen, J.; Xiao, Y.; He, L. Chemical-protein interactome and its application in off-target identification. Interdiscip. Sci. Comput. Life Sci. 2012, 3, 22–30. [Google Scholar] [CrossRef]
- Cheng, F.; Zhou, Y.; Li, J.; Li, W.; Liu, G.; Tang, Y. Prediction of chemical-protein interactions: multitarget-QSAR versus computational chemogenomic methods. Mol. Biosyst. 2012, 8, 2373–2384. [Google Scholar] [CrossRef]
- Sawada, R.; Iwata, H.; Mizutani, S.; Yamanishi, Y. Target-Based Drug Repositioning Using Large-Scale Chemical-Protein Interactome Data. J. Chem. Inf. Model. 2015, 55, 2717–2730. [Google Scholar] [CrossRef]
- Luo, H.; Chen, J.; Shi, L.; Mikailov, M.; Zhu, H.; Wang, K.; He, L.; Yang, L. DRAR-CPI: A server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome. Nucleic Acids Res. 2011, 39, 492–498. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Bender, A.; Davies, J.W. In silico target fishing: Predicting biological targets from chemical structure. Drug Discov. Today Technol. 2006, 3, 413–421. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Bauer, R.A.; Bourne, P.E.; Formella, A.; Frömmel, C.; Gille, C.; Goede, A.; Guerler, A.; Hoppe, A.; Knapp, E.-W.; Pöschel, T.; et al. Superimpose: A 3D structural superposition server. Nucleic Acids Res. 2008, 36, W47–W54. [Google Scholar] [CrossRef]
- Li, H.; Gao, Z.; Kang, L.; Zhang, H.; Yang, K.; Yu, K.; Luo, X.; Zhu, W.; Chen, K.; Shen, J.; et al. TarFisDock: A web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006, 34, W219–W224. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Zhi, D.G. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins 2001, 43, 217–226. [Google Scholar] [CrossRef]
- Viña, D.; Uriarte, E.; Orallo, F.; González-Díaz, H. Alignment-free prediction of a drug-target complex network based on parameters of drug connectivity and protein sequence of receptors. Mol. Pharm. 2009, 6, 825–835. [Google Scholar] [CrossRef]
- Fang, J.; Li, Y.; Liu, R.; Pang, X.; Li, C.; Yang, R.; He, Y.; Lian, W.; Liu, A.-L.; Du, G.-H. Discovery of multitarget-directed ligands against Alzheimer’s disease through systematic prediction of chemical-protein interactions. J. Chem. Inf. Model. 2015, 55, 149–164. [Google Scholar] [CrossRef]
- Antanasijević, D.; Antanasijević, J.; Trišović, N.; Ušćumlić, G.; Pocajt, V. From Classification to Regression Multitasking QSAR Modeling Using a Novel Modular Neural Network: Simultaneous Prediction of Anticonvulsant Activity and Neurotoxicity of Succinimides. Mol. Pharm. 2017, 14, 4476–4484. [Google Scholar] [CrossRef] [PubMed]
- Porwal, A.; Carranza, E.J.M.; Hale, M. Bayesian network classifiers for mineral potential mapping. Comput. Geosci. 2006, 32, 1–16. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Gilson, M.K. Virtual Screening of Molecular Databases Using a Support Vector Machine. J. Chem. Inf. Model. 2005, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G.; et al. Therapeutic target database update 2018: Enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2018, 46, D1121–D1127. [Google Scholar]
- Gaulton, A.; Hersey, A.; Nowotka, M.L.; Patricia Bento, A.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular Similarity Measures. Chemoinformatics 2004, 275, 1–50. [Google Scholar]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Vapnik, V.; Sterin, A. On structural risk minimization or overall risk in a problem of pattern recognition. Autom. Remote Control 1977, 10, 1495e1503. [Google Scholar]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L.; Karapetyan, K.; Dracheva, S.; Shoemaker, B.A.; et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2012, 40, D400–D412. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Qiu, Z.; Su, Y.; He, Y. The N-Terminal T-T Motif of a Third-Generation HIV-1 Fusion Inhibitor Is Not Required for Binding Affinity and Antiviral Activity. J. Med. Chem. 2015, 58, 6378–6388. [Google Scholar] [CrossRef] [PubMed]
- Nugent, R.A.; Schlachter, S.T.; Murphy, M.J.; Cleek, G.J.; Poel, T.J.; Wishka, D.G.; Graber, D.R.; Yagi, Y.; Keiser, B.J.; Olmsted, R.A.; et al. Pyrimidine thioethers: A novel class of HIV-1 reverse transcriptase inhibitors with activity against BHAP-resistant HIV. J. Med. Chem. 1998, 41, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Thaisrivongs, S.; Romero, D.L.; Tommasi, R. a; Janakiraman, M.N.; Strohbach, J.W.; Turner, S.R.; Biles, C.; Morge, R.R.; Johnson, P.D.; Aristoff, P. a; et al. Structure-based design of HIV protease inhibitors: 5,6-dihydro-4-hydroxy-2-pyrones as effective, nonpeptidic inhibitors. J. Med. Chem. 1996, 39, 4630–4642. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Müh, U.; Hanzelka, B.L.; Bartels, D.J.; Wei, Y.; Rao, B.G.; Brennan, D.L.; Tigges, A.M.; Swenson, L.; Kwong, A.D.; et al. Phenotypic and structural analyses of hepatitis C virus NS3 protease Arg155 variants: Sensitivity to telaprevir (VX-950) and interferon alpha. J. Biol. Chem. 2007, 282, 22619–22628. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.Z.; Zhang, Y.-K.; Li, X.; Liu, Y.; Zhang, S.; Zhou, Y.; Plattner, J.J.; Baker, S.J.; Liu, L.; Duan, M.; et al. Synthesis and biological evaluations of P4-benzoxaborole-substituted macrocyclic inhibitors of HCV NS3 protease. Bioorg. Med. Chem. Lett. 2010, 20, 7317–7322. [Google Scholar] [CrossRef] [PubMed]
- Neelamkavil, S.F.; Agrawal, S.; Bara, T.; Bennett, C.; Bhat, S.; Biswas, D.; Brockunier, L.; Buist, N.; Burnette, D.; Cartwright, M.; et al. Discovery of MK-8831, A Novel Spiro-Proline Macrocycle as a Pan-Genotypic HCV-NS3/4a Protease Inhibitor. ACS Med. Chem. Lett. 2016, 7, 111–116. [Google Scholar] [CrossRef]
- Meguellati, A.; Ahmed-Belkacem, A.; Nurisso, A.; Yi, W.; Brillet, R.; Berqouch, N.; Chavoutier, L.; Fortuné, A.; Pawlotsky, J.-M.; Boumendjel, A.; et al. New pseudodimeric aurones as palm pocket inhibitors of Hepatitis C virus RNA-dependent RNA polymerase. Eur. J. Med. Chem. 2016, 115, 217–229. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Koontz, D.; Bassit, L.; Hernandez-Santiago, B.I.; Detorio, M.; Rapp, K.L.; Amblard, F.; Bondada, L.; Grier, J.; Coats, S.J.; et al. Anti-human immunodeficiency virus activity, cross-resistance, cytotoxicity, and intracellular pharmacology of the 3′-azido-2′,3′- dideoxypurine nucleosides. Antimicrob. Agents Chemother. 2009, 53, 3715–3719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kobayashi, M.; Yoshinaga, T.; Seki, T.; Wakasa-Morimoto, C.; Brown, K.W.; Ferris, R.; Foster, S.A.; Hazen, R.J.; Miki, S.; Suyama-Kagitani, A.; et al. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents Chemother. 2011, 55, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.; Métifiot, M.; Chung, S.; Cuzzucoli Crucitti, G.; Maddali, K.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Scipione, L.; et al. Basic quinolinonyl diketo acid derivatives as inhibitors of HIV integrase and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, R.G.; Furfine, E.S.; Hazen, R.J.; Miller, J.F.; Reynolds, D.J.; Sammond, D.M.; Spaltenstein, A.; Wheelan, P.; Wright, L.L. Synthesis and antiviral activities of novel N-alkoxy-arylsulfonamide-based HIV protease inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 3560–3564. [Google Scholar] [CrossRef] [PubMed]
- Bold, G.; Fässler, A.; Capraro, H.G.; Cozens, R.; Klimkait, T.; Lazdins, J.; Mestan, J.; Poncioni, B.; Rösel, J.; Stover, D.; et al. New aza-dipeptide analogues as potent and orally absorbed HIV-1 protease inhibitors: Candidates for clinical development. J. Med. Chem. 1998, 41, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Eissenstat, M.; Guerassina, T.; Gulnik, S.; Afonina, E.; Silva, A.M.; Ludtke, D.; Yokoe, H.; Yu, B.; Erickson, J. Enamino-oxindole HIV protease inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5078–5083. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.V.; O’Brien, J.L.; Gorey-Feret, L.J.; Smith, A.B. Structure-based design and synthesis of HIV-1 protease inhibitors employing β-d-mannopyranoside scaffolds. Bioorg. Med. Chem. Lett. 2002, 12, 1763–1766. [Google Scholar] [CrossRef]
- Liu, H.; Xu, L.; Hui, H.; Vivian, R.; Callebaut, C.; Murray, B.P.; Hong, A.; Lee, M.S.; Tsai, L.K.; Chau, J.K.; et al. Structure-activity relationships of diamine inhibitors of cytochrome P450 (CYP) 3A as novel pharmacoenhancers, part I: Core region. Bioorganic Med. Chem. Lett. 2014, 24, 989–994. [Google Scholar] [CrossRef]
- Tamamura, H.; Omagari, A.; Hiramatsu, K.; Gotoh, K.; Kanamoto, T.; Xu, Y.; Kodama, E.; Matsuoka, M.; Hattori, T.; Yamamoto, N.; et al. Development of specific CXCR4 inhibitors possessing high selectivity indexes as well as complete stability in serum based on an anti-HIV peptide T140. Bioorg. Med. Chem. Lett. 2001, 11, 1897–1902. [Google Scholar] [CrossRef]
- Dong, M.; Zhang, J.; Peng, X.; Lu, H.; Yun, L.; Jiang, S.; Dai, Q. Tricyclononene carboxamide derivatives as novel anti-HIV-1 agents. Eur. J. Med. Chem. 2010, 45, 4096–4103. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Sechi, M.; Bacchi, A.; Carcelli, M.; Compari, C.; Duce, E.; Fisicaro, E.; Rogolino, D.; Gates, P.; Derudas, M.; Al-Mawsawi, L.Q.; et al. From ligand to complexes: Inhibition of human immunodeficiency virus type 1 integrase by beta-diketo acid metal complexes. J. Med. Chem. 2006, 49, 4248–4260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pais, G.C.G.; Svarovskaia, E.S.; Marchand, C.; Johnson, A.A.; Karki, R.G.; Nicklaus, M.C.; Pathak, V.K.; Pommier, Y.; Burke, T.R. Azido-containing aryl beta-diketo acid HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1215–1219. [Google Scholar] [CrossRef]
- Zeng, L.-F.; Jiang, X.-H.; Sanchez, T.; Zhang, H.-S.; Dayam, R.; Neamati, N.; Long, Y.-Q. Novel dimeric aryldiketo containing inhibitors of HIV-1 integrase: Effects of the phenyl substituent and the linker orientation. Bioorg. Med. Chem. 2008, 16, 7777–7787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Marchand, C.; Pommier, Y.; Burke, T.R. Design and synthesis of photoactivatable aryl diketo acid-containing HIV-1 integrase inhibitors as potential affinity probes. Bioorg. Med. Chem. Lett. 2004, 14, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, I.; Avolio, S.; Colarusso, S.; Gennari, N.; Narjes, F.; Pacini, B.; Ponzi, S.; Harper, S. Active site inhibitors of HCV NS5B polymerase. The development and pharmacophore of 2-thienyl-5,6-dihydroxypyrimidine-4-carboxylic acid. Bioorg. Med. Chem. Lett. 2004, 14, 5085–5088. [Google Scholar] [CrossRef] [PubMed]
- Summa, V.; Petrocchi, A.; Pace, P.; Matassa, V.G.; De Francesco, R.; Altamura, S.; Tomei, L.; Koch, U.; Neuner, P. Discovery of alpha,gamma-diketo acids as potent selective and reversible inhibitors of hepatitis C virus NS5b RNA-dependent RNA polymerase. J. Med. Chem. 2004, 47, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.; Gurukumar, K.R.; Basu, A.; Patel, M.R.; Kaushik-Basu, N.; Talele, T.T. Synthesis and SAR optimization of diketo acid pharmacophore for HCV NS5B polymerase inhibition. Eur. J. Med. Chem. 2011, 46, 5138–5145. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R.; Fermeglia, M.; Ferrone, M.; Paneni, M.S.; Costi, R.; Artico, M.; Roux, A.; Gabriele, M.; Tardif, K.D.; Siddiqui, A.; et al. Simple but highly effective three-dimensional chemical-feature-based pharmacophore model for diketo acid derivatives as hepatitis C virus RNA-dependent RNA polymerase inhibitors. J. Med. Chem. 2005, 48, 6304–6314. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, A.; Chopra, R.; Lim, K.; Ciszewski, G.; Shi, M.; Curran, K.J.; Sukits, S.F.; Svenson, K.; Bard, J.; Ellingboe, J.W.; et al. Discovery of proline sulfonamides as potent and selective hepatitis C virus NS5b polymerase inhibitors. Evidence for a new NS5b polymerase binding site. J. Med. Chem. 2006, 49, 3052–3055. [Google Scholar] [CrossRef]
- Vara Prasad, J.V.N.; Boyer, F.E.; Domagala, J.M.; Ellsworth, E.L.; Gajda, C.; Hagen, S.E.; Markoski, L.J.; Tait, B.D.; Lunney, E.A.; Tummino, P.J.; et al. Nonpeptidic HIV protease inhibitors: 6-alkyl-5, 6-dihydropyran-2-ones possessing achiral 3-(4-amino/carboxamide-2-t-butyl, 5-methylphenyl thio) moiety: Antiviral activities and pharmacokinetic properties. Bioorg. Med. Chem. Lett. 1999, 9, 1481–1486. [Google Scholar] [CrossRef]
- Hao, W.; Herlihy, K.J.; Zhang, N.J.; Fuhrman, S.A.; Doan, C.; Patick, A.K.; Duggal, R. Development of a novel dicistronic reporter-selectable hepatitis C virus replicon suitable for high-throughput inhibitor screening. Antimicrob. Agents Chemother. 2007, 51, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, J.; Chen, Z.; Wang, F.; Huang, W.; Hong, Z.; Lin, J. Multistage virtual screening and identification of novel HIV-1 protease inhibitors by integrating SVM, shape, pharmacophore and docking methods. Eur. J. Med. Chem. 2015, 101, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Billamboz, M.; Bailly, F.; Lion, C.; Calmels, C.; Andréola, M.L.; Witvrouw, M.; Christ, F.; Debyser, Z.; De Luca, L.; Chimirri, A.; et al. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: Influence of the alkylation of position 4. Eur. J. Med. Chem. 2011, 46, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- Adrià, C.M.; Garcia-Vallvé, S.; Pujadas, G. DecoyFinder, a tool for finding decoy molecules. J. Cheminform. 2012, 4, P2. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz information miner: Version 2 and Beyond. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef]
- Grisoni, F.; Consonni, V.; Todeschini, R. Impact of Molecular Descriptors on Computational Models. Methods Mol. Biol. 2018, 1825, 171–209. [Google Scholar]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef]
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E. Recent Developments of the Chemistry Development Kit (CDK)—An Open-Source Java Library for Chemo- and Bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, L. Improving SVM through a Risk Decision Rule Running on MATLAB. J. Softw. 2012, 7, 2252–2257. [Google Scholar] [CrossRef]
- Yang, C.; Odvody, G.N.; Fernandez, C.J.; Landivar, J.A.; Minzenmayer, R.R.; Nichols, R.L. Evaluating unsupervised and supervised image classification methods for mapping cotton root rot. Precis. Agric. 2015, 16, 201–215. [Google Scholar] [CrossRef]
- Bouzalmat, A.; Kharroubi, J.; Zarghili, A. Face Recognition Using SVM Based on LDA. Int. J. Comput. Sci. Issues 2013, 10, 171–179. [Google Scholar]
- Ballabio, D.; Grisoni, F.; Todeschini, R. Multivariate comparison of classification performance measures. Chemom. Intell. Lab. Syst. 2018, 174, 33–44. [Google Scholar] [CrossRef]
- Glide. Schrödinger, Version 6.7; LLC: New York, NY, USA, 2015. [Google Scholar]
- Fader, L.D.; Malenfant, E.; Parisien, M.; Carson, R.; Bilodeau, F.; Landry, S.; Pesant, M.; Brochu, C.; Morin, S.; Chabot, C.; et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.A.; Andrews Iii, C.W.; Hopkins, A.L.; Lowell, G.S.; Schaller, L.T.; Cowan, J.R.; Gonzales, S.S.; Koszalka, G.W.; Hazen, R.J.; Boone, L.R.; et al. Design of non-nucleoside inhibitors of HIV-1 reverse transcriptase with improved drug resistance properties. 2. J. Med. Chem. 2004, 47, 5923–5936. [Google Scholar] [CrossRef]
- Ganguly, A.K.; Alluri, S.S.; Wang, C.-H.; Antropow, A.; White, A.; Caroccia, D.; Biswas, D.; Kang, E.; Zhang, L.-K.; Carroll, S.S.; et al. Structural optimization of cyclic sulfonamide based novel HIV-1 protease inhibitors to picomolar affinities guided by X-ray crystallographic analysis. Tetrahedron 2014, 70, 2894–2904. [Google Scholar] [CrossRef]
- Love, R.A.; Parge, H.E.; Yu, X.; Hickey, M.J.; Diehl, W.; Gao, J.; Wriggers, H.; Ekker, A.; Wang, L.; Thomson, J.A.; et al. Crystallographic identification of a noncompetitive inhibitor binding site on the hepatitis C virus NS5B RNA polymerase enzyme. J. Virol. 2003, 77, 7575–7581. [Google Scholar] [CrossRef]
- Bressanelli, S.; Tomei, L.; Rey, F.A.; De Francesco, R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 2002, 76, 3482–3492. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Object | Target | Training Set | Test Set | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Inhibitors | Decoys | Total | Tanimoto Similarity Index | Inhibitors | Decoys | Total | Tanimoto Similarity Index | ||
| HIV-1 | CXCR4 | 113 | 339 | 452 | 0.152 | 44 | 132 | 176 | 0.158 |
| CCR5 | 255 | 766 | 1021 | 0.161 | 115 | 344 | 459 | 0.160 | |
| GP120 | 208 | 624 | 832 | 0.142 | 85 | 255 | 340 | 0.147 | |
| GP41 | 14 | 42 | 56 | 0.172 | 7 | 21 | 28 | 0.189 | |
| RT | 2279 | 6837 | 9116 | 0.124 | 977 | 2931 | 3908 | 0.124 | |
| IN | 1617 | 4850 | 6467 | 0.134 | 598 | 1795 | 2393 | 0.135 | |
| PR | 2837 | 8509 | 11,346 | 0.141 | 1274 | 3824 | 5098 | 0.141 | |
| Gag-pol | 69 | 205 | 274 | 0.147 | 32 | 98 | 130 | 0.146 | |
| Protein tat | 302 | 906 | 1208 | 0.118 | 136 | 408 | 544 | 0.117 | |
| PKC | 20 | 58 | 78 | 0.162 | 9 | 29 | 38 | 0.169 | |
| CYP3A | 10 | 30 | 40 | 0.202 | 5 | 15 | 20 | 0.215 | |
| HCV | NS5B | 649 | 1948 | 2597 | 0.141 | 253 | 758 | 1011 | 0.141 |
| NS4B | 13 | 37 | 50 | 0.178 | 6 | 20 | 26 | 0.192 | |
| NS3/4A | 334 | 1000 | 1334 | 0.145 | 136 | 410 | 546 | 0.145 | |
| NS5A | 27 | 80 | 107 | 0.182 | 13 | 40 | 53 | 0.182 | |
| Object | Target | MACCS | ECFP6 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NB | SVM | NB | SVM | ||||||
| AUC | NER | AUC | NER | AUC | NER | AUC | NER | ||
| HIV-1 | CXCR4 | 0.992 | 0.962 | 0.998 | 0.978 | 0.994 | 0.969 | 0.997 | 0.973 |
| CCR5 | 0.977 | 0.878 | 0.997 | 0.990 | 0.986 | 0.936 | 0.996 | 0.976 | |
| GP120 | 0.845 | 0.799 | 0.926 | 0.851 | 0.880 | 0.775 | 0.923 | 0.737 | |
| GP41 | 0.941 | 0.964 | 0.958 | 0.964 | 0.963 | 0.964 | 0.934 | 0.786 | |
| RT | 0.838 | 0.763 | 0.994 | 0.960 | 0.898 | 0.797 | 0.994 | 0.955 | |
| IN | 0.930 | 0.863 | 0.996 | 0.976 | 0.949 | 0.878 | 0.994 | 0.960 | |
| PR | 0.980 | 0.908 | 0.999 | 0.987 | 0.982 | 0.925 | 0.997 | 0.987 | |
| Gag-pol | 0.966 | 0.942 | 0.984 | 0.949 | 0.967 | 0.925 | 0.980 | 0.935 | |
| Protein tat | 0.834 | 0.745 | 0.909 | 0.792 | 0.870 | 0.805 | 0.901 | 0.739 | |
| PKC | 0.991 | 0.991 | 1 | 0.975 | 0.994 | 0.925 | 0.988 | 0.925 | |
| CYP3A | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0.950 | |
| HCV | NS3/4A | 0.990 | 0.853 | 1 | 0.993 | 0.994 | 0.953 | 1 | 0.987 |
| NS4B | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0.885 | |
| NS5A | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| NS5B | 0.924 | 0.863 | 1 | 0.987 | 0.946 | 0.801 | 1 | 0.984 | |
| Object | Target | MACCS | ECFP6 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NB | SVM | NB | SVM | ||||||
| AUC | NER | AUC | NER | AUC | NER | AUC | NER | ||
| HIV-1 | CXCR4 | 1 | 0.989 | 1 | 0.989 | 1 | 0.989 | 1 | 0.989 |
| CCR5 | 0.981 | 0.891 | 0.999 | 0.990 | 0.995 | 0.926 | 1 | 0.978 | |
| GP120 | 0.881 | 0.784 | 0.940 | 0.861 | 0.883 | 0.775 | 0.910 | 0.759 | |
| GP41 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| RT | 0.837 | 0.745 | 0.995 | 0.964 | 0.899 | 0.799 | 0.994 | 0.963 | |
| IN | 0.934 | 0.856 | 0.997 | 0.979 | 0.947 | 0.876 | 0.996 | 0.972 | |
| PR | 0.975 | 0.897 | 1 | 0.991 | 0.982 | 0.932 | 0.999 | 0.987 | |
| Gag-pol | 0.977 | 0.938 | 0.988 | 0.969 | 0.975 | 0.923 | 1 | 0.969 | |
| Protein tat | 0.833 | 0.771 | 0.920 | 0.810 | 0.864 | 0.803 | 0.926 | 0.749 | |
| PKC | 1 | 1 | 1 | 0.889 | 1 | 0.944 | 1 | 0.889 | |
| CYP3A | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0.900 | |
| HCV | NS3/4A | 0.986 | 0.836 | 1 | 0.996 | 0.987 | 0.946 | 1 | 0.971 |
| NS4B | 0.842 | 0.833 | 0.925 | 0.833 | 0.867 | 0.833 | 0.908 | 0.833 | |
| NS5A | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| NS5B | 0.907 | 0.837 | 1 | 0.984 | 0.944 | 0.788 | 1 | 0.988 | |
| Drug Name | Predicted Target | Known Activity | Reference |
|---|---|---|---|
| Enfuvirtide | GP41 | IC50 = 90.2 nM | [35] |
| Delavirdine | RT | IC50 = 0.17 μM | [36] |
| Didanosine | RT | IC50 = 4.6 μM | [42] |
| Lamivudine | RT | IC50 = 0.60 μM | [42] |
| Stavudine | RT | IC50 = 1.9 μM | [42] |
| Zalcitabine | RT | IC50 = 0.13 μM | [42] |
| Zidovudine | RT | IC50 = 0.13 μM | [42] |
| Dolutegravir | RT | Inactive | [50] |
| Dolutegravir | IN | IC50 = 2.7 nM | [43] |
| Elvitegravir | RT | IC50 = 91 μM | [44] |
| Elvitegravir | IN | IC50 = 28 nM | [44] |
| Amprenavir | PR | IC50 = 0.15 μM | [45] |
| Atazanavir | RT | IC50 = 26 nM | [46] |
| Atazanavir | PR | IC50 = 3.5 nM | [47] |
| Darunavir | PR | IC50 = 3.0 nM | [47] |
| Indinavir | RT | Inconclusive | [51] |
| Indinavir | PR | IC50 < 0.01 μM | [51] |
| Nelfinavir | PR | IC50 = 0.53 nM | [48] |
| Ritonavir | PR | IC50 = 0.6 nM | [49] |
| Ritonavir | CYP3A | IC50 = 0.11 μM | [49] |
| Tipranavir | PR | IC50 = 0.03 μM | [37] |
| Cobicistat | PR | IC50 = 0.15 μM | [49] |
| Cobicistat | CYP3A | IC50 > 30 μM | [49] |
| Telaprevir | NS3/4A | IC50 = 0.2 μM | [38] |
| Simeprevir | NS3/4A | IC50 = 10 nM | [39] |
| Grazoprevir | NS3/4A | IC50 = 0.07 nM | [40] |
| Dasabuvir | NS5B | IC50 = 0.6 μM | [41] |
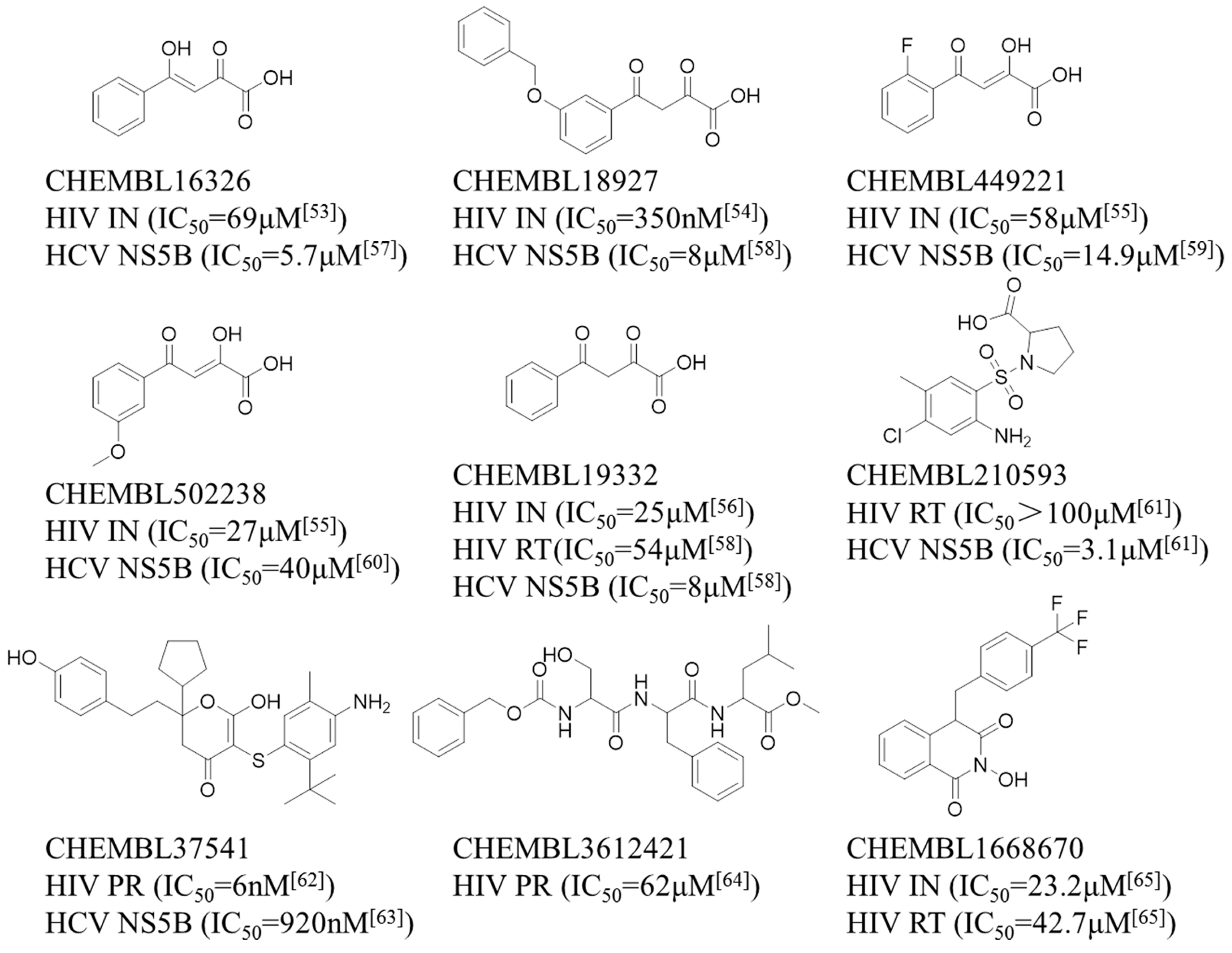
| CHEMBL ID | PR/Pred 1 | PR/Exp 2 | IN/Pred 3 | IN/Exp 4 | RT/Pred 5 | RT/Exp 6 | NS5B/Pred 7 | NS5B/Exp 8 |
|---|---|---|---|---|---|---|---|---|
| CHEMBL16326 | false | — 9 | true | active | false | — | true | active |
| CHEMBL18927 | false | — | true | active | false | — | true | active |
| CHEMBL449221 | false | — | true | active | false | — | true | active |
| CHEMBL502238 | false | — | true | active | false | — | true | active |
| CHEMBL19332 | false | — | true | active | false | active | true | active |
| CHEMBL210593 | false | — | false | — | true | active | true | active |
| CHEMBL37541 | true | active | false | — | false | — | true | active |
| CHEMBL3612421 | true | active | false | — | false | — | true | — |
| CHEMBL1668670 | false | — | true | active | true | active | true | — |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Li, W.; Du, T.; Hong, Z.; Lin, J. Targeting HIV/HCV Coinfection Using a Machine Learning-Based Multiple Quantitative Structure-Activity Relationships (Multiple QSAR) Method. Int. J. Mol. Sci. 2019, 20, 3572. https://doi.org/10.3390/ijms20143572
Wei Y, Li W, Du T, Hong Z, Lin J. Targeting HIV/HCV Coinfection Using a Machine Learning-Based Multiple Quantitative Structure-Activity Relationships (Multiple QSAR) Method. International Journal of Molecular Sciences. 2019; 20(14):3572. https://doi.org/10.3390/ijms20143572
Chicago/Turabian StyleWei, Yu, Wei Li, Tengfei Du, Zhangyong Hong, and Jianping Lin. 2019. "Targeting HIV/HCV Coinfection Using a Machine Learning-Based Multiple Quantitative Structure-Activity Relationships (Multiple QSAR) Method" International Journal of Molecular Sciences 20, no. 14: 3572. https://doi.org/10.3390/ijms20143572
APA StyleWei, Y., Li, W., Du, T., Hong, Z., & Lin, J. (2019). Targeting HIV/HCV Coinfection Using a Machine Learning-Based Multiple Quantitative Structure-Activity Relationships (Multiple QSAR) Method. International Journal of Molecular Sciences, 20(14), 3572. https://doi.org/10.3390/ijms20143572