Chemi-Net: A Molecular Graph Convolutional Network for Accurate Drug Property Prediction

Abstract
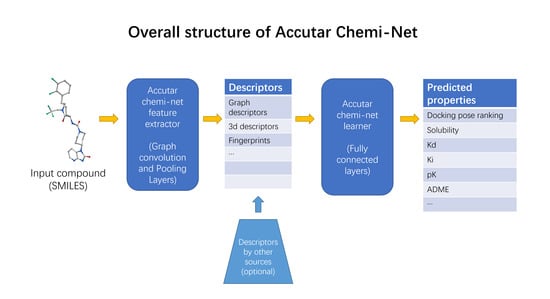
:
1. Introduction
2. Results
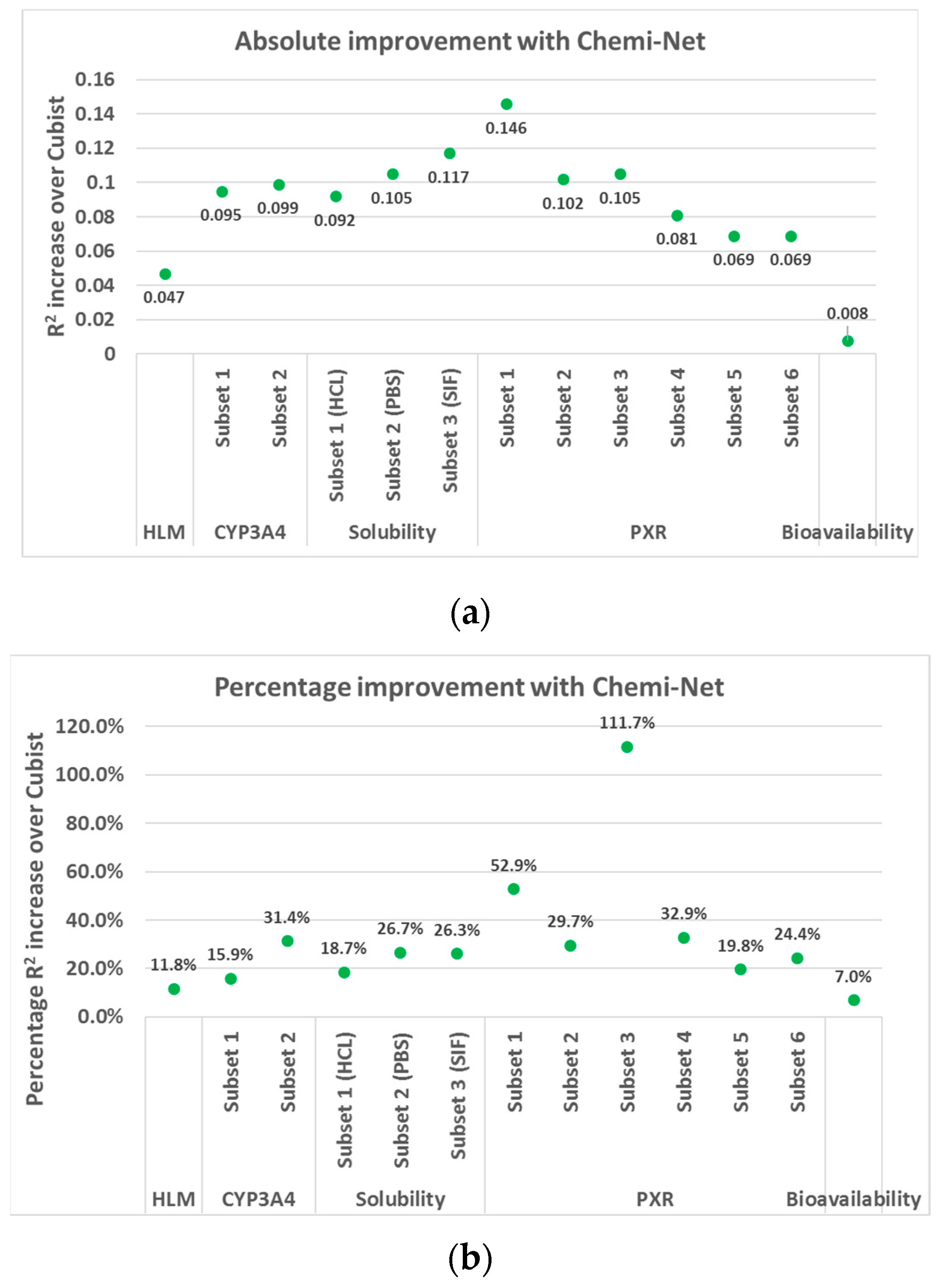
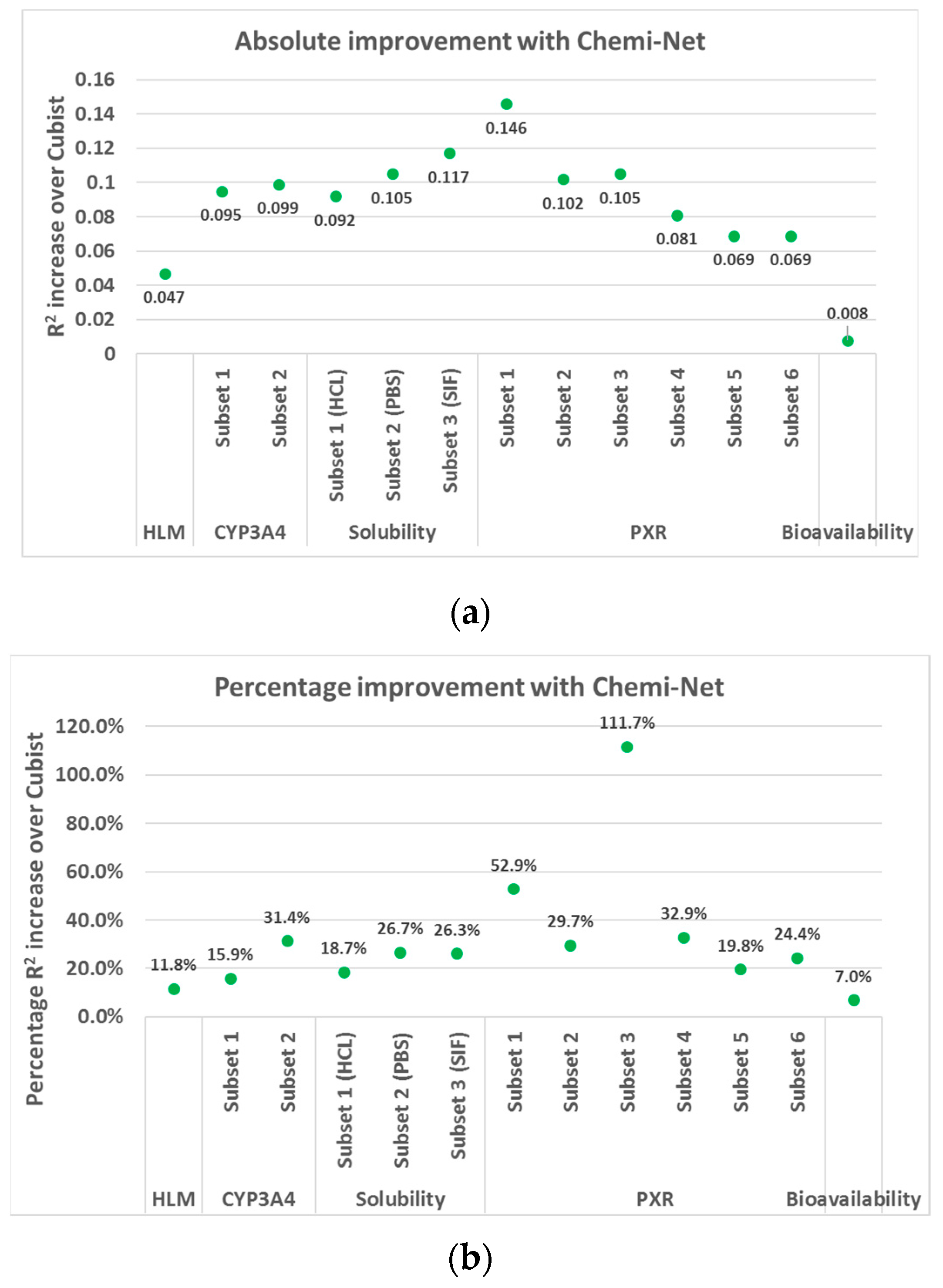
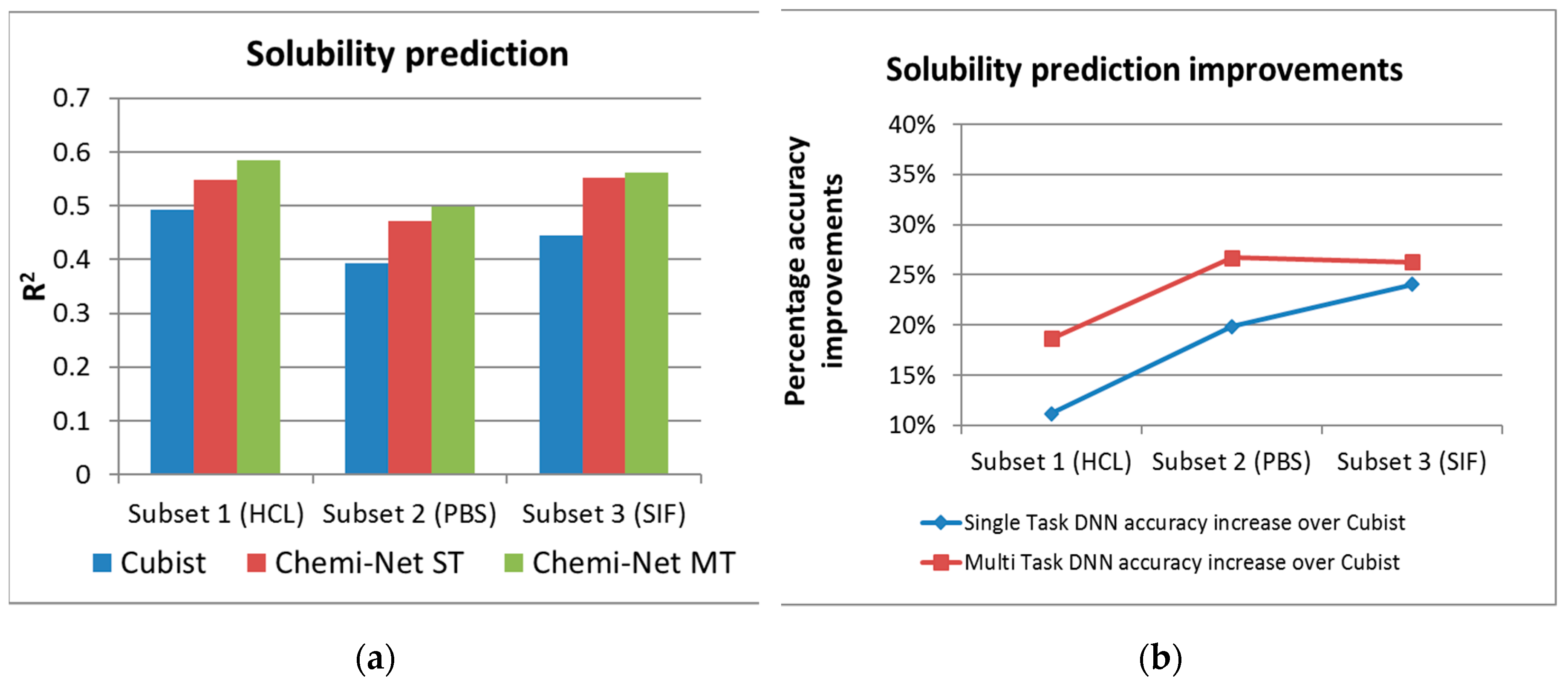
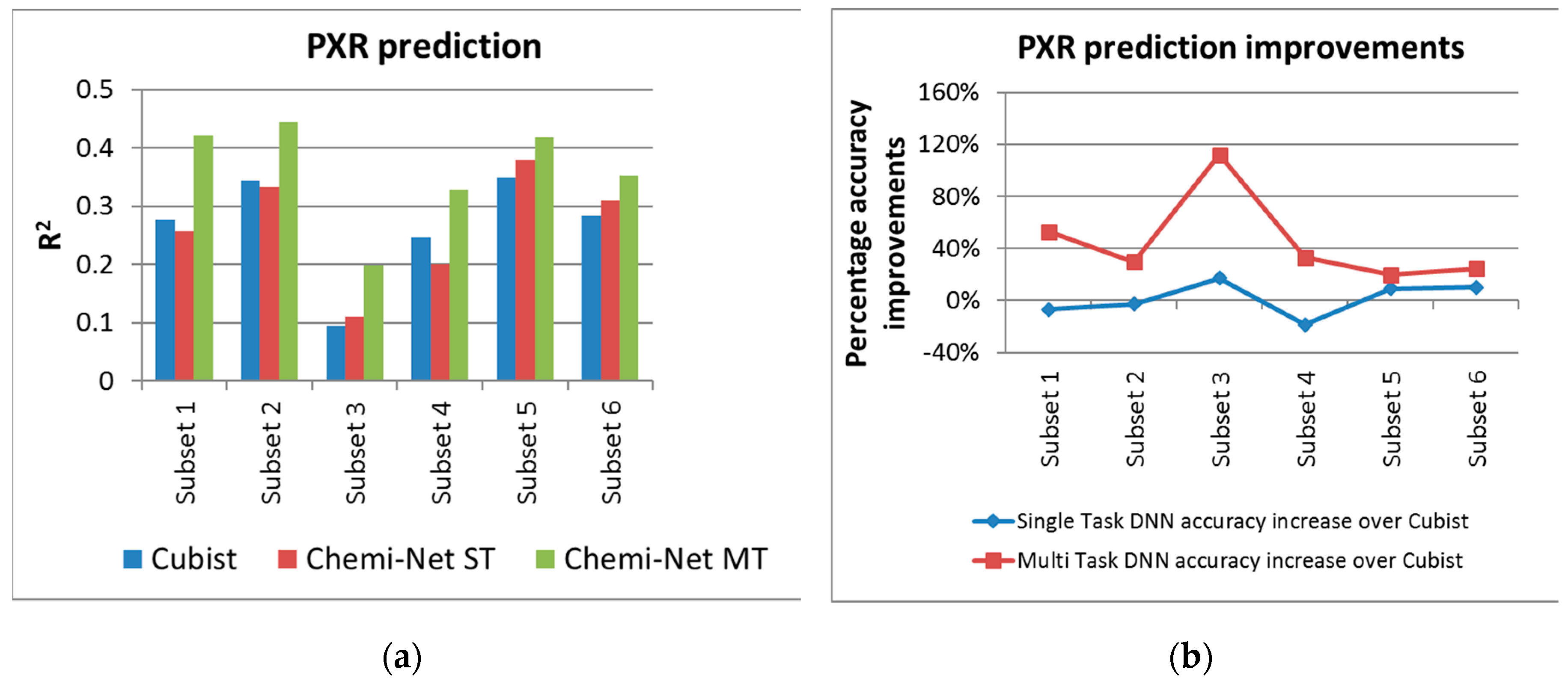
2.1. MT-DNN Method of Chemi-Net Improves Predictive Accuracy Comparing to Cubist
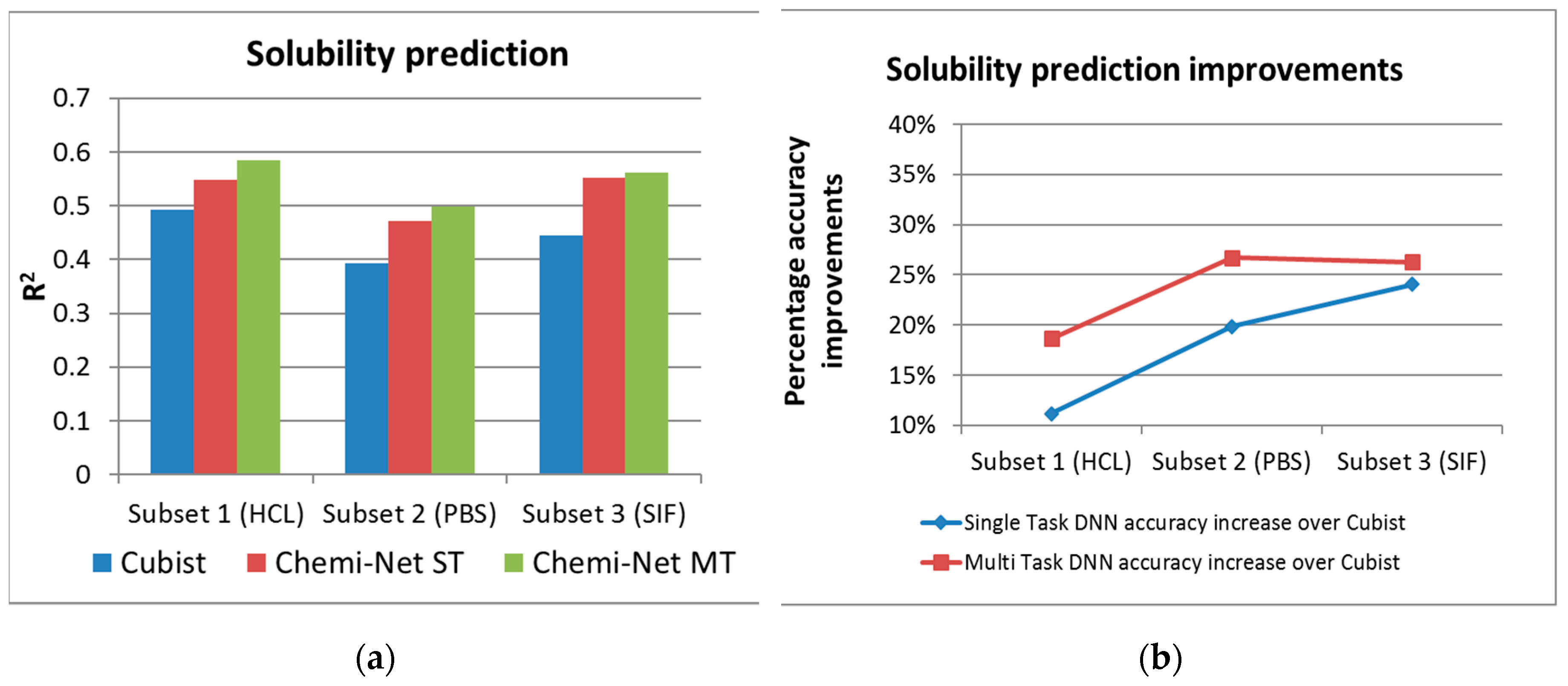
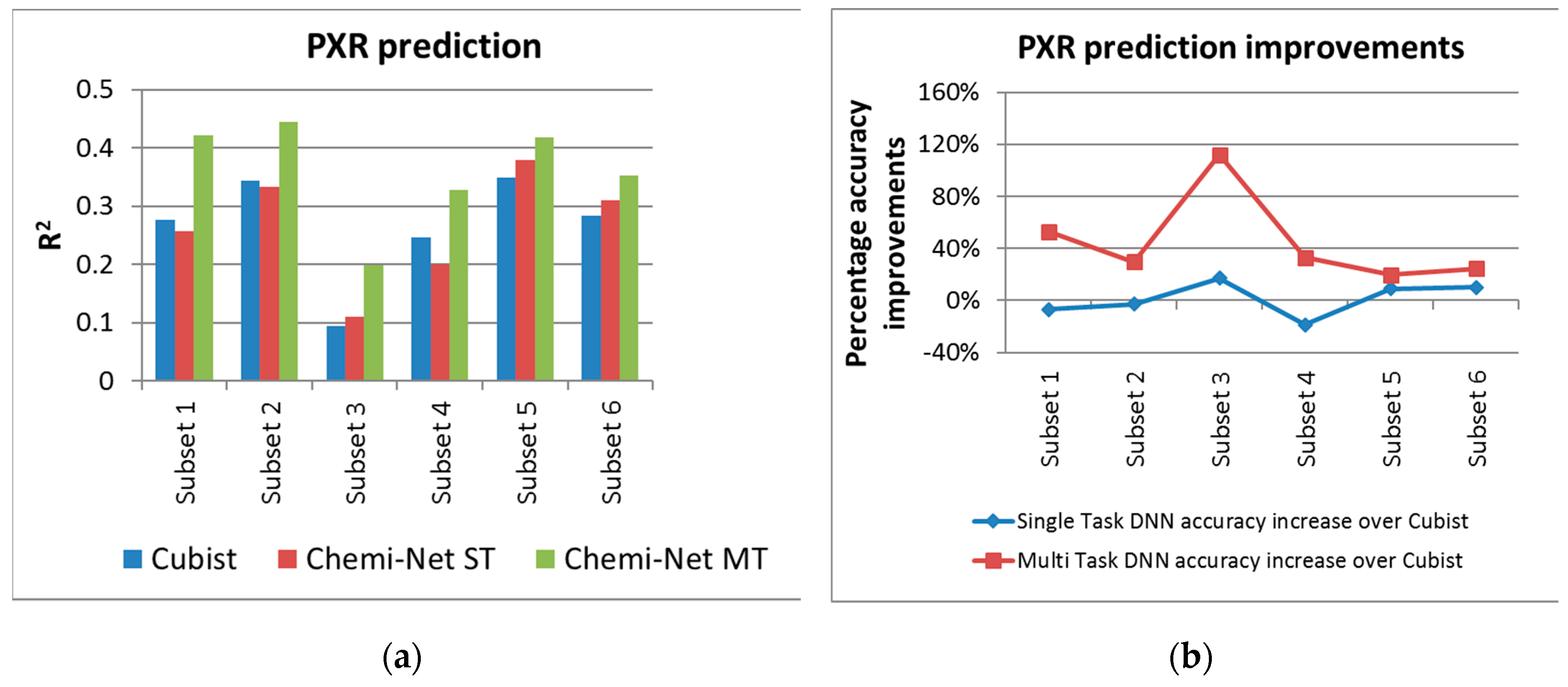
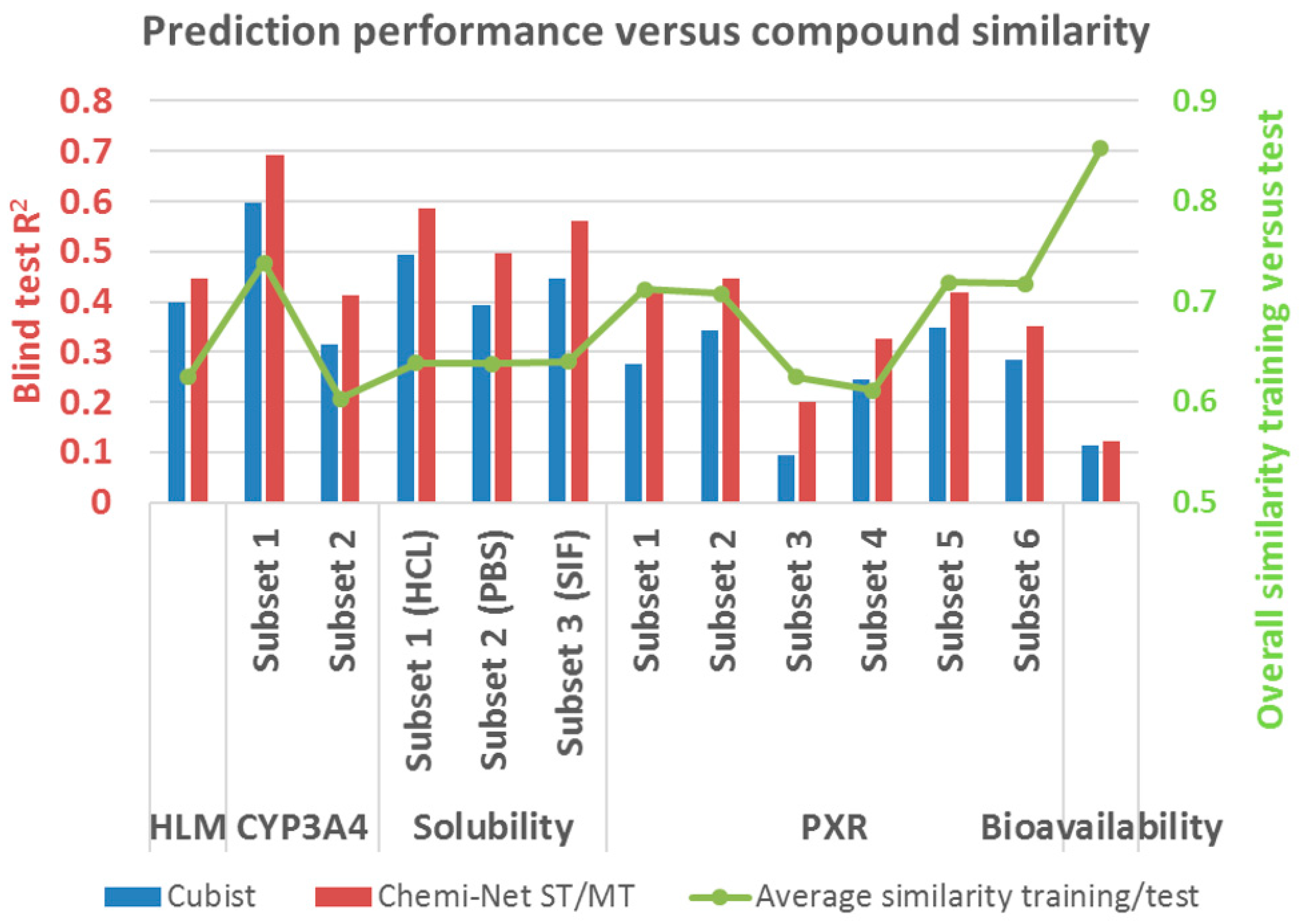
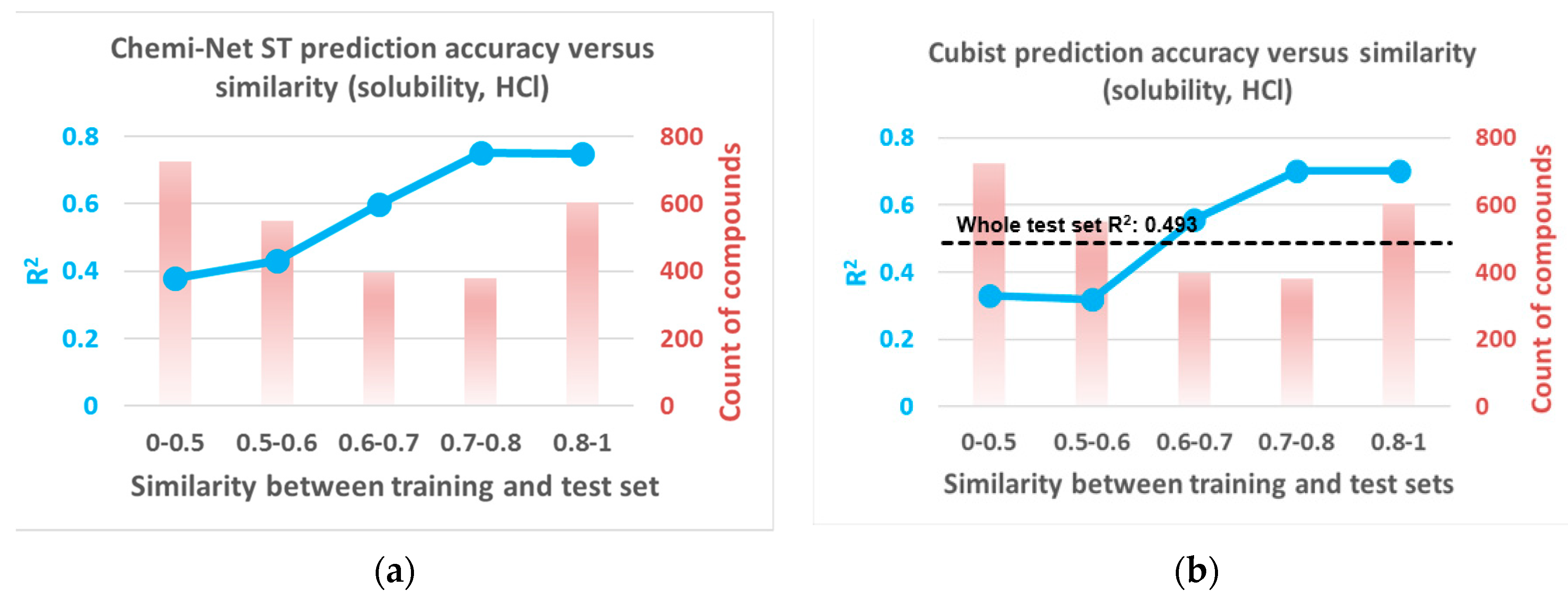
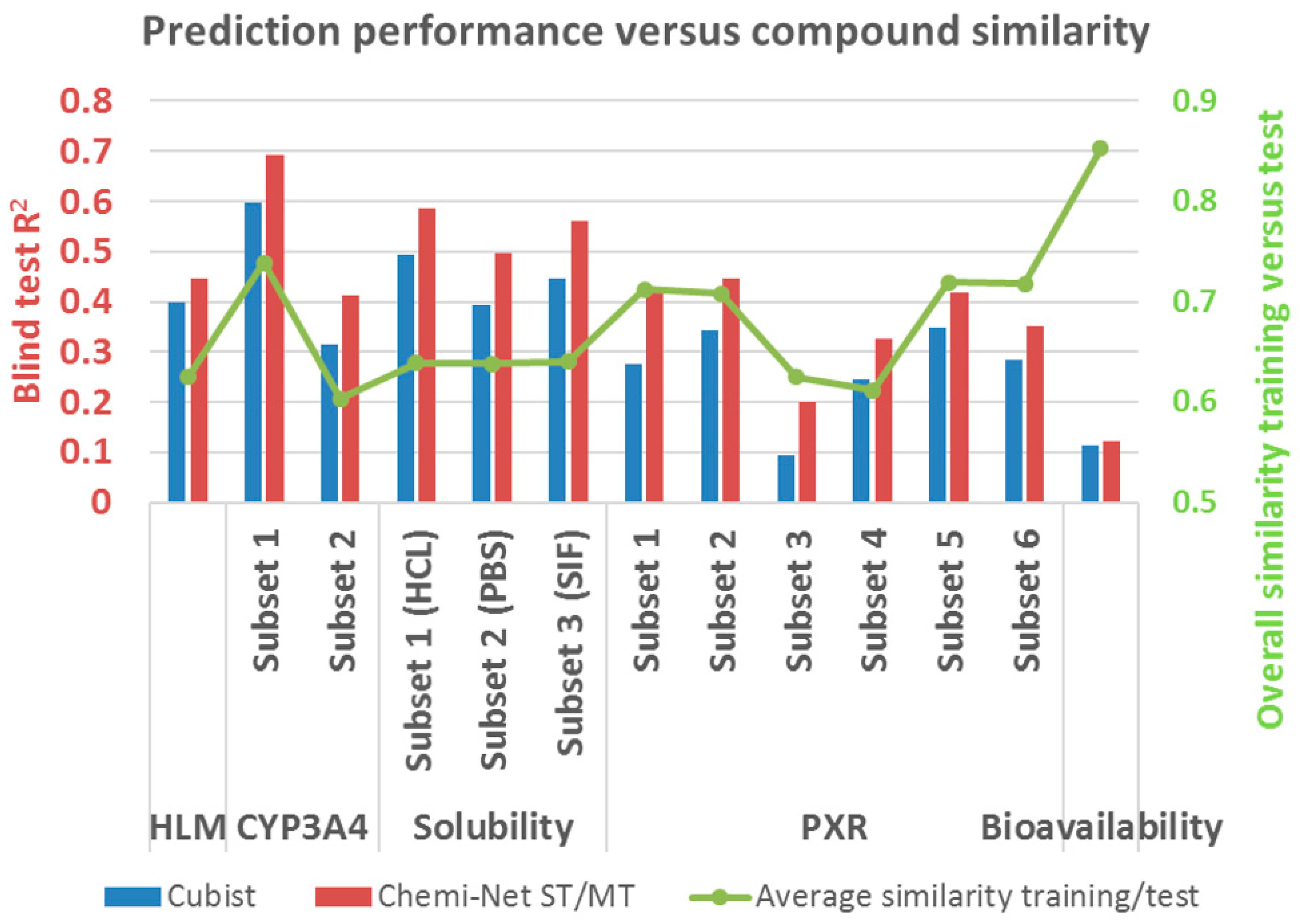
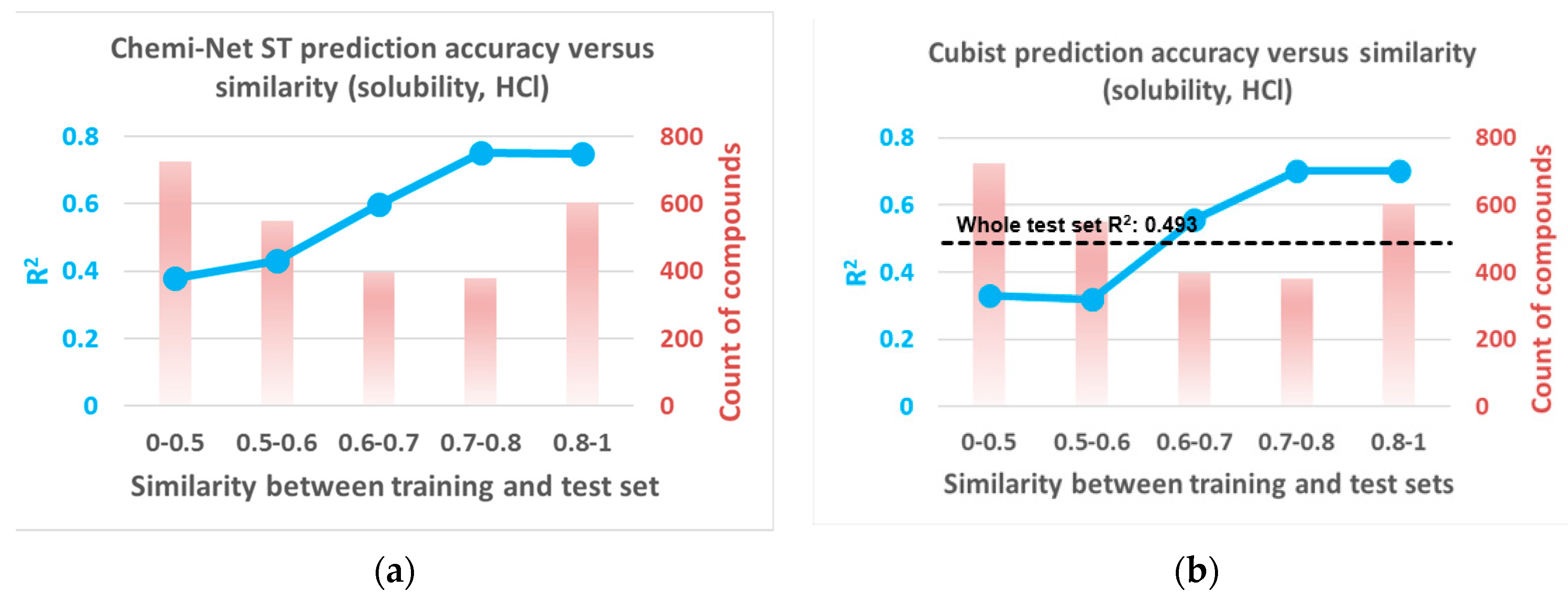
2.2. Prediction Performance and Compound Similarity
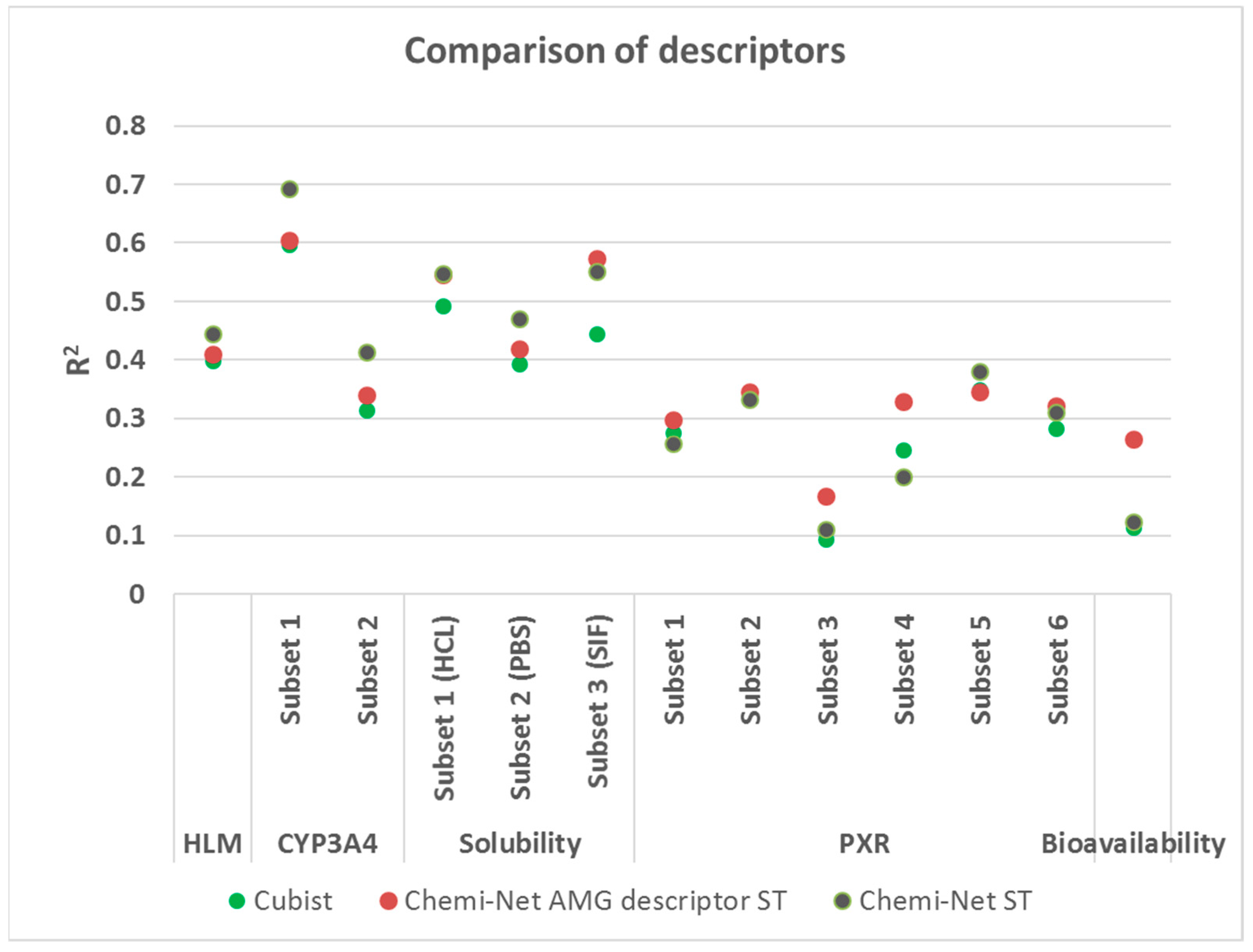
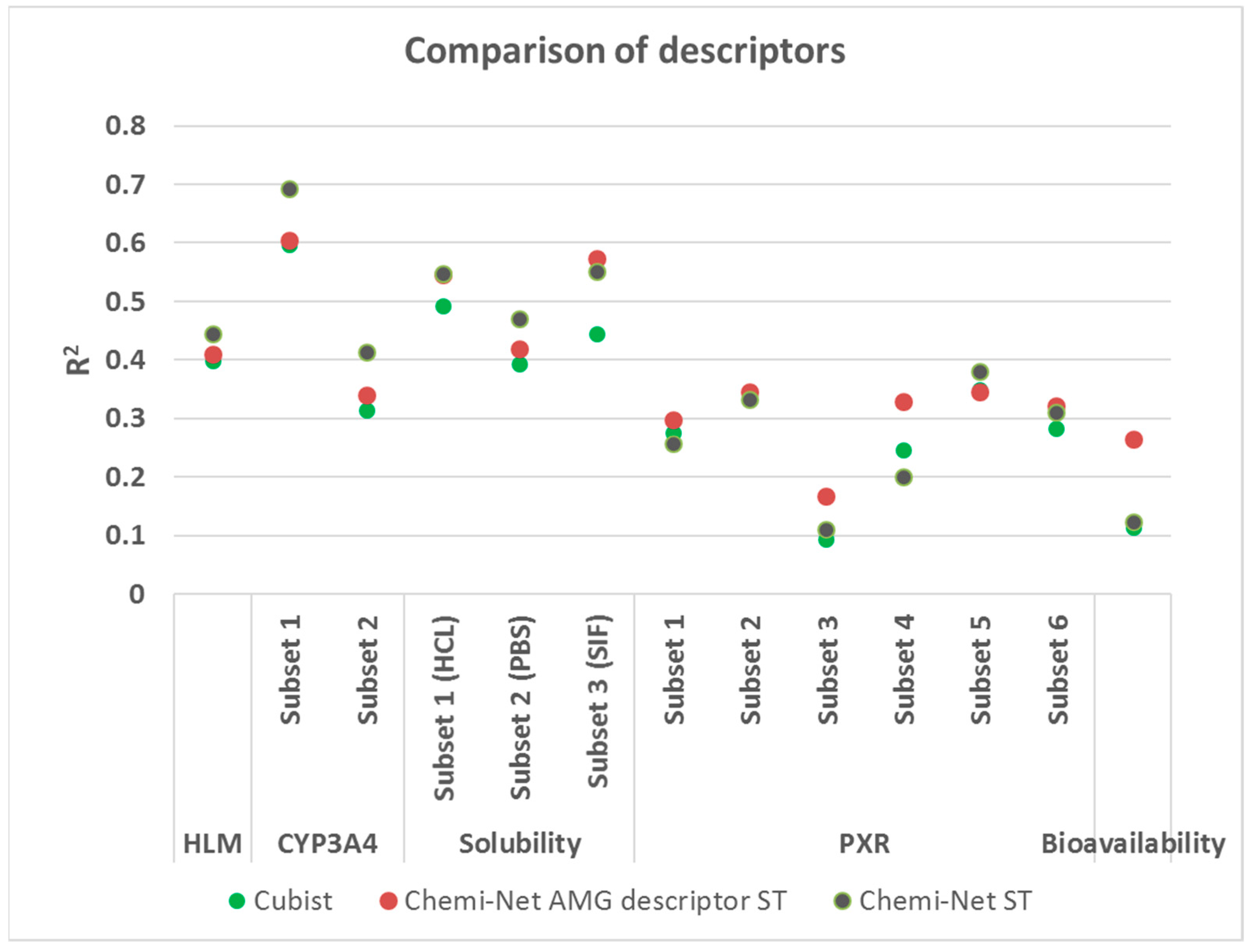
2.3. Comparison between Chemi-Net’s Descriptors and Amgen’s Traditional Property and Molecular Keys Descriptors
2.4. Chemi-Net’s Performance on Public Data Sets
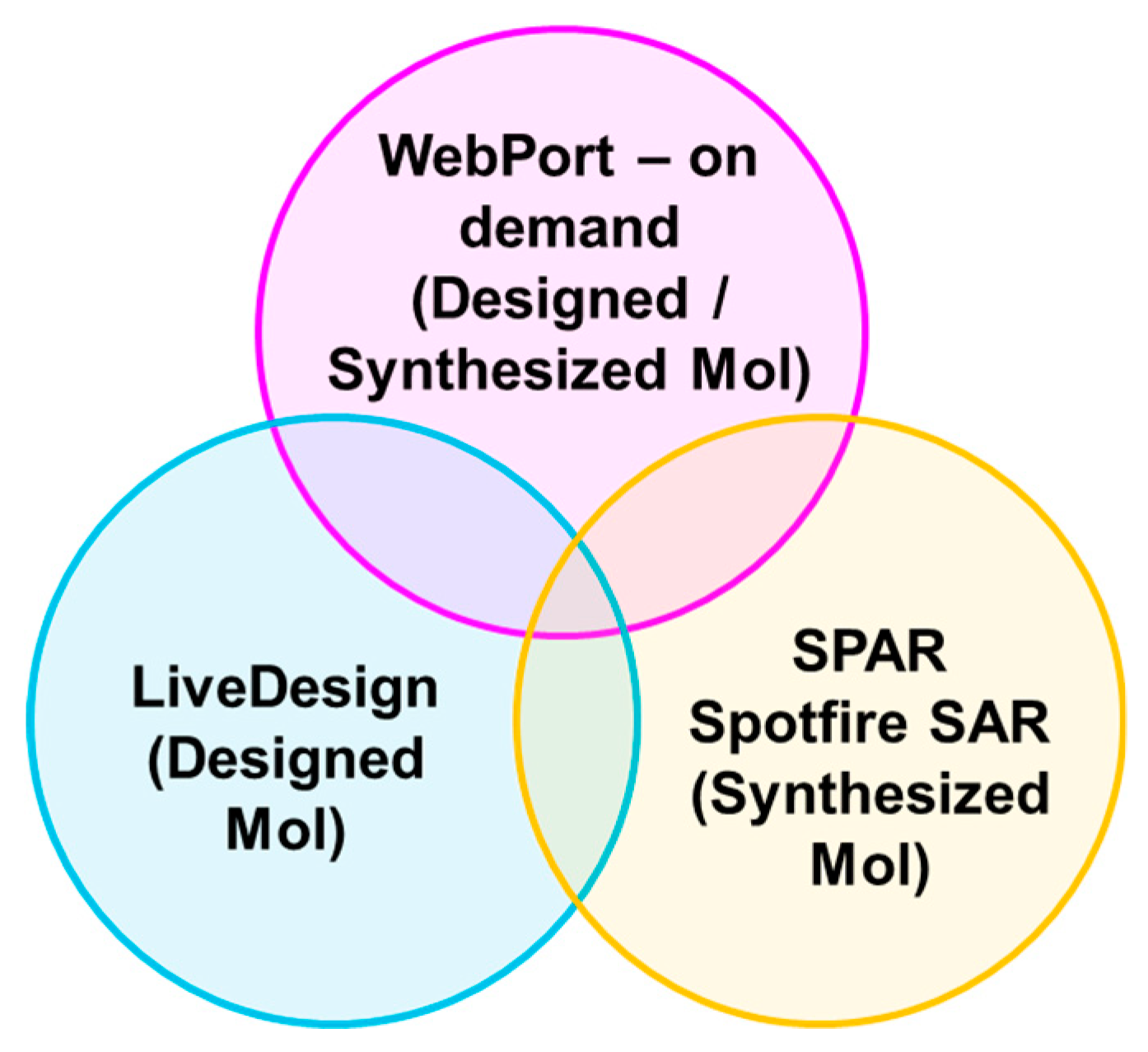
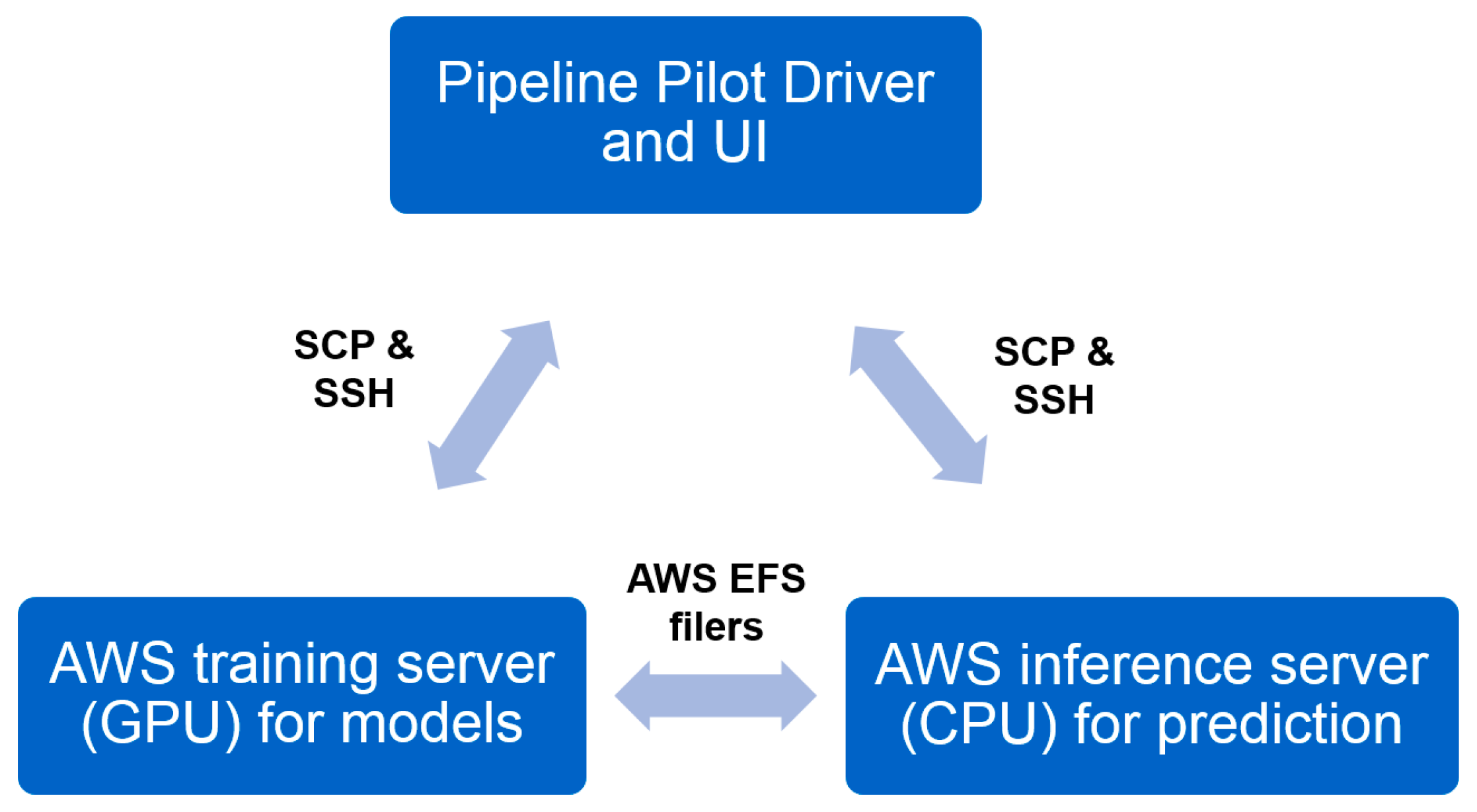
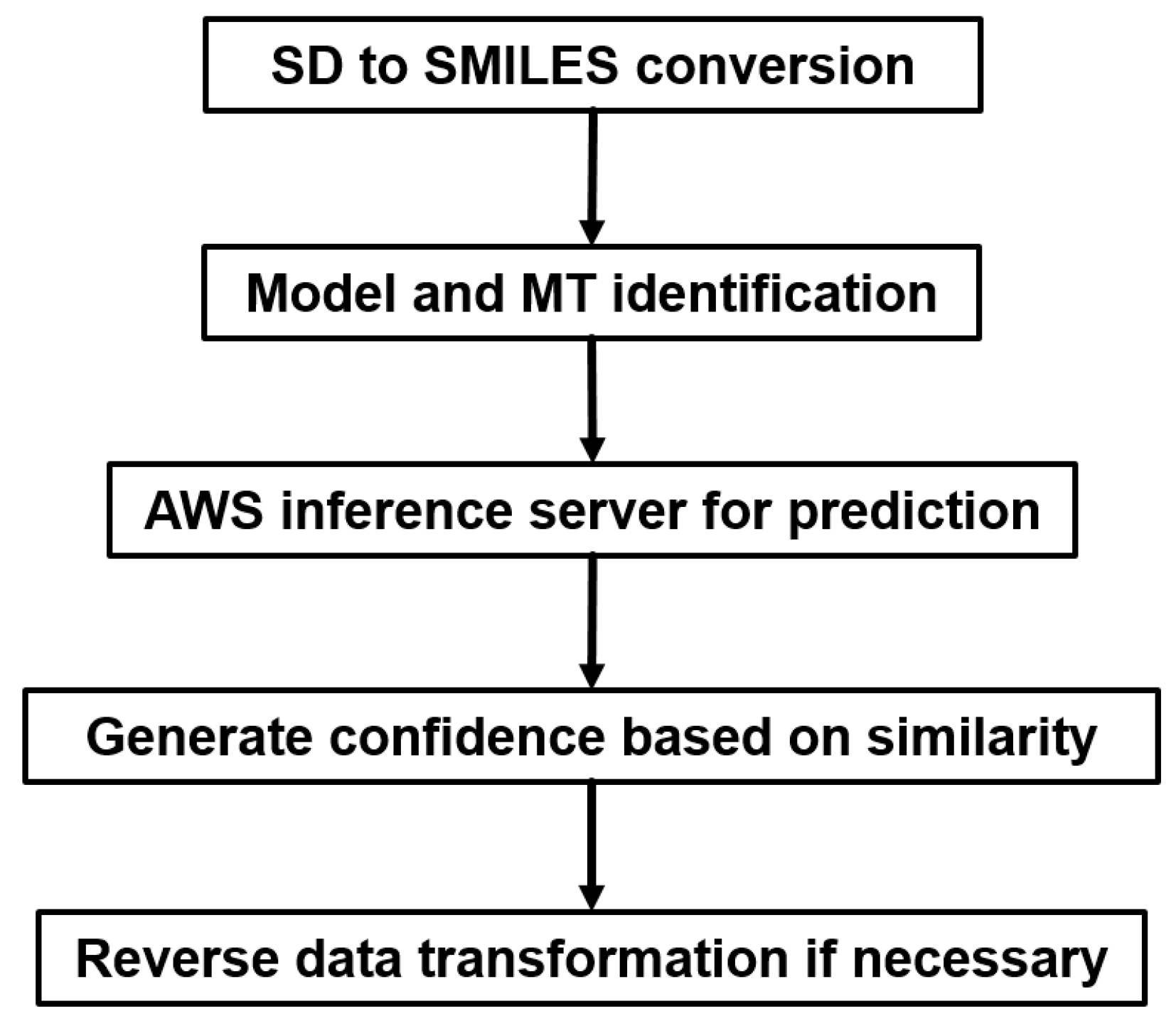
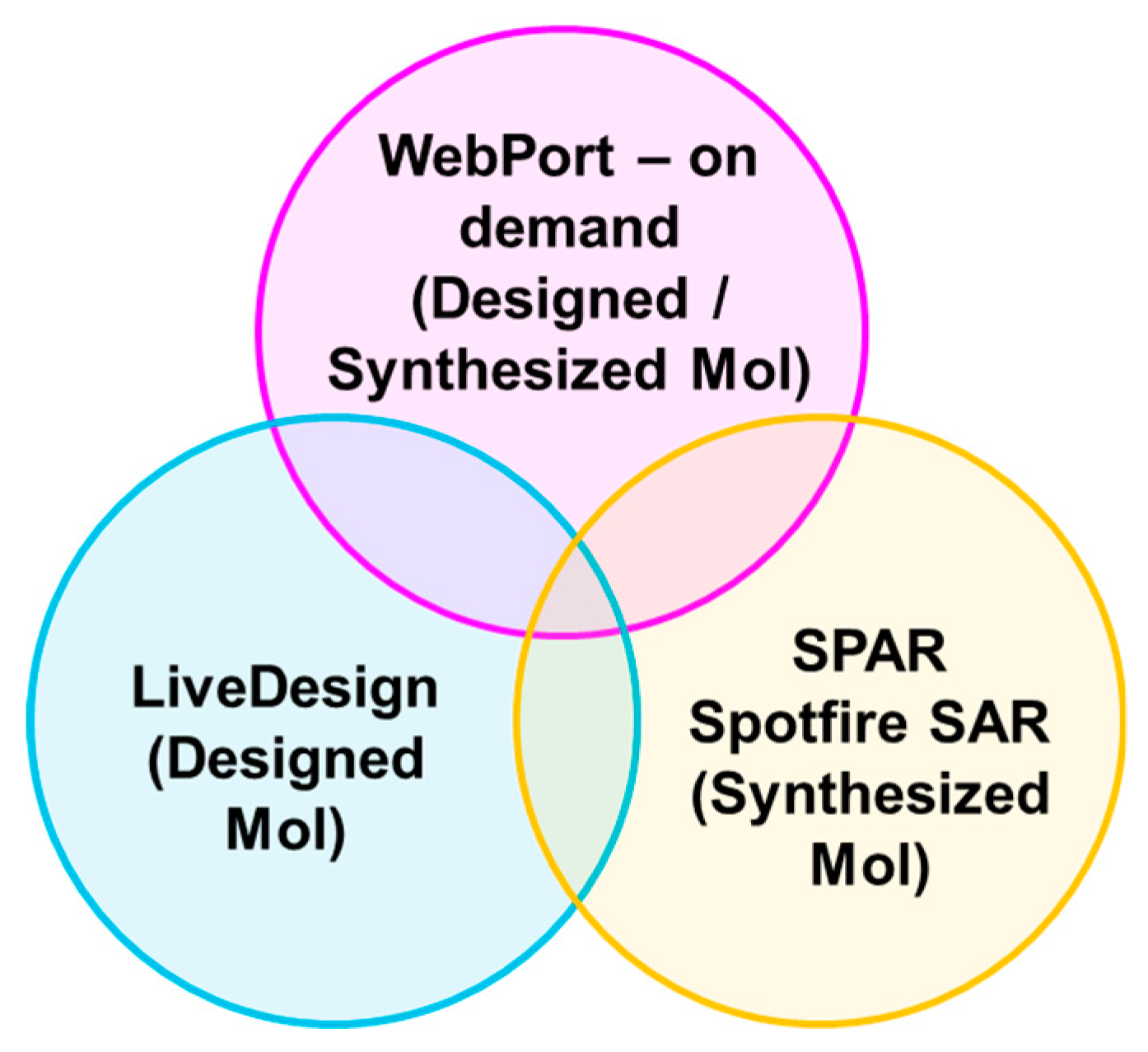
2.5. Industrial Implementation of Chemi-Net for ADME Prediction
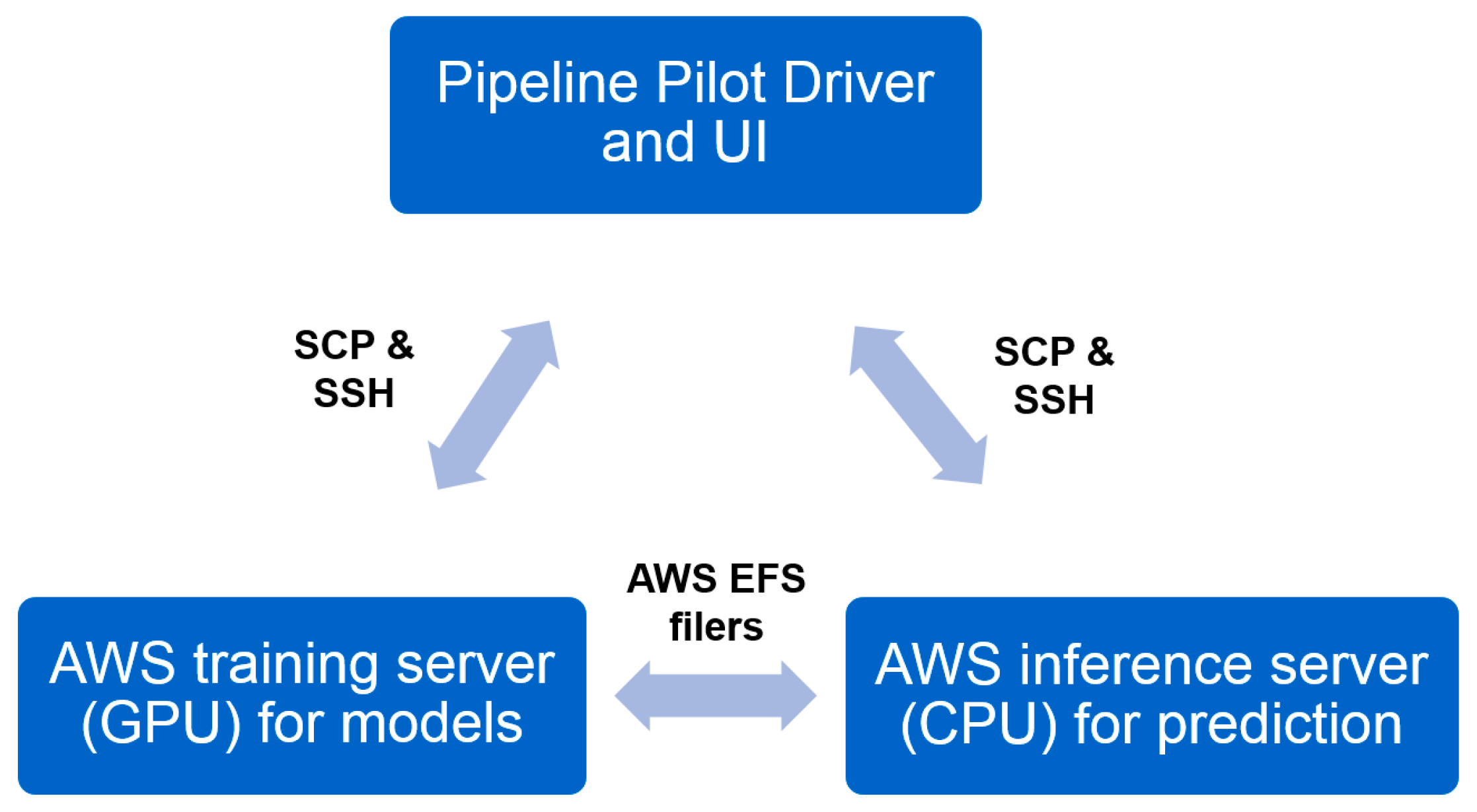
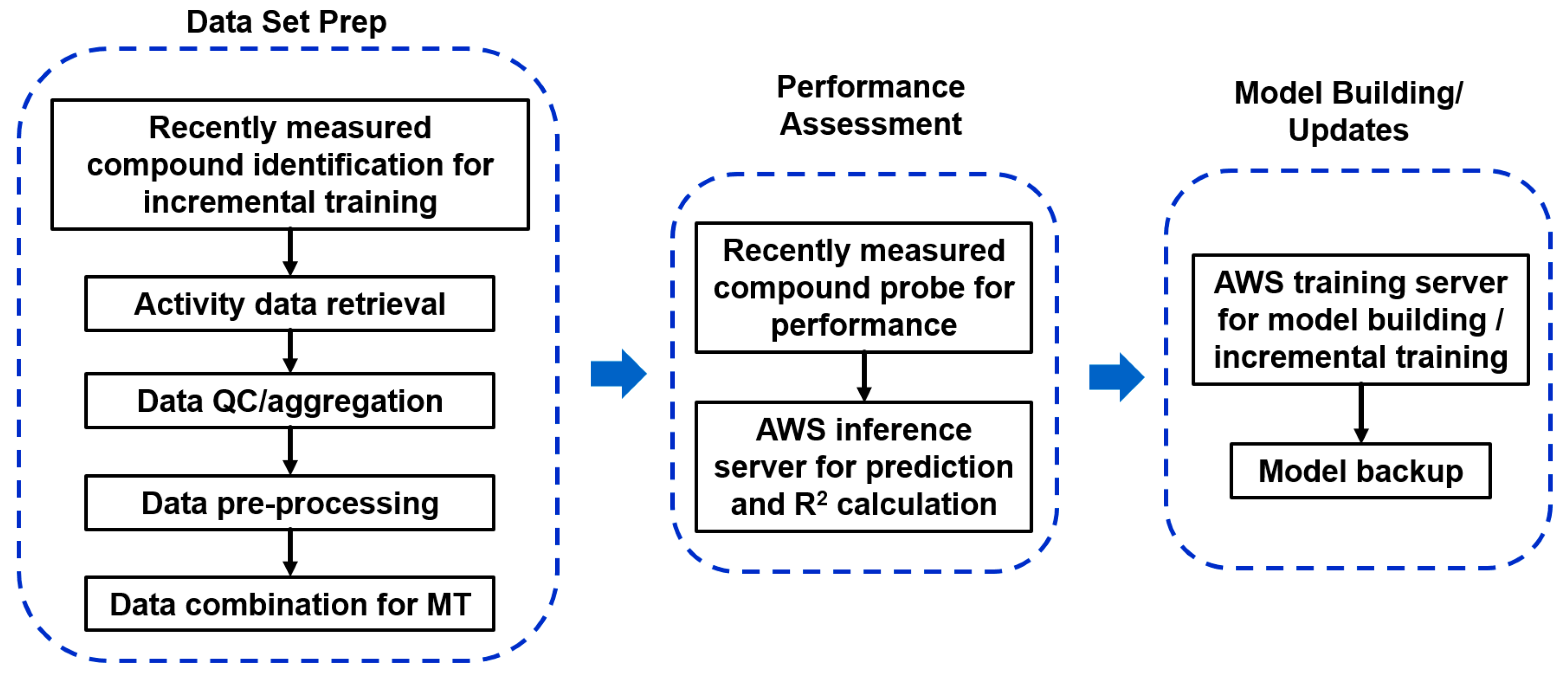
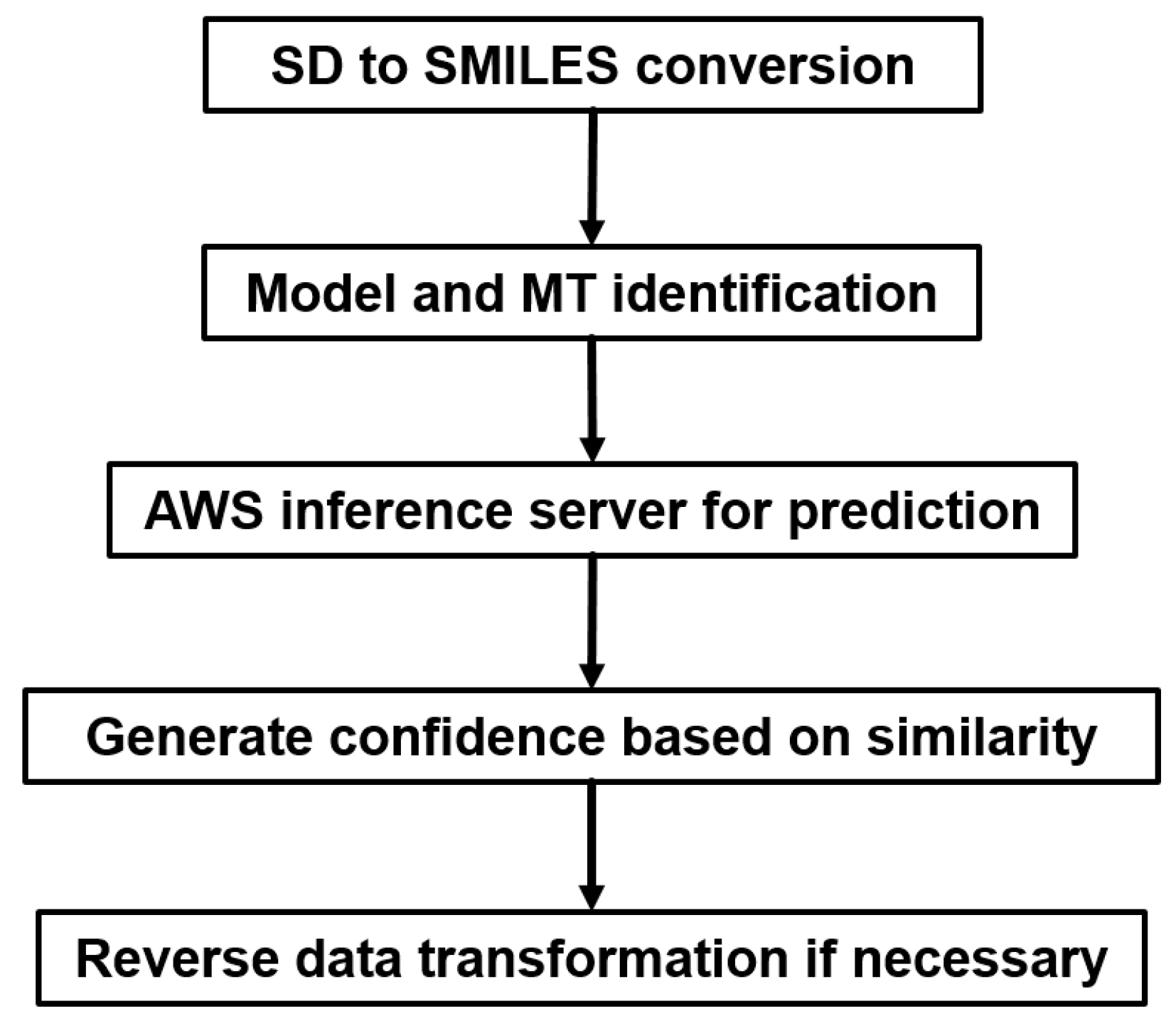
2.5.1. Heavy Computing with on Demand Prediction Request
2.5.2. Large Data Set Assembly and Processing
2.5.3. Incorporation to Amgen’s Existing ADME Prediction Service
2.5.4. Additional Features
3. Discussion
4. Materials and Methods
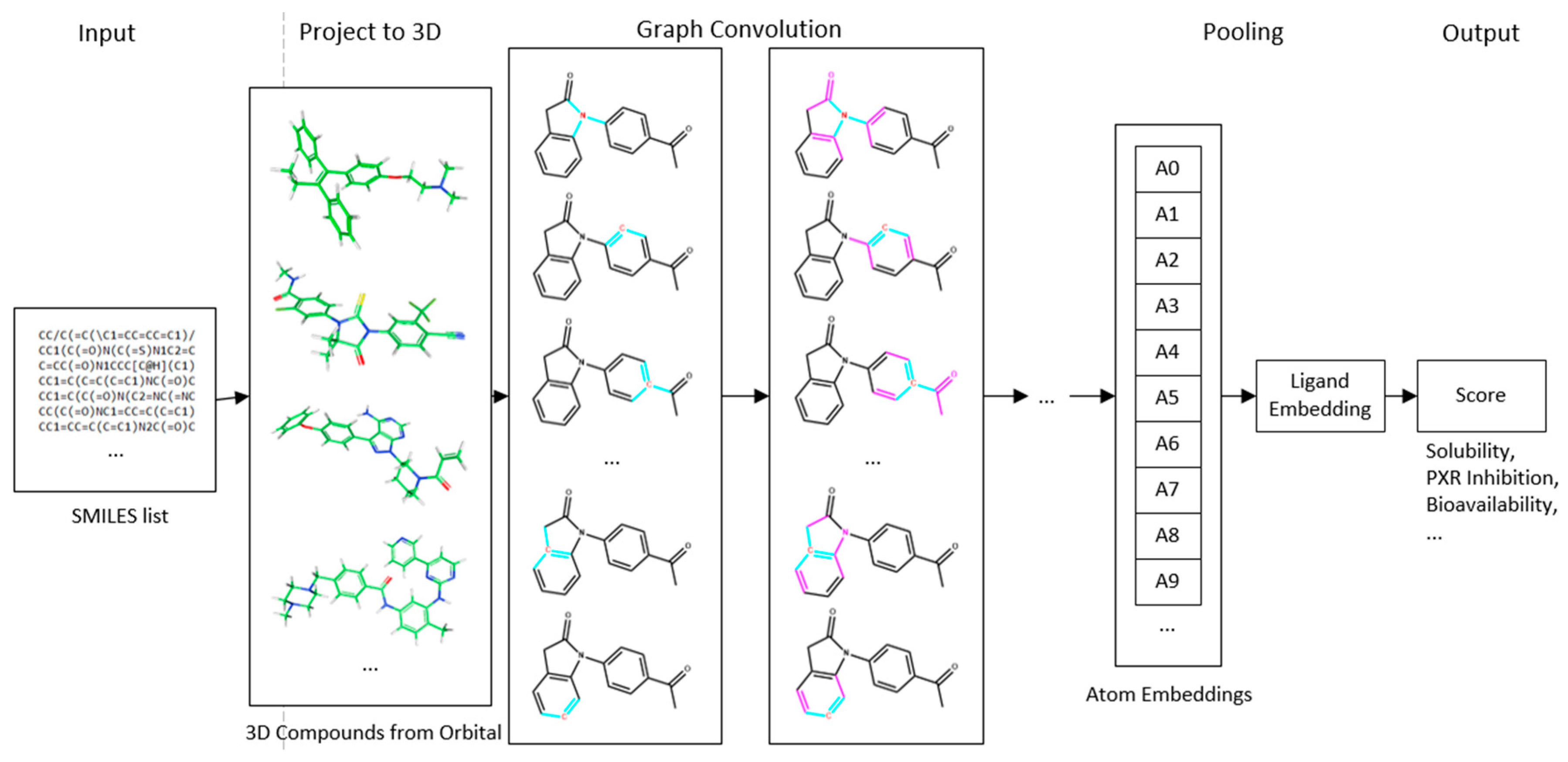
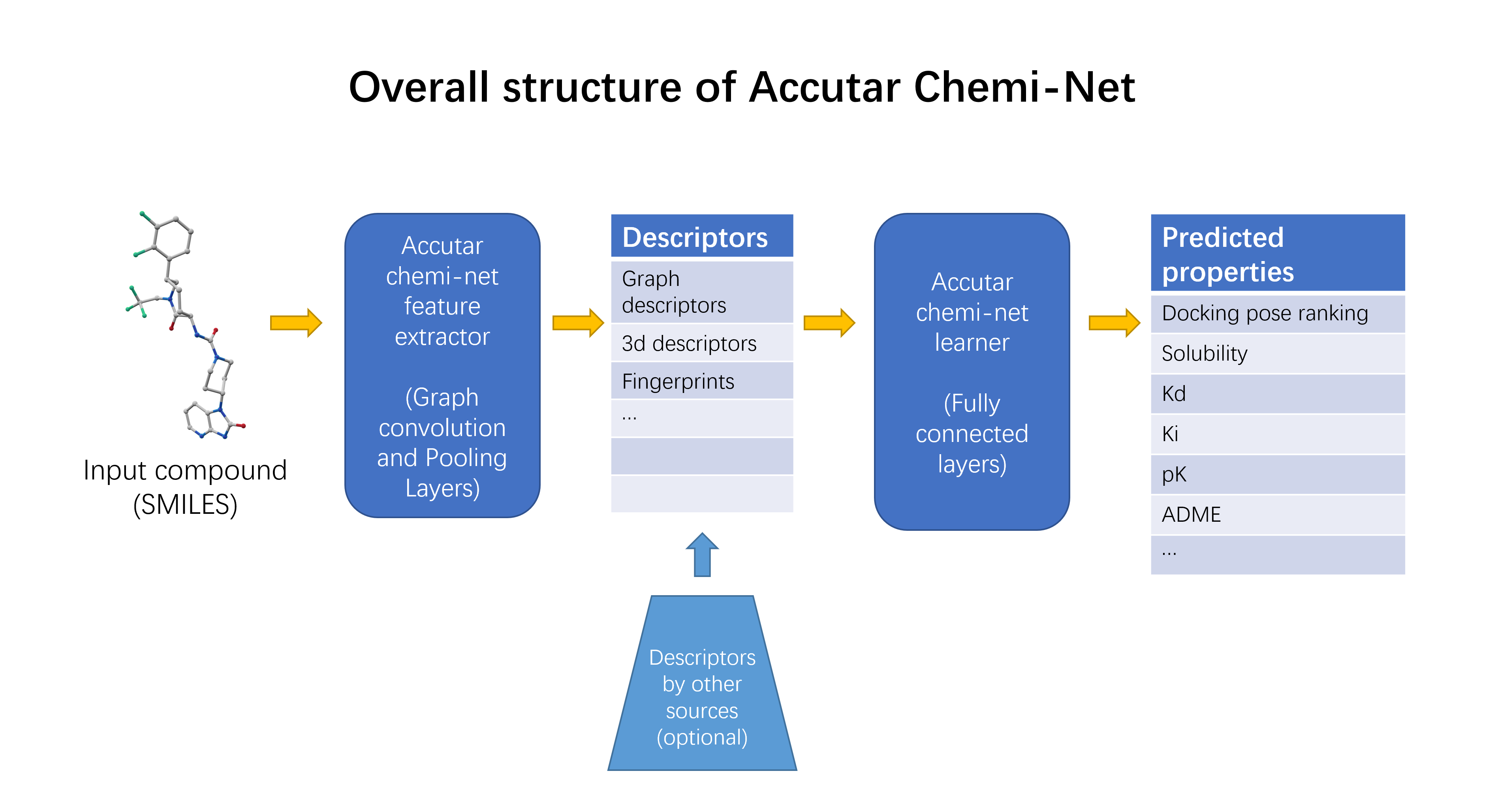
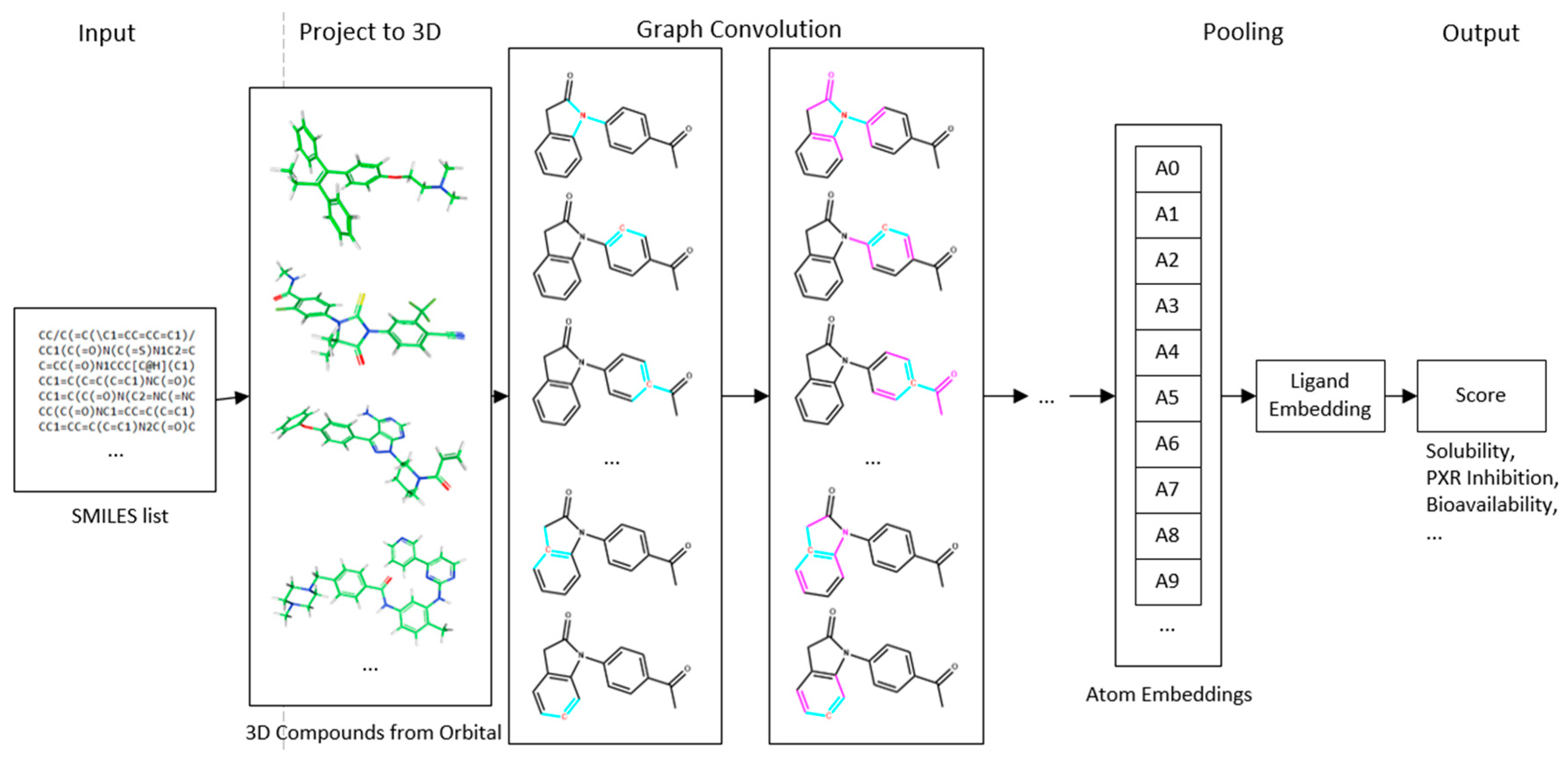
4.1. Deep Neural Network-Based Model
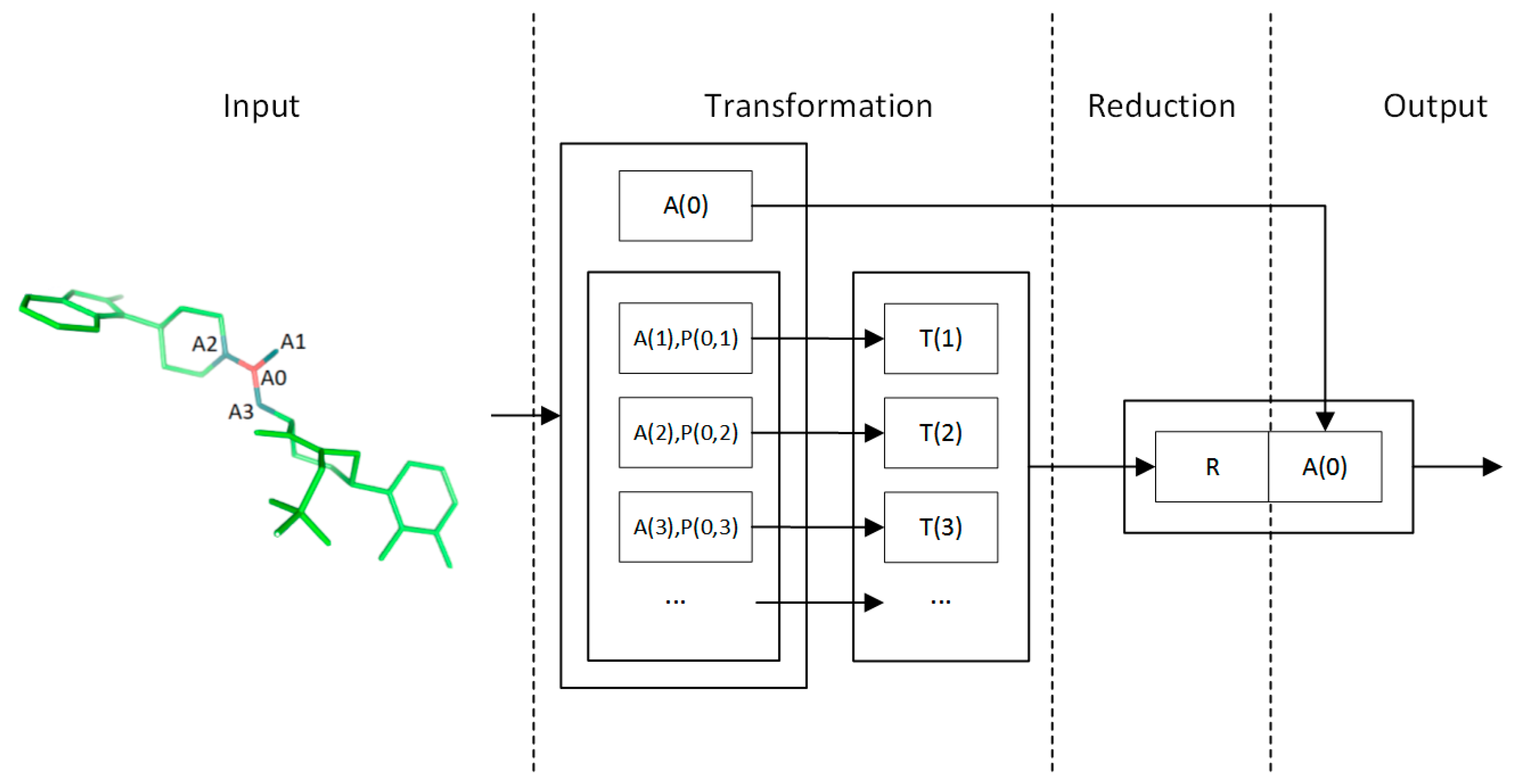
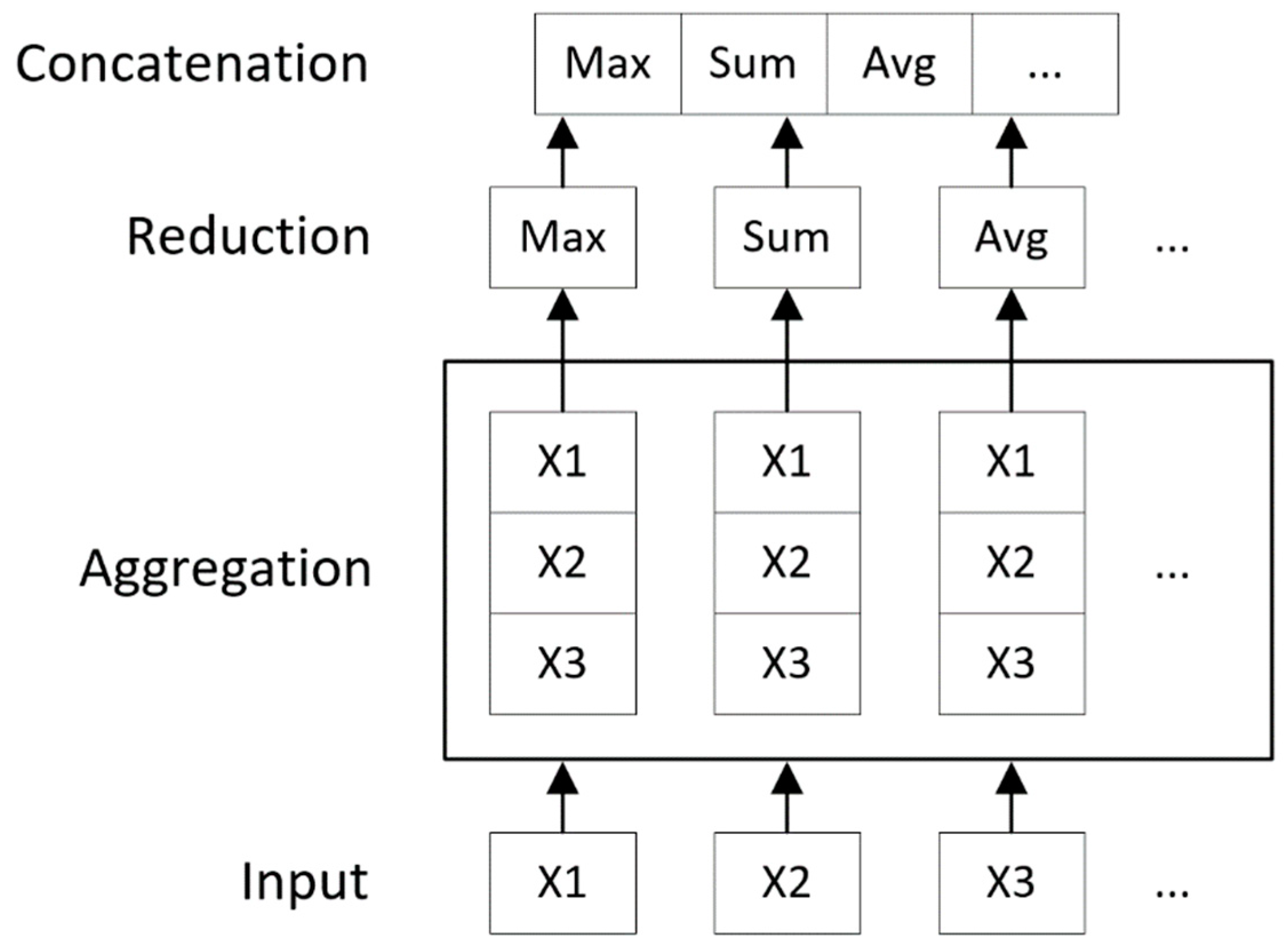
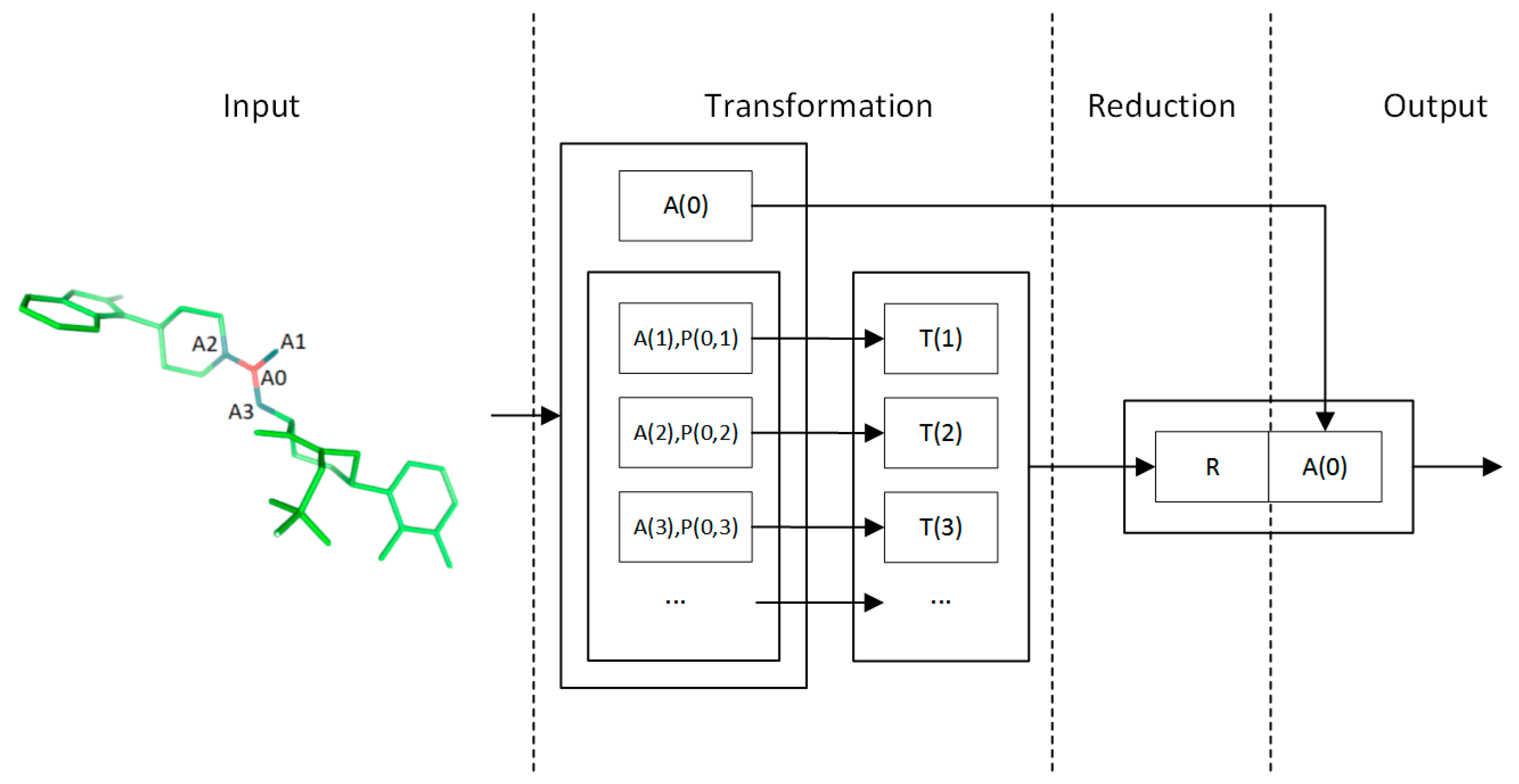
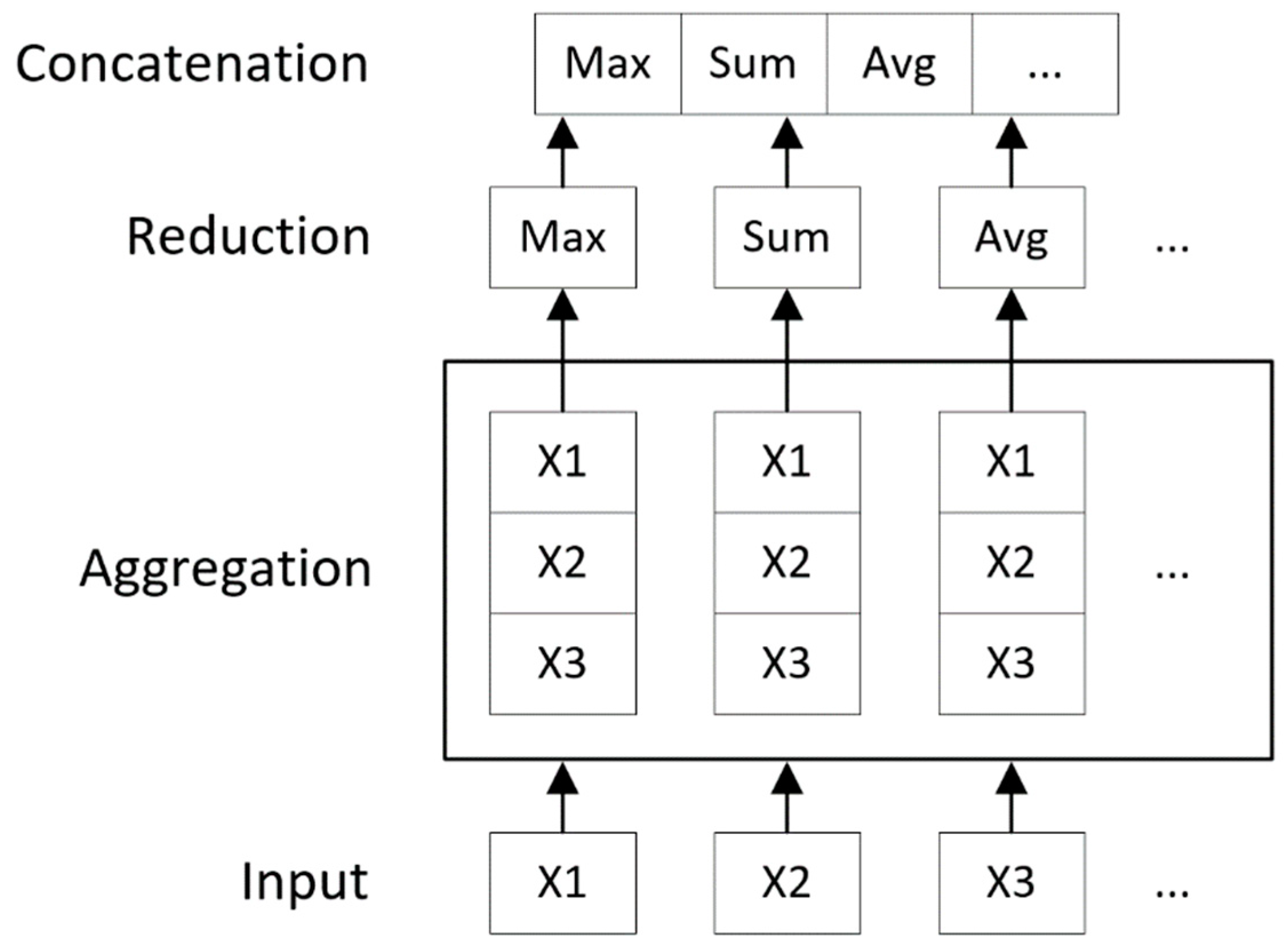
4.2. Convolution Operator
4.3. Input Quantization
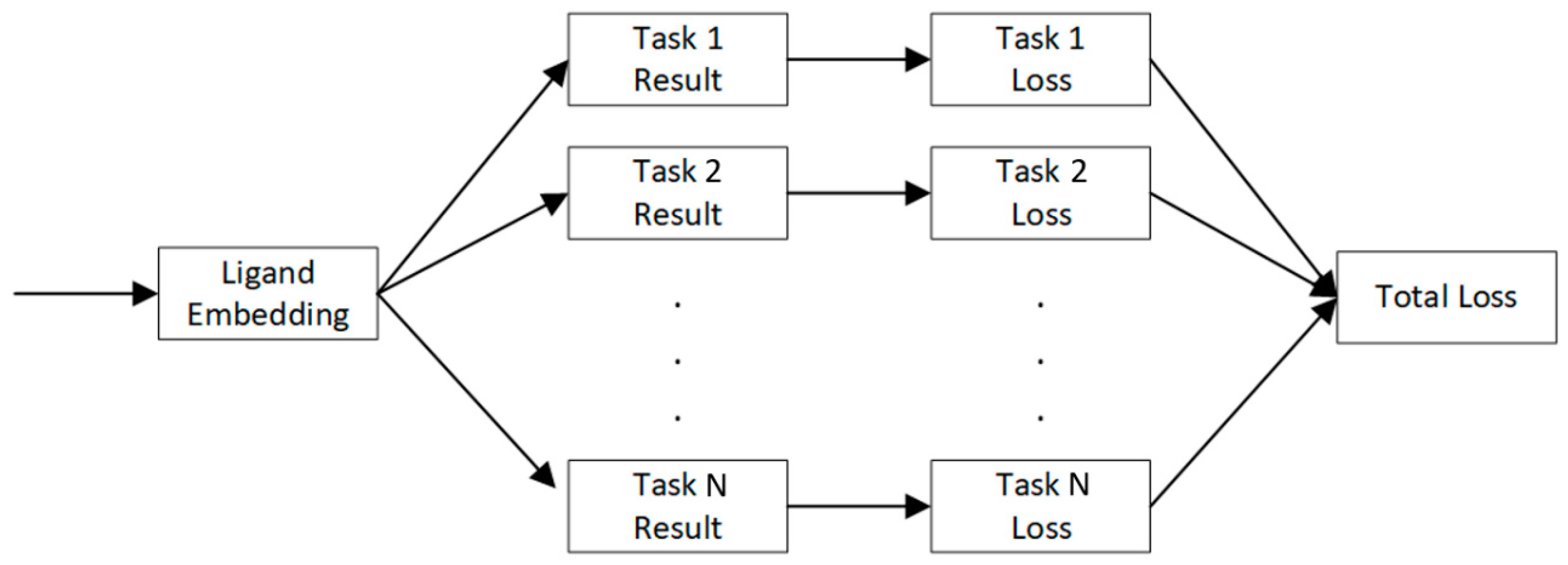
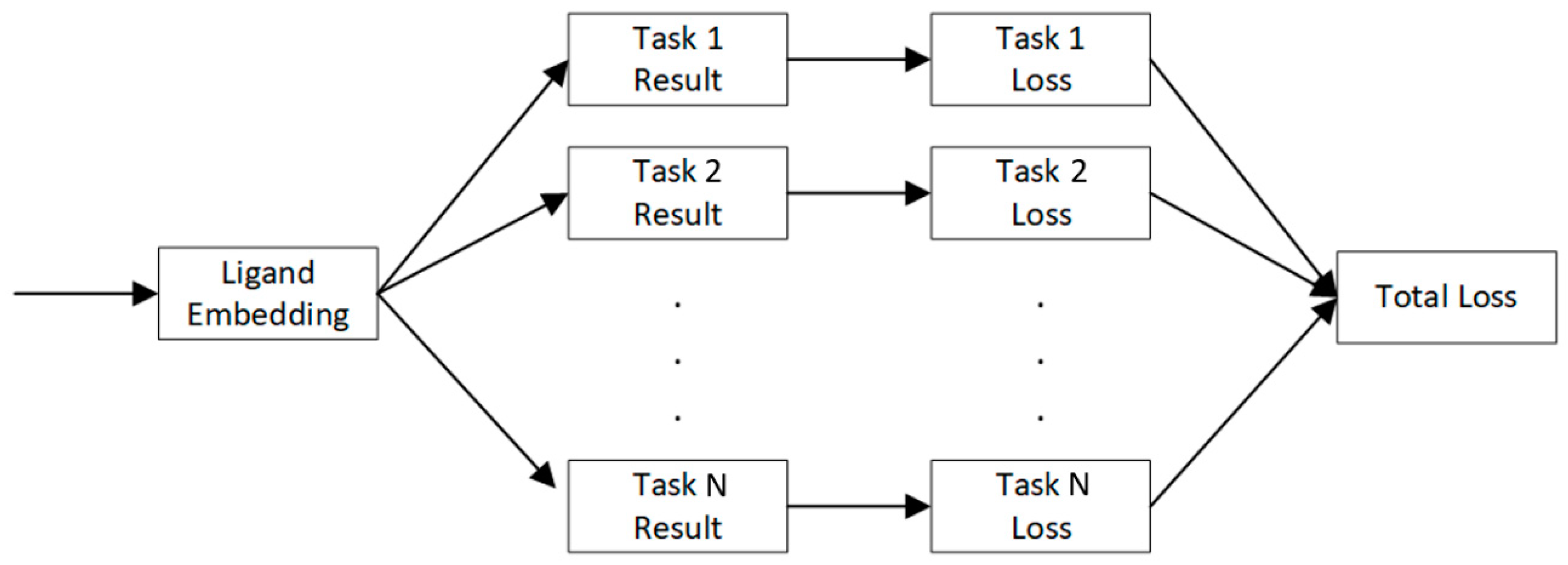
4.4. Multi-Task Learning
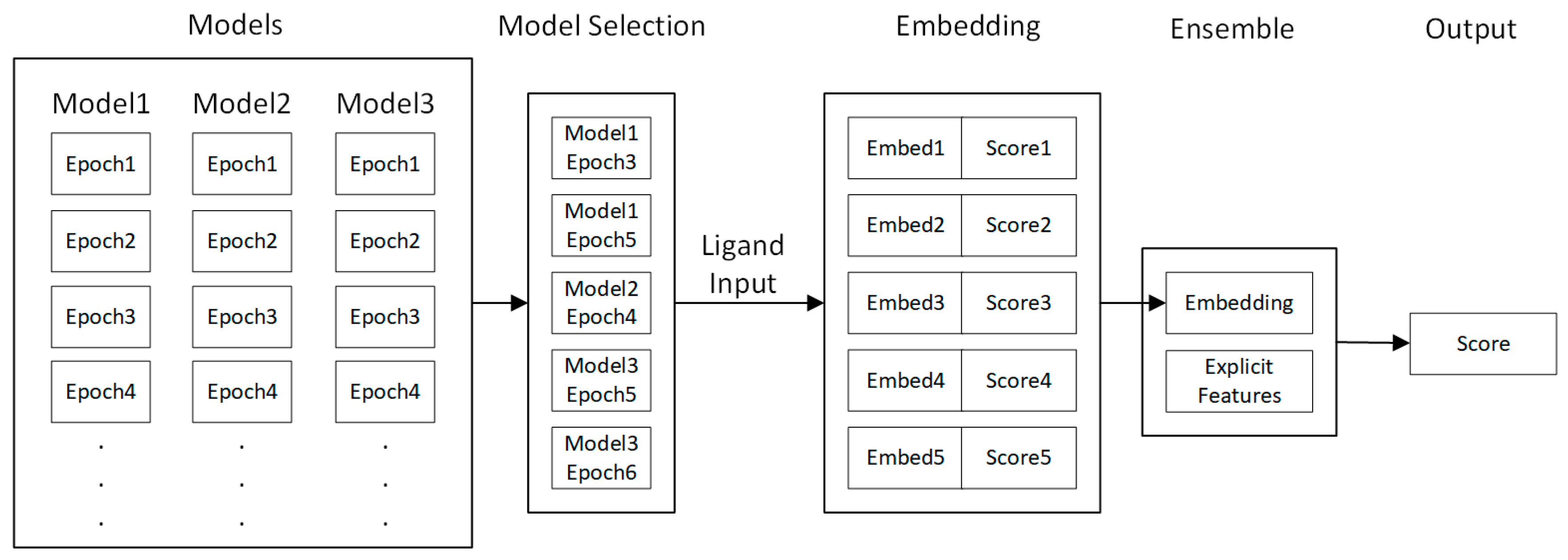
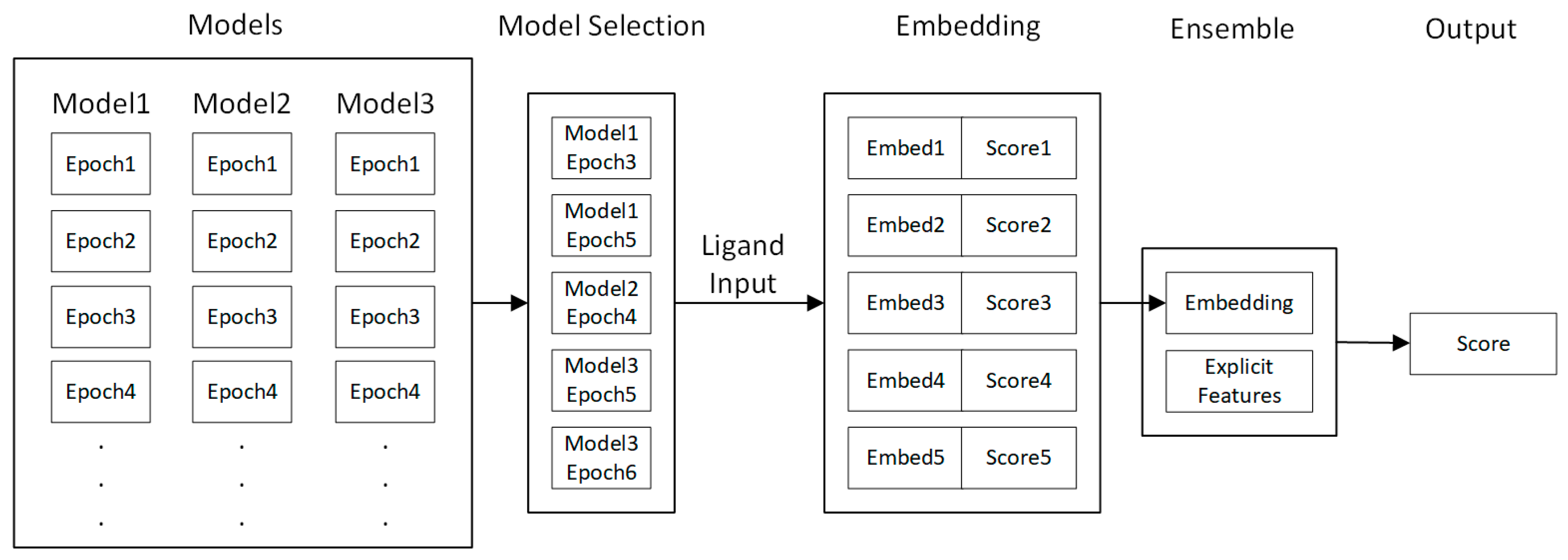
4.5. Fine-Tuning
4.6. Benchmark Method: Cubist
4.7. Benchmark Descriptors
4.8. Data Sets
4.9. Model Training and Test Procedure
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Kearnes, S.; Goldman, B.; Pande, V. Modeling industrial ADMET data with multitask networks. arXiv 2016, arXiv:1606.08793. [Google Scholar]
- Korotcov, A.; Tkachenko, V.; Russo, D.P.; Ekins, S. Comparison of deep learning with multiple machine learning methods and metrics using diverse drug discovery datasets. Mol. Pharm. 2017, 14, 4462–4475. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2017, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, J.; Liaw, A.; Sheridan, R.P.; Svetnik, V. Demystifying multitask deep neural networks for quantitative structure-activity relationships. J. Chem. Inf. Model. 2017, 57, 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- Ramsundar, B.; Liu, B.; Wu, Z.; Verras, A.; Tudor, M.; Sheridan, R.P.; Pande, V. Is multitask deep learning practical for pharma? J. Chem. Inf. Model. 2017, 57, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, T.T. An Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation: New York, NY, USA, 1958; pp. 1–10. [Google Scholar]
- Wenzel, J.; Matter, H.; Schmidt, F. Predictive multitask deep neural network models for ADME-tox properties: Learning from large data sets. J. Chem. Inf. Model. 2019, 59, 1253–1268. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalt, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acid Res. 2015, 42, W612–W620. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. Match 2006, 56, 237–248. [Google Scholar]
- Molecular Operating Environment. Available online: https://www.chemcomp.com (accessed on 3 July 2019).
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 21 December 2018).
- Silver, D.; Schrittwieser, J.; Simonyan, K.; Antonoglou, I.; Huang, A.; Guez, A.; Hubert, T.; Baker, L.; Lai, M.; Bolton, A.; et al. Mastering the game of go without human knowledge. Nature 2017, 550, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Szegedy, C.; Liu, W.; Jia, Y.; Sermanet, P.; Reed, S.; Anguelov, D.; Erhan, D.; Vanhoucke, V.; Rabinovich, A. Going deeper with convolutions. In Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; pp. 1–9. [Google Scholar]
- Wu, S.; Zhong, S.; Liu, Y. Deep residual learning for image steganalysis. Multimed. Tools Appl. 2017, 77, 10437–10453. [Google Scholar] [CrossRef]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. arXiv 2015, arXiv:1502.03167v3. [Google Scholar]
- RulequesResearch. Available online: http://www.rulequest.com (accessed on 21 May 2019).
- Gao, H.; Yao, L.; Mathieu, H.W.; Zhang, Y.; Maurer, T.S.; Troutman, M.D.; Scott, D.O.; Ruggeri, R.B.; Lin, J. In silico modeling of nonspecific binding to human liver microsomes. Drug Metab. Dispos. 2008, 36, 2130–2135. [Google Scholar] [CrossRef]
- Toms, J.D.; Lesperance, M.L. Piecewise regression: A tool for identifying ecological thresholds. Ecology 2003, 84, 2034–2041. [Google Scholar] [CrossRef]
- Kier, L.B. An index of flexibility from molecular shape descriptors. Prog. Clin. Biol. Res. 1989, 291, 105–109. [Google Scholar]
- Kier, L.B. Indexes of molecular shape from chemical graphs. Med. Res. Rev. 1987, 7, 417–440. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. Molecular Structure Description; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Lee, P.H.; Cucurull-Sanchez, L.; Lu, J.; Du, Y.J. Development of in silico models for human liver microsomal stability. J. Comput. Aided. Mol. Des. 2007, 21, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Subset | Train Size | Test Size | Cubist | Chemi-Net ST-DNN | Chemi-Net MT-DNN |
|---|---|---|---|---|---|---|
| HLM | 1 | 69,176 | 17,294 | 0.39 | 0.445 | |
| CYP450 | 1 | 3019 | 755 | 0.597 | 0.692 | |
| 2 | 71,695 | 17,924 | 0.315 | 0.414 | ||
| Solubility | 1 (HCl) | 10,650 | 2659 | 0.493 | 0.548 | 0.585 |
| 2 (PBS) | 10,650 | 2664 | 0.393 | 0.471 | 0.498 | |
| 3 (SIF) | 10,650 | 2645 | 0.445 | 0.552 | 0.562 | |
| PXR | 1 @ 2 uM | 19,902 | 4981 | 0.276 | 0.257 | 0.422 |
| 2 @ 10 uM | 17,414 | 4256 | 0.343 | 0.333 | 0.445 | |
| 3 @ 2 uM | 8883 | 2223 | 0.094 | 0.11 | 0.199 | |
| 4 @ 10 uM | 8205 | 2054 | 0.246 | 0.2 | 0.327 | |
| 5 @ 10 uM | 10,047 | 2511 | 0.349 | 0.38 | 0.418 | |
| 6 @ 2 uM | 10,047 | 2536 | 0.283 | 0.311 | 0.352 | |
| Bioavailability | 1 | 183 | 46 | 0.115 | 0.123 |
| Task | Total Compounds | Training Set Size | Ext. Validation Set Size | Chemi-Net MT [R2] | Wenzel et al.’s MT [R2] |
|---|---|---|---|---|---|
| Human microsomal clearance | 5348 | 4821 | 527 | 0.620 | 0.574 |
| Rat microsomal clearance | 2166 | 1967 | 199 | 0.786 | 0.783 |
| Mouse microsomal clearance | 790 | 734 | 56 | 0.325 | 0.486 |
| Caco-2_Papp permeability | 2582 | 2336 | 246 | 0.560 | 0.542 |
| Atom Feature | Description | Size |
|---|---|---|
| Atom type | One hot vector specifying the type of this atom | 23 |
| Radius | vdW radius and covalent radius of the atom | 2 |
| In rings | For each size of ring (3-8), the number of rings that include this atom | 6 |
| In aromatic ring | Whether this atom is part of an aromatic ring | 1 |
| Charge | Electrostatic charge of this atom | 1 |
| Pair Feature | Description | Size |
|---|---|---|
| Bond type | One hot vector of {Single, Double, None} | 3 |
| Distance | Euclidean distance between this atom pair | 1 |
| Same ring | Whether the atoms are in the same ring | 1 |
| Dataset | Subset | Train Size | Test Size |
|---|---|---|---|
| Human microsomal intrinsic clearance log10 rate) (µL/min/mg protein) | 1 | 69,176 | 17,294 |
| Human CYP450 inhibition log10(IC50 µM) | 1 | 3019 | 755 |
| 2 | 71,695 | 17,924 | |
| Solubility log10 (µM) | 1 (HCl) | 10,650 | 2659 |
| 2 (PBS) | 10,650 | 2664 | |
| 3 (SIF) | 10,650 | 2645 | |
| PXR induction (POC) | 1 @ 2 uM | 19,902 | 4981 |
| 2 @ 10 uM | 17,414 | 4256 | |
| 3 @ 2 uM | 8883 | 2223 | |
| 4 @ 10 uM | 8205 | 2054 | |
| 5 @ 10 uM | 10,047 | 2511 | |
| 6 @ 2 uM | 10,047 | 2536 | |
| Bioavailability | 1 | 183 | 46 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Sun, X.; Jia, L.; Ma, J.; Xing, H.; Wu, J.; Gao, H.; Sun, Y.; Boulnois, F.; Fan, J. Chemi-Net: A Molecular Graph Convolutional Network for Accurate Drug Property Prediction. Int. J. Mol. Sci. 2019, 20, 3389. https://doi.org/10.3390/ijms20143389
Liu K, Sun X, Jia L, Ma J, Xing H, Wu J, Gao H, Sun Y, Boulnois F, Fan J. Chemi-Net: A Molecular Graph Convolutional Network for Accurate Drug Property Prediction. International Journal of Molecular Sciences. 2019; 20(14):3389. https://doi.org/10.3390/ijms20143389
Chicago/Turabian StyleLiu, Ke, Xiangyan Sun, Lei Jia, Jun Ma, Haoming Xing, Junqiu Wu, Hua Gao, Yax Sun, Florian Boulnois, and Jie Fan. 2019. "Chemi-Net: A Molecular Graph Convolutional Network for Accurate Drug Property Prediction" International Journal of Molecular Sciences 20, no. 14: 3389. https://doi.org/10.3390/ijms20143389
APA StyleLiu, K., Sun, X., Jia, L., Ma, J., Xing, H., Wu, J., Gao, H., Sun, Y., Boulnois, F., & Fan, J. (2019). Chemi-Net: A Molecular Graph Convolutional Network for Accurate Drug Property Prediction. International Journal of Molecular Sciences, 20(14), 3389. https://doi.org/10.3390/ijms20143389