Contribution of p38 MAPK Pathway to Norcantharidin-Induced Programmed Cell Death in Human Oral Squamous Cell Carcinoma

 and
and 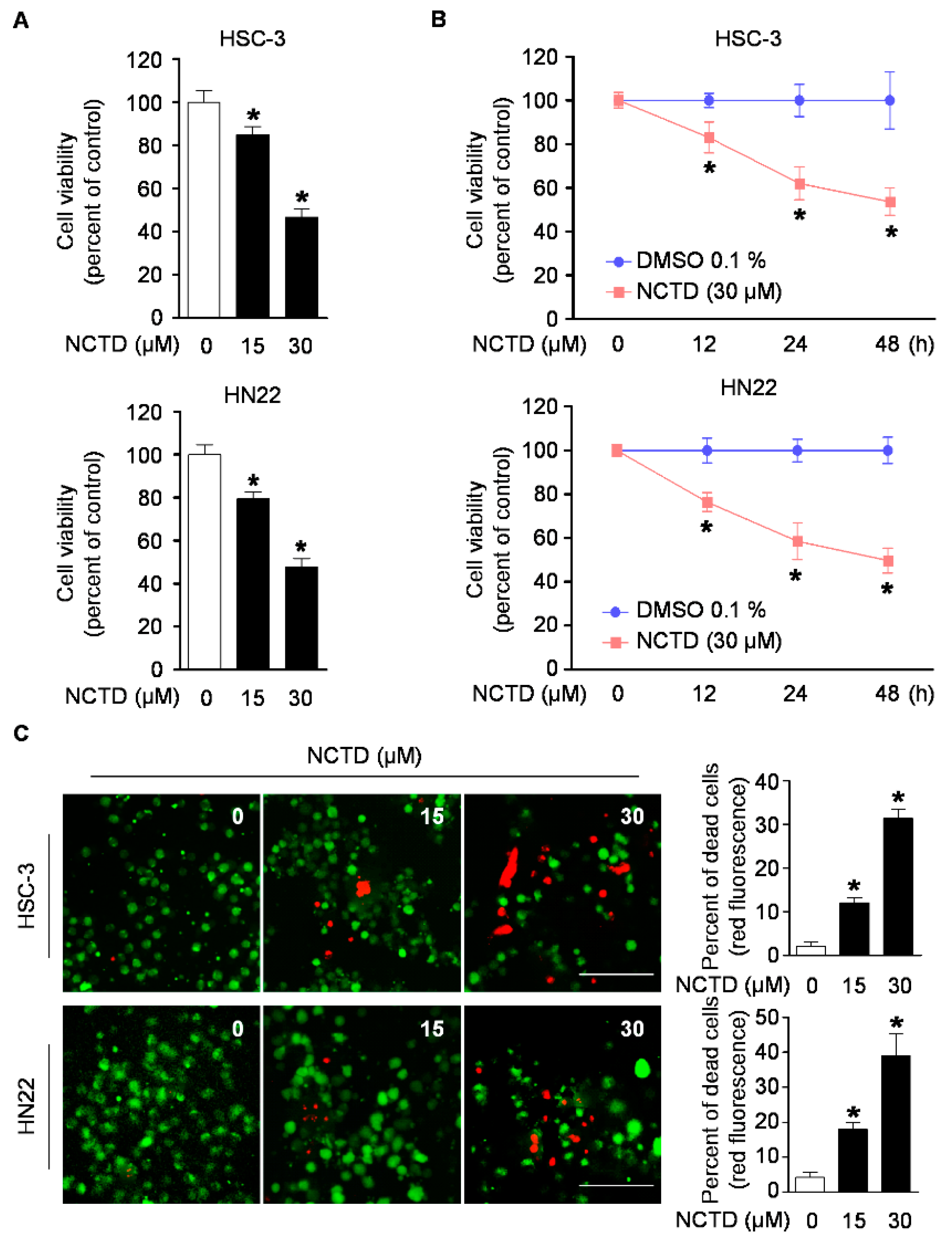{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. NCTD Inhibits Cell Viability and Induces Cell Death in Human OSCC Cell Lines
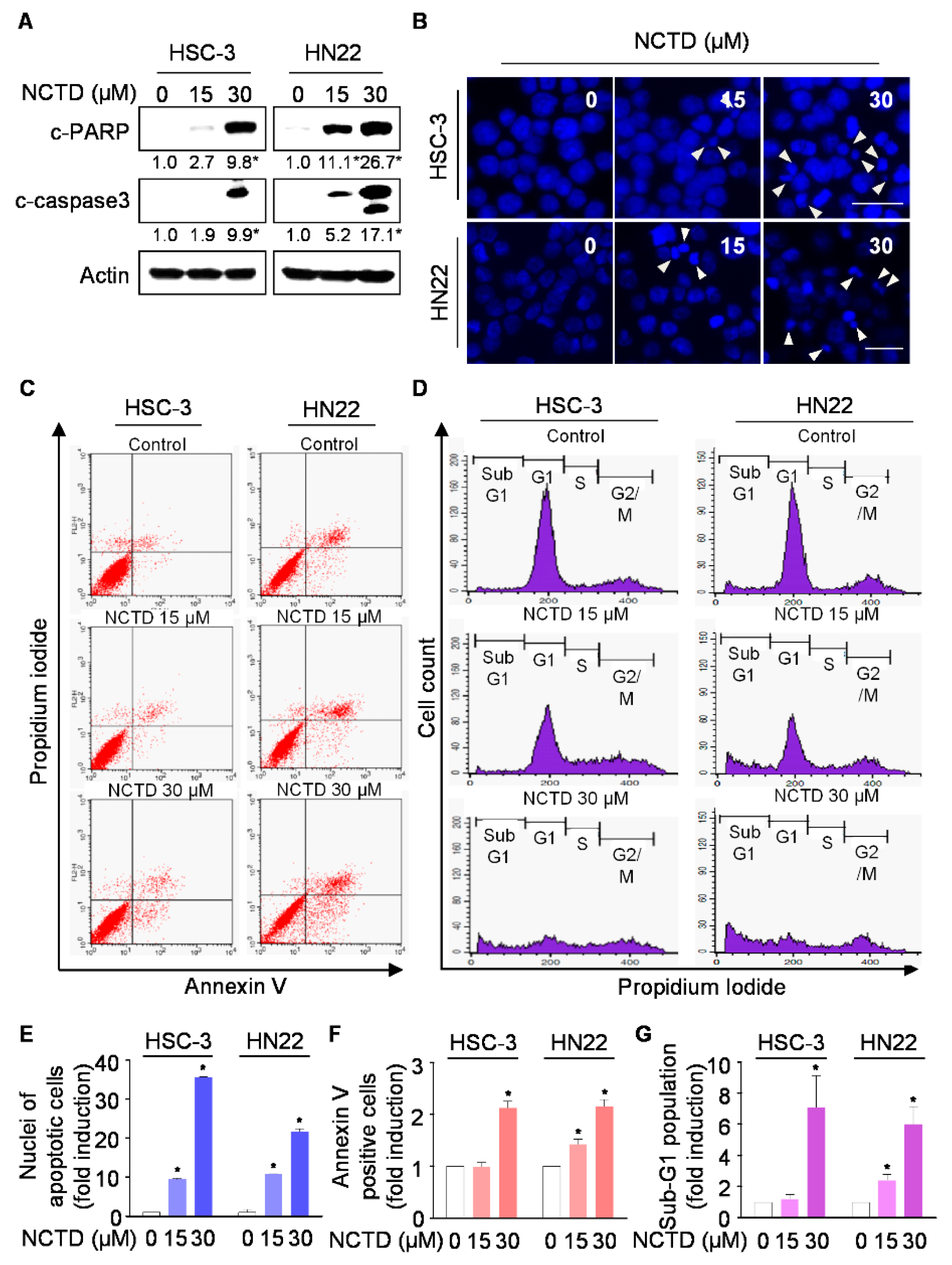
2.2. NCTD Induces Programmed Cell Death (Apoptosis) in OSCC Cell Lines
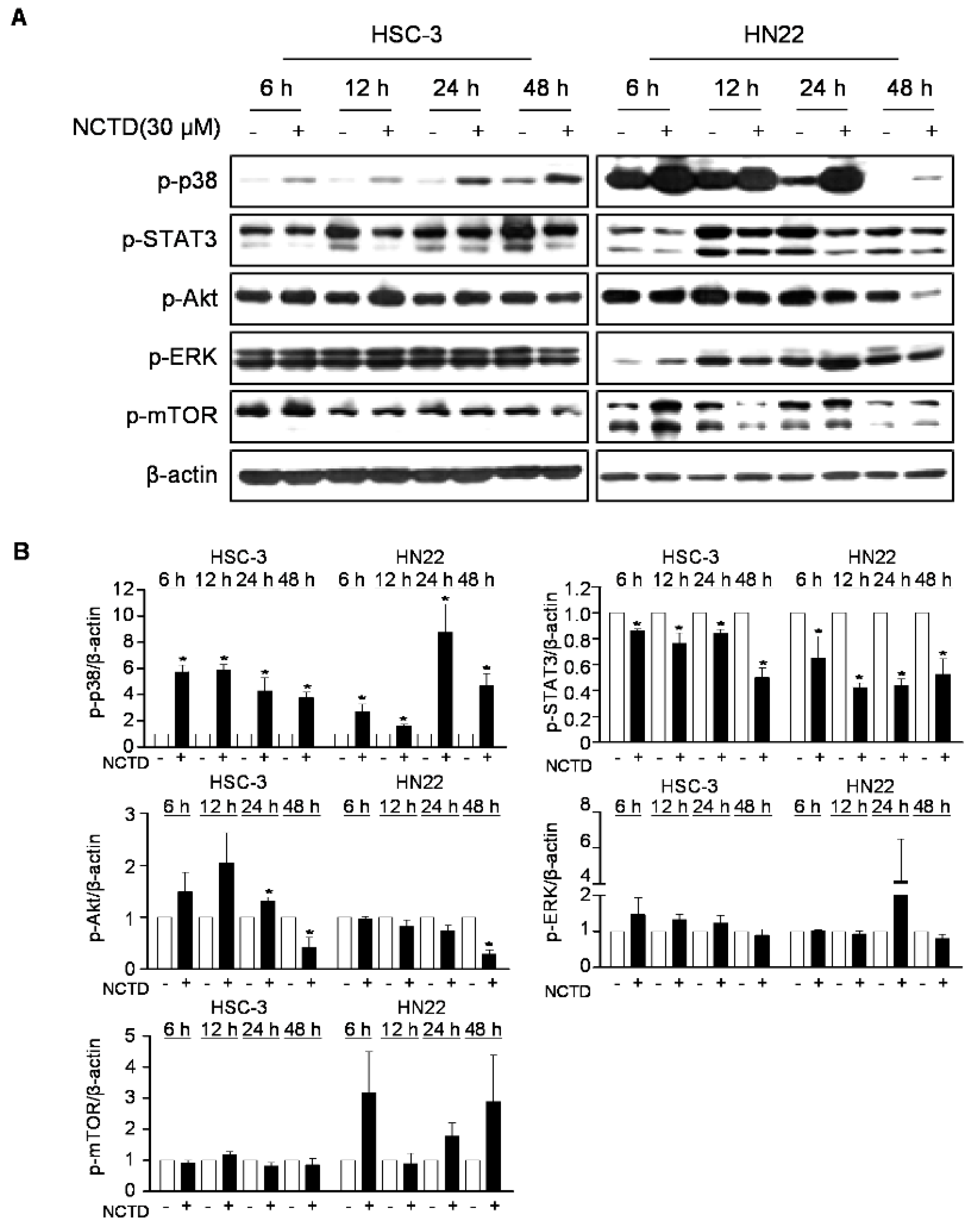
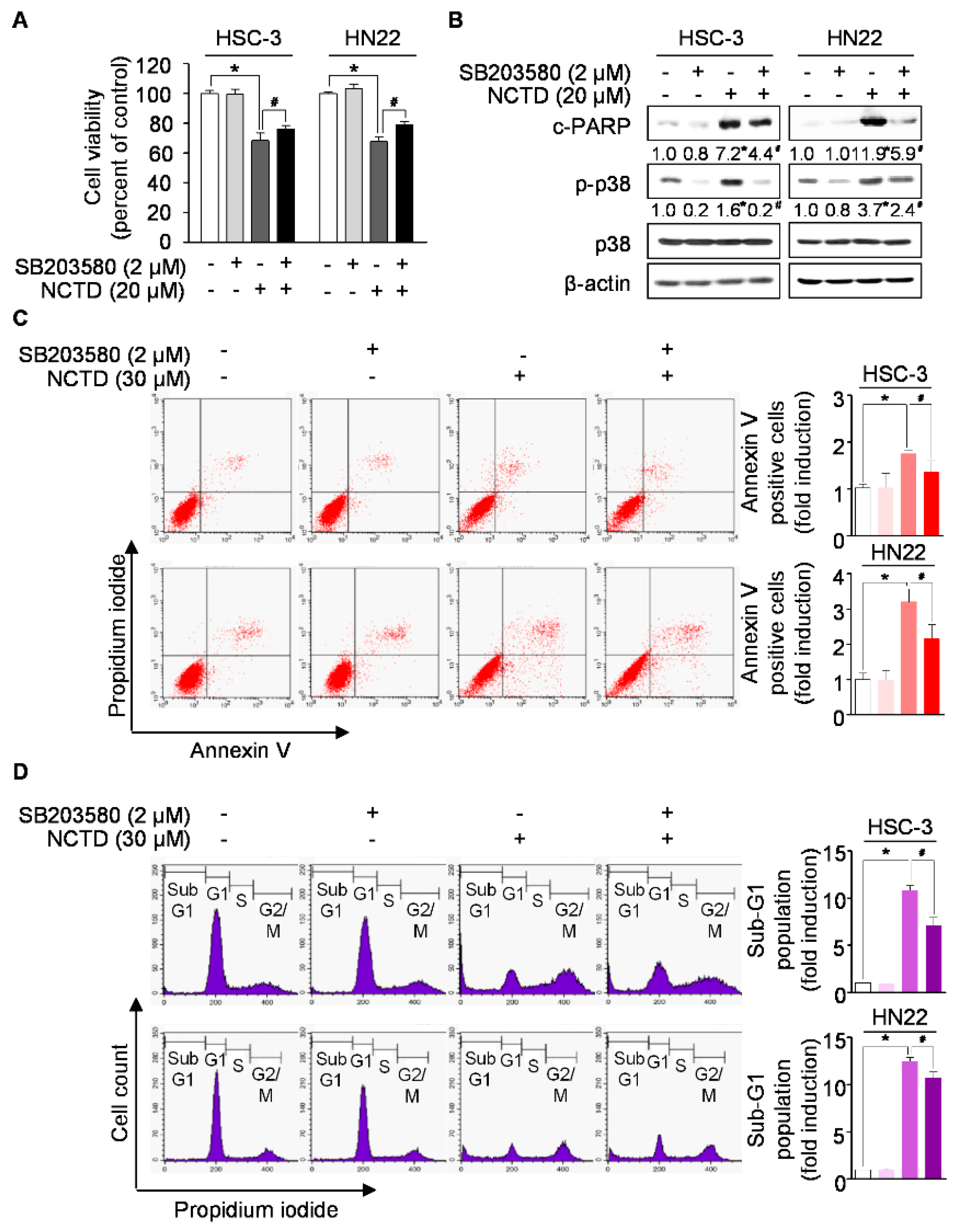
2.3. p38 MAPK is Involved in NCTD-Induced Programmed Cell Death in OSCC Cell Lines
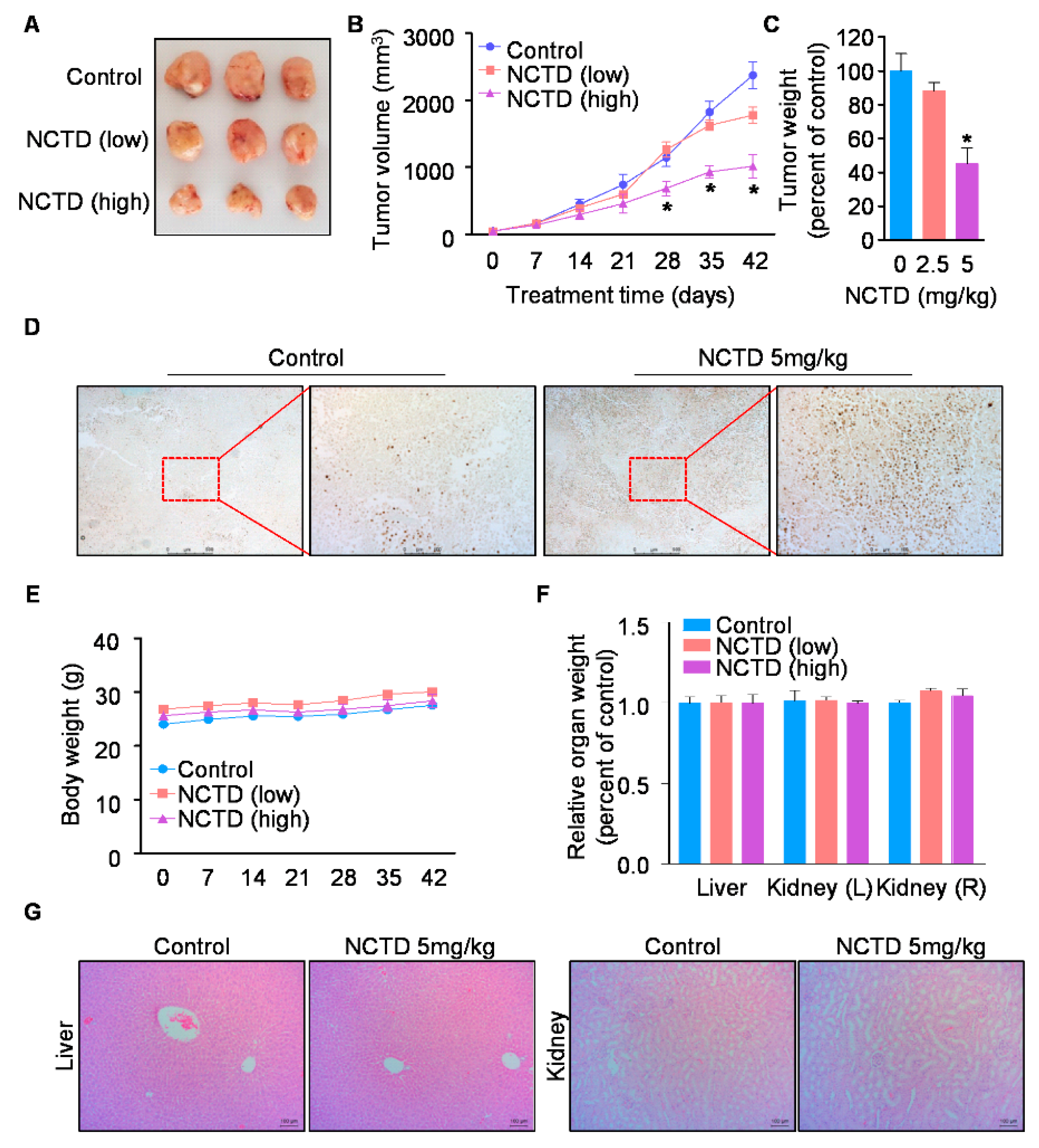
2.4. NCTD Inhibits the Xenograft Tumor Growth of Human OSCC by Inducing Programmed Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemical Treatment
4.2. Trypan Blue Exclusion Assay
4.3. Live/Dead Assay
4.4. Western Blot Analysis
4.5. DAPI Staining
4.6. Annexin V/PI Staining
4.7. Cell Cycle Analysis
4.8. Construction of STAT3 Over-Expression Vector and Transient Transfection
4.9. Nude Mouse Xenograft Assay
4.10. TUNEL Assay
4.11. Histopathological Examination of Organs
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Rivera, C. Essentials of oral cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11884–11894. [Google Scholar] [PubMed]
- Mallery, S.R.; Wang, D.; Santiago, B.; Pei, P.; Schwendeman, S.P.; Nieto, K.; Spinney, R.; Tong, M.; Koutras, G.; Han, B.; et al. Benefits of multifaceted chemopreventives in the suppression of the oral squamous cell carcinoma (oscc) tumorigenic phenotype. Cancer Prev. Res. (Phila) 2017, 10, 76–88. [Google Scholar] [CrossRef]
- Mascitti, M.; Rubini, C.; De Michele, F.; Balercia, P.; Girotto, R.; Troiano, G.; Lo Muzio, L.; Santarelli, A. American joint committee on cancer staging system 7th edition versus 8th edition: Any improvement for patients with squamous cell carcinoma of the tongue? Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2018, 126, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Molinolo, A.A.; Amornphimoltham, P.; Squarize, C.H.; Castilho, R.M.; Patel, V.; Gutkind, J.S. Dysregulated molecular networks in head and neck carcinogenesis. Oral. Oncol. 2009, 45, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.H.; Khadka, S.; Shin, J.A.; Jung, J.Y.; Ryu, M.H.; Yu, H.J.; Lee, H.N.; Jang, B.; Yang, I.H.; Won, D.H.; et al. Nitidine chloride acts as an apoptosis inducer in human oral cancer cells and a nude mouse xenograft model via inhibition of stat3. Oncotarget 2017, 8, 91306–91315. [Google Scholar] [CrossRef] [PubMed]
- Macha, M.A.; Matta, A.; Kaur, J.; Chauhan, S.S.; Thakar, A.; Shukla, N.K.; Gupta, S.D.; Ralhan, R. Prognostic significance of nuclear pstat3 in oral cancer. Head Neck 2011, 33, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The stat3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral. Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 map kinase signal transduction in solid tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef]
- Bradham, C.; McClay, D.R. P38 mapk in development and cancer. Cell Cycle 2006, 5, 824–828. [Google Scholar] [CrossRef]
- Gkouveris, I.; Nikitakis, N.; Sklavounou, A. P38 expression and modulation of stat3 signaling in oral cancer. Pathol. Oncol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jung, J.Y.; Shim, J.H.; Kim, J.; Choi, K.H.; Shin, J.A.; Choi, E.S.; Lee, S.O.; Chintharlapalli, S.; Kwon, K.H.; et al. Apoptotic effect of tolfenamic acid in kb human oral cancer cells: Possible involvement of the p38 mapk pathway. J. Clin. Biochem. Nutr. 2010, 47, 74–80. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, K.H.; Kim, H.K.; Kim, J.H.; Choi, E.S.; Shin, J.A.; Lee, S.O.; Chintharlapalli, S.; Safe, S.; Abdelrahim, M.; Kong, G.; et al. The p38 mapk pathway is critical for 5,5′-dibromodiindolylmethane-induced apoptosis to prevent oral squamous carcinoma cells. Eur. J. Cancer Prev. 2010, 19, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.Y.; Liang, S.S.; Han, L.Y.; Chou, H.L.; Chou, C.K.; Lin, S.R.; Chiu, C.C. Cardiotoxin iii inhibits proliferation and migration of oral cancer cells through mapk and mmp signaling. Sci. World J. 2013, 2013, 650946. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, E.; Sankari, L.S.; Malathi, L.; Krupaa, J.R. Naturally occurring products in cancer therapy. J. Pharm. Bioallied Sci. 2015, 7, S181–S183. [Google Scholar] [CrossRef] [PubMed]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.A.; Ryu, M.H.; Kwon, K.H.; Choi, B.; Cho, S.D. Down-regulation of akt by methanol extracts of impatiens balsamina l. Promotes apoptosis in human oral squamous cell carcinoma cell lines. J. Oral. Pathol. Med. 2015, 44, 420–428. [Google Scholar] [CrossRef]
- Cho, N.P.; Han, H.S.; Leem, D.H.; Choi, I.S.; Jung, J.Y.; Kim, H.J.; Moon, K.S.; Choi, K.H.; Soh, Y.; Kong, G.; et al. Sulforaphane enhances caspase-dependent apoptosis through inhibition of cyclooxygenase-2 expression in human oral squamous carcinoma cells and nude mouse xenograft model. Oral. Oncol. 2009, 45, 654–660. [Google Scholar] [CrossRef]
- Wang, G.S. Medical uses of mylabris in ancient china and recent studies. J. Ethnopharmacol. 1989, 26, 147–162. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chang, W.M.; Liu, Y.W.; Lee, C.Y.; Jang, Y.H.; Kuo, C.D.; Liao, H.F. A small-molecule metastasis inhibitor, norcantharidin, downregulates matrix metalloproteinase-9 expression by inhibiting sp1 transcriptional activity in colorectal cancer cells. Chem. Biol. Interact. 2009, 181, 440–446. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Q.; Liu, K.; Yagasaki, K.; Zhang, G. Suppression of growth of highly-metastatic human breast cancer cells by norcantharidin and its mechanisms of action. Cytotechnology 2009, 59, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Chen, C.M.; Lin, C.L.; Cheng, C.W.; Lee, C.H.; Hsieh, Y.H. Norcantharidin induces mitochondrial-dependent apoptosis through mcl-1 inhibition in human prostate cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Dai, B.; Zhu, Y.; Ye, D. Norcantharidin induces autophagy-related prostate cancer cell death through beclin-1 upregulation by mir-129-5p suppression. Tumour Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Fan, Y.Z.; Chen, C.Q.; Zhao, Z.M.; Sun, W. Norcantharidin: A potential antiangiogenic agent for gallbladder cancers in vitro and in vivo. Int. J. Oncol. 2012, 40, 1501–1514. [Google Scholar] [PubMed]
- Papadopoulos, N.G.; Dedoussis, G.V.; Spanakos, G.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. An improved fluorescence assay for the determination of lymphocyte-mediated cytotoxicity using flow cytometry. J. Immunol. Methods 1994, 177, 101–111. [Google Scholar] [CrossRef]
- McCluskey, A.; Sim, A.T.; Sakoff, J.A. Serine-threonine protein phosphatase inhibitors: Development of potential therapeutic strategies. J. Med. Chem. 2002, 45, 1151–1175. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Rauh, R.; Kahl, S.; Tomicic, M.; Bochzelt, H.; Tome, M.E.; Briehl, M.M.; Bauer, R.; Kaina, B. Molecular modes of action of cantharidin in tumor cells. Biochem. Pharmacol. 2005, 69, 811–818. [Google Scholar] [CrossRef]
- Karras, D.J.; Farrell, S.E.; Harrigan, R.A.; Henretig, F.M.; Gealt, L. Poisoning from “spanish fly” (cantharidin). Am. J. Emerg. Med. 1996, 14, 478–483. [Google Scholar] [CrossRef]
- Till, J.S.; Majmudar, B.N. Cantharidin poisoning. South Med. J. 1981, 74, 444–447. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Chao, K.S.; Liao, H.F.; Chen, Y.J. Norcantharidin, derivative of cantharidin, for cancer stem cells. Evid. Based Complement. Alternat. Med. 2013, 2013, 838651. [Google Scholar] [CrossRef]
- Yeh, C.B.; Hsieh, M.J.; Hsieh, Y.H.; Chien, M.H.; Chiou, H.L.; Yang, S.F. Antimetastatic effects of norcantharidin on hepatocellular carcinoma by transcriptional inhibition of mmp-9 through modulation of nf-kb activity. PLoS ONE 2012, 7, e31055. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.Y.; Chen, M.F.; Tsai, C.H.; Hu, D.N.; Chang, F.R.; Wu, Y.C. Involvement of caspase and mapk activities in norcantharidin-induced colorectal cancer cell apoptosis. Toxicol. In Vitro 2010, 24, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Zhu, Y.; Li, X.F.; Tang, L.P.; Su, Z.Q.; Wang, X.Q.; Li, C.Y.; Yang, H.M.; Zheng, G.J.; Feng, B. Norcantharidin alone or in combination with crizotinib induces autophagic cell death in hepatocellular carcinoma by repressing c-met-mtor signaling. Oncotarget 2017, 8, 114945–114955. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.Y.; Chen, M.F.; Kao, Y.H.; Hu, D.N.; Chang, F.R.; Wu, Y.C. Norcantharidin induces apoptosis of breast cancer cells: Involvement of activities of mitogen activated protein kinases and signal transducers and activators of transcription. Toxicol. In Vitro 2011, 25, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Davis, R.J.; Flavell, R.A. Map kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by jnk and p38 mapk pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, J.C.; Jiang, Y.Y.; Xia, M.Y.; Tashiro, S.; Onodera, S.; Ikejima, T. P38-nf-kappab-promoted mitochondria-associated apoptosis and g2/m cell cycle arrest in norcantharidin-treated hela cells. J. Asian Nat. Prod. Res. 2012, 14, 1008–1019. [Google Scholar] [CrossRef]
- An, W.W.; Gong, X.F.; Wang, M.W.; Tashiro, S.; Onodera, S.; Ikejima, T. Norcantharidin induces apoptosis in hela cells through caspase, mapk, and mitochondrial pathways. Acta. Pharmacol. Sin. 2004, 25, 1502–1508. [Google Scholar]
- Chen, Y.J.; Tsai, Y.M.; Kuo, C.D.; Ku, K.L.; Shie, H.S.; Liao, H.F. Norcantharidin is a small-molecule synthetic compound with anti-angiogenesis effect. Life Sci. 2009, 85, 642–651. [Google Scholar] [CrossRef]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and approaches of targeting stat3 for cancer treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef]
- Gao, Y.; Li, W.; Liu, R.; Guo, Q.; Li, J.; Bao, Y.; Zheng, H.; Jiang, S.; Hua, B. Norcantharidin inhibits il-6-induced epithelialmesenchymal transition via the jak2/stat3/twist signaling pathway in hepatocellular carcinoma cells. Oncol. Rep. 2017, 38, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dong, J.; Deng, L. Overview of cantharidin and its analogues. Curr. Med. Chem. 2018, 25, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, B.; Zhang, K.; Zhang, Y.; Wang, J.; Qi, N.; Yang, S.; He, H.; Tang, X. Preclinical evaluations of norcantharidin-loaded intravenous lipid microspheres with low toxicity. Expert Opin. Drug Deliv. 2012, 9, 1449–1462. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, C.-H.; Hong, K.-O.; Jin, B.; Lee, W.; Jung, Y.C.; Lee, H.; Shin, J.-A.; Cho, S.-D.; Hong, S.D. Contribution of p38 MAPK Pathway to Norcantharidin-Induced Programmed Cell Death in Human Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 3487. https://doi.org/10.3390/ijms20143487
Ahn C-H, Hong K-O, Jin B, Lee W, Jung YC, Lee H, Shin J-A, Cho S-D, Hong SD. Contribution of p38 MAPK Pathway to Norcantharidin-Induced Programmed Cell Death in Human Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2019; 20(14):3487. https://doi.org/10.3390/ijms20143487
Chicago/Turabian StyleAhn, Chi-Hyun, Kyoung-Ok Hong, Bohwan Jin, WonWoo Lee, Yun Chan Jung, Hakmo Lee, Ji-Ae Shin, Sung-Dae Cho, and Seong Doo Hong. 2019. "Contribution of p38 MAPK Pathway to Norcantharidin-Induced Programmed Cell Death in Human Oral Squamous Cell Carcinoma" International Journal of Molecular Sciences 20, no. 14: 3487. https://doi.org/10.3390/ijms20143487
APA StyleAhn, C.-H., Hong, K.-O., Jin, B., Lee, W., Jung, Y. C., Lee, H., Shin, J.-A., Cho, S.-D., & Hong, S. D. (2019). Contribution of p38 MAPK Pathway to Norcantharidin-Induced Programmed Cell Death in Human Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences, 20(14), 3487. https://doi.org/10.3390/ijms20143487