Hydrogel-Encapsulated Mesoporous Silica-Coated Gold Nanoshells for Smart Drug Delivery



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
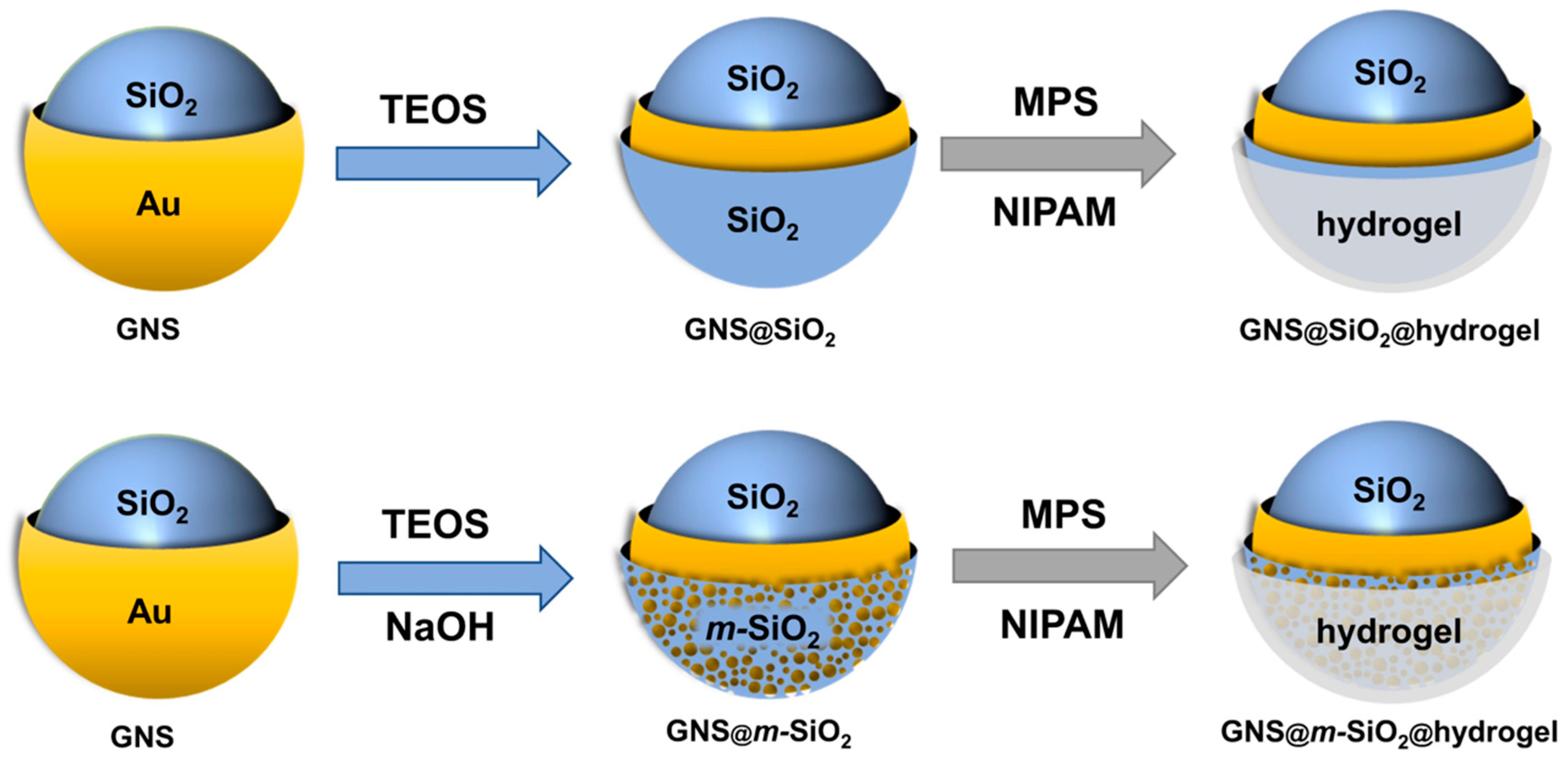
2.1. Synthetic Strategy
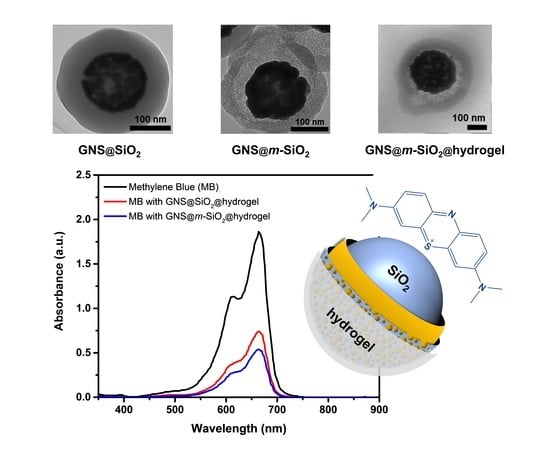
2.2. Characterization of the Composite Nanoparticles
2.3. Thermal Response of the Composite Nanoparticles
2.4. Loading and Release Capacity of the Mesoporous Layer in the Composite Nanoparticles
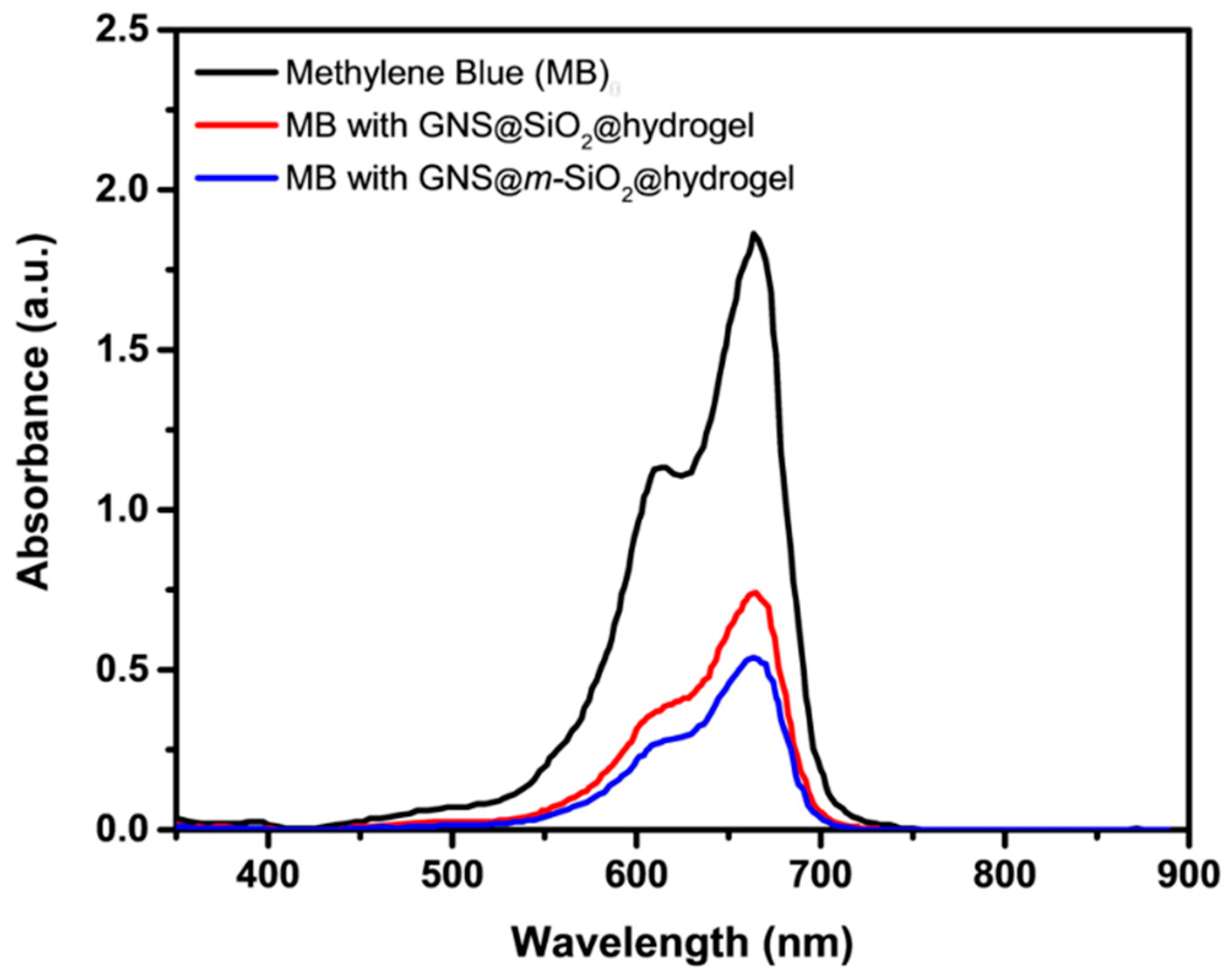
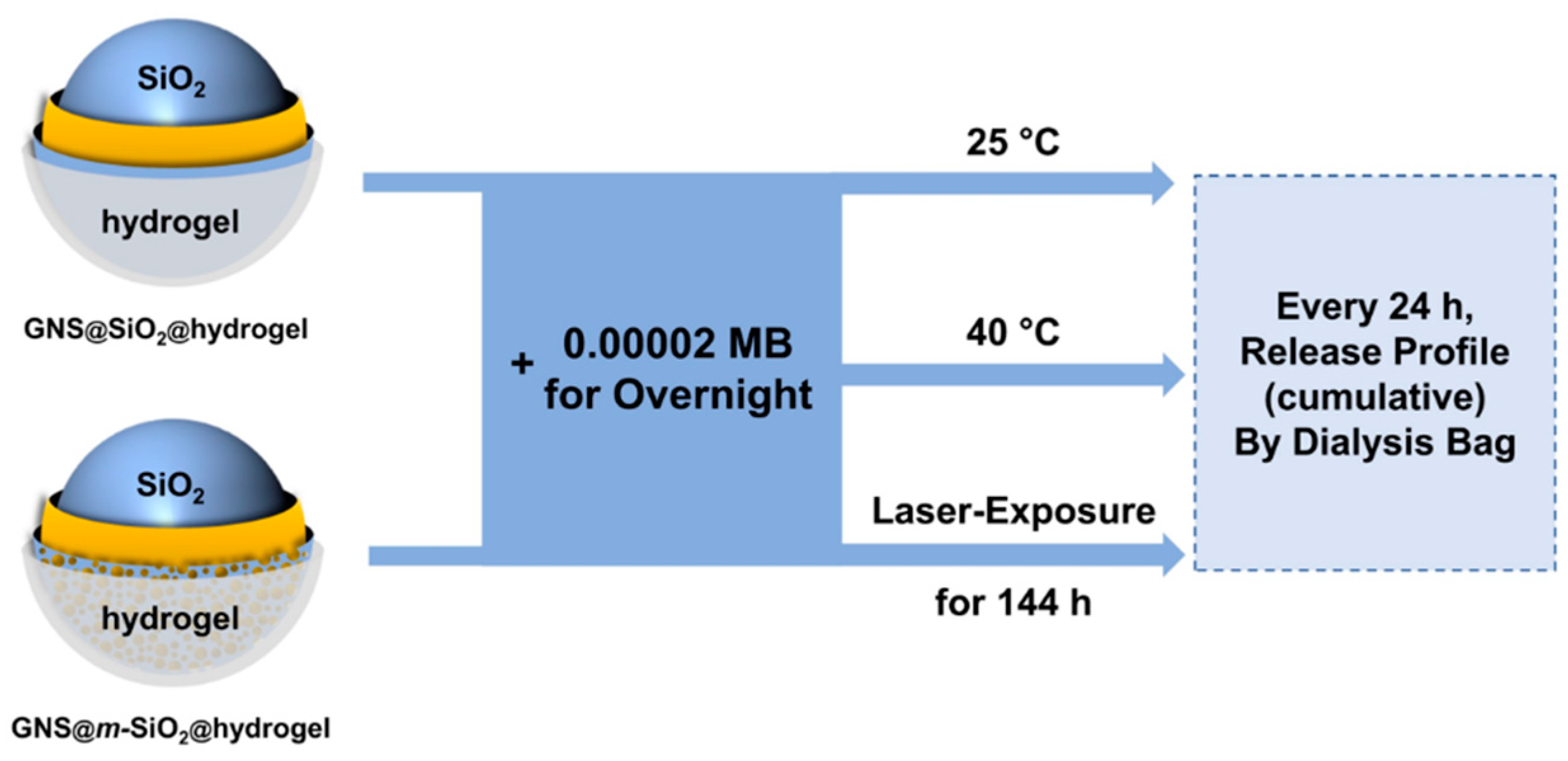
2.5. Photothermal Release Behavior of the Mesoporous Nanoparticles
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Gold Nanoshells
3.3. Synthesis of the Mesoporous Silica Shell
3.4. Synthesis of the Hydrogel Layer
3.5. Loading and Release of Methylene Blue (MB)
3.6. Characterization Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Suzuki, M.; Shinkai, M.; Kamihira, M.; Kobayashi, T. Preparation and characteristics of magnetite-labelled antibody with the use of poly(ethylene glycol) derivatives. Biotechnol. Appl. Biochem. 1995, 21, 335–345. [Google Scholar] [PubMed]
- Tiefenauer, L.X.; Kuehne, G.; Andres, R.Y. Antibody-magnetite nanoparticles: In vitro characterization of a potential tumor-specific contrast agent for magnetic resonance imaging. Bioconj. Chem. 1993, 4, 347–352. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Case, B.L.; Liu, Y.-S.; Caraway, J.W. Poly(N-isopropylacrylamide) soluble polymer supports in catalysis and synthesis. Macromolecules 1998, 31, 6053–6062. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Lee, D.S.; Kim, S.W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388, 860–862. [Google Scholar] [CrossRef] [PubMed]
- Quaroni, L.; Chumanov, G. Preparation of polymer-coated functionalized silver nanoparticles. J. Am. Chem. Soc. 1999, 121, 10642–10643. [Google Scholar] [CrossRef]
- Teranishi, T.; Miyake, M. Size control of palladium nanoparticles and their crystal structures. Chem. Mater. 1998, 10, 594–600. [Google Scholar] [CrossRef]
- Yue, Q.; Li, J.; Luo, W.; Zhang, Y.; Elzatahry, A.A.; Wang, X.; Wang, C.; Li, W.; Cheng, X.; Alghamdi, A.; et al. An interface coassembly in biliquid phase: Toward core–shell magnetic mesoporous silica microspheres with tunable pore size. J. Am. Chem. Soc. 2015, 137, 13282–13289. [Google Scholar] [CrossRef]
- Li, W.-P.; Liao, P.-Y.; Su, C.-H.; Yeh, C.-S. Formation of oligonucleotide-gated silica shell-coated Fe3O4-Au core–shell nanotrisoctahedra for magnetically targeted and near-infrared light-responsive theranostic platform. J. Am. Chem. Soc. 2014, 136, 10062–10075. [Google Scholar] [CrossRef]
- Wang, D.-W.; Zhu, X.-M.; Lee, S.-F.; Chan, H.-M.; Li, H.-W.; Kong, S.K.; Yu, J.C.; Cheng, C.H.K.; Wang, Y.-X.J.; Leung, K.C.-F. Folate-conjugated Fe3O4@SiO2@gold nanorods@mesoporous SiO2 hybrid nanomaterial: A theranostic agent for magnetic resonance imaging and photothermal therapy. J. Mater. Chem. B 2013, 1, 2934–2942. [Google Scholar] [CrossRef]
- Jain, P.K.; Huang, X.; El-Sayed, I.H.; El-Sayed, M.A. Noble metals on the nanoscale: Optical and photothermal properties and some applications in imaging, sensing, biology, and medicine. Acc. Chem. Res. 2008, 41, 1578–1586. [Google Scholar] [CrossRef]
- Vongsavat, V.; Vittur, B.M.; Bryan, W.W.; Kim, J.-H.; Lee, T.R. Ultrasmall hollow gold–silver nanoshells with extinctions strongly red-shifted to the near-infrared. ACS Appl. Mater. Interfaces 2011, 3, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Jackson, J.B.; Halas, N.J.; Lee, T.R. Preparation and characterization of gold nanoshells coated with self-assembled monolayers. Langmuir 2002, 18, 4915–4920. [Google Scholar] [CrossRef]
- Cao, Y.; Jin, R.; Mirkin, C.A. DNA-modified core−shell Ag/Au nanoparticles. J. Am. Chem. Soc. 2001, 123, 7961–7962. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, M.M.; Hansen, J.L.; Pedersen, T.G.; Gaiduk, P.; Larsen, A.N. Tuning the plasmon resonance of metallic tin nanocrystals In Si-based materials. Appl. Phys. A 2010, 100, 31–37. [Google Scholar] [CrossRef]
- Day, E.S.; Morton, J.G.; West, J.L. Nanoparticles for thermal cancer therapy. J. Biomech. Eng. 2009, 131, 074001–074005. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.R.; Matthias, K.; Matthias, E.; Mark, C. Near-infrared optical properties of ex vivo human skin and subcutaneous tissues measured using the monte carlo inversion technique. Phys. Med. Biol. 1998, 43, 2465–2478. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Susha, A.S.; Caruso, F. Metallodielectric opals of layer-by-layer processed coated colloids. Adv. Mater. 2002, 14, 1160–1164. [Google Scholar] [CrossRef]
- Chen, C.-W.; Serizawa, T.; Akashi, M. Preparation of platinum colloids on polystyrene nanospheres and their catalytic properties in hydrogenation. Chem. Mater. 1999, 11, 1381–1389. [Google Scholar] [CrossRef]
- Siiman, O.; Burshteyn, A. Preparation, microscopy, and flow cytometry with excitation into surface plasmon resonance bands of gold or silver nanoparticles on aminodextran-coated polystyrene beads. J. Phys. Chem. B 2000, 104, 9795–9810. [Google Scholar] [CrossRef]
- Feng, W.; Tan, W.B.; Yong, Z.; Xianping, F.; Minquan, W. Luminescent nanomaterials for biological labelling. Nanotechnology 2006, 17, R1. [Google Scholar]
- Holmes, K.L.; Lantz, L.M. Chapter 9 Protein Labeling with Fluorescent Probes. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2001; Volume 63, pp. 185–204. [Google Scholar]
- Lin, Y.; Weissleder, R.; Tung, C.-H. Novel near-infrared cyanine fluorochromes: synthesis, properties, and bioconjugation. Bioconj. Chem. 2002, 13, 605–610. [Google Scholar] [CrossRef]
- Frangioni, J.V. In vivo near-infrared fluorescence imaging. Curr. Opin. Chem. Biol. 2003, 7, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.; Lee, J.-H. Synergistically integrated nanoparticles as multimodal probes for nanobiotechnology. Acc. Chem. Res. 2008, 41, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liong, M.; Li, Z.; Zink, J.I.; Tamanoi, F. Biocompatibility, biodistribution, and drug-delivery efficiency of mesoporous silica nanoparticles for cancer therapy in animals. Small 2010, 6, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.P.; Padera, R.F.; Langer, R.; Kohane, D.S. The biocompatibility of mesoporous silicates. Biomaterials 2008, 29, 4045–4055. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Vivero-Escoto, J.L.; Trewyn, B.G.; Lin, V.S.Y. Mesoporous silica nanoparticles: Structural design and applications. J. Mater. Chem. 2010, 20, 7924–7937. [Google Scholar] [CrossRef]
- Baeza, A.; Guisasola, E.; Ruiz-Hernández, E.; Vallet-Regí, M. Magnetically triggered multidrug release by hybrid mesoporous silica nanoparticles. Chem. Mater. 2012, 24, 517–524. [Google Scholar] [CrossRef]
- Pelton, R. Temperature-sensitive aqueous microgels. Adv. Colloid Interface Sci. 2000, 85, 1–33. [Google Scholar] [CrossRef]
- Park, H.; Srisombat, L.-O.; Jamison, A.; Liu, T.; Marquez, M.; Park, H.; Lee, S.; Lee, T.-C.; Lee, T. Temperature-responsive hydrogel-coated gold nanoshells. Gels 2018, 4, 28. [Google Scholar] [CrossRef]
- Schild, H.G.; Tirrell, D.A. Microcalorimetric detection of lower critical solution temperatures in aqueous polymer solutions. J. Phys. Chem. 1990, 94, 4352–4356. [Google Scholar] [CrossRef]
- Saunders, B.R.; Vincent, B. thermal and osmotic deswelling of poly(NIPAM) microgel particles. J. Chem. Soc. Faraday Trans. 1996, 92, 3385–3389. [Google Scholar] [CrossRef]
- Zhou, S.; Chu, B. Synthesis and volume phase transition of poly(methacrylic acid-co-N-isopropylacrylamide) microgel particles in water. J. Phys. Chem. B 1998, 102, 1364–1371. [Google Scholar] [CrossRef]
- Winnik, F.M. Phase transition of aqueous poly-(N-isopropylacrylamide) solutions: A study by non-radiative energy transfer. Polymer 1990, 31, 2125–2134. [Google Scholar] [CrossRef]
- Pelton, R.H.; Pelton, H.M.; Morphesis, A.; Rowell, R.L. Particle sizes and electrophoretic mobilities of poly(N-isopropylacrylamide) latex. Langmuir 1989, 5, 816–818. [Google Scholar] [CrossRef]
- Snowden, M.J.; Chowdhry, B.Z.; Vincent, B.; Morris, G.E. Colloidal copolymer microgels of N-isopropylacrylamide and acrylic acid: pH, ionic strength and temperature effects. J. Chem. Soc. Faraday Trans. 1996, 92, 5013–5016. [Google Scholar] [CrossRef]
- Yoshida, R.; Sakai, K.; Okano, T.; Sakurai, Y. Modulating the phase transition temperature and thermosensitivity in N-isopropylacrylamide copolymer gels. J. Biomater. Sci. Polym. Ed. 1995, 6, 585–598. [Google Scholar] [CrossRef]
- Dick, K.; Dhanasekaran, T.; Zhang, Z.; Meisel, D. Size-dependent melting of silica-encapsulated gold nanoparticles. J. Am. Chem. Soc. 2002, 124, 2312–2317. [Google Scholar] [CrossRef]
- Graf, C.; Vossen, D.L.J.; Imhof, A.; Van Blaaderen, A. A general method to coat colloidal particles with silica. Langmuir 2003, 19, 6693–6700. [Google Scholar] [CrossRef]
- Ge, J.; Zhang, Q.; Zhang, T.; Yin, Y. Core–satellite nanocomposite catalysts protected by a porous silica shell: Controllable reactivity, high stability, and magnetic recyclability. Angew. Chem. 2008, 47, 8924–8928. [Google Scholar] [CrossRef]
- Saunders, B.R.; Vincent, B. Microgel particles as model colloids: Theory, properties and applications. Adv. Colloid Interface Sci. 1999, 80, 1–25. [Google Scholar] [CrossRef]
- Saunders, B.R.; Crowther, H.M.; Vincent, B. Poly[(methyl methacrylate)-co-(methacrylic acid)] microgel particles: Swelling control using pH, cononsolvency, and osmotic deswelling. Macromolecules 1997, 30, 482–487. [Google Scholar] [CrossRef]
- Neyret, S.; Vincent, B. The properties of polyampholyte microgel particles prepared by microemulsion polymerization. Polymer 1997, 38, 6129–6134. [Google Scholar] [CrossRef]
- Singhana, B.; Slattery, P.; Chen, A.; Wallace, M.; Melancon, M.P. Light-activatable gold nanoshells for drug delivery applications. AAPS Pharm. Sci. Tech. 2014, 15, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.C.; Wu, X.Y.; Pelton, R.H. Viscometry—A useful tool for studying conformational changes of poly(N-isopropylacrylamide) in solutions. Polymer 1992, 33, 436–438. [Google Scholar] [CrossRef]
- Hu, M.; Chen, J.; Li, Z.-Y.; Au, L.; Hartland, G.V.; Li, X.; Marquez, M.; Xia, Y. Gold nanostructures: Engineering their plasmonic properties for biomedical applications. Chem. Soc. Rev. 2006, 35, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, M.; Mizutani, S.-Y.; Nomura, S. Thermal properties of copolymer gels containing N-isopropylacrylamide. Macromolecules 1996, 29, 2019–2024. [Google Scholar] [CrossRef]
- Kato, E. Volume-phase transition of N-isopropylacrylamide gels induced by hydrostatic pressure. J. Chem. Phys. 1997, 106, 3792–3797. [Google Scholar] [CrossRef]
- Choi, D.-G.; Yu, H.K.; Jang, S.G.; Yang, S.-M. Arrays of binary and ternary particles and their replica pores on patterned microchannels. Chem. Mater. 2003, 15, 4169–4171. [Google Scholar] [CrossRef]
- Ash, C.; Dudec, M.; Donne, K.; Bashford, T. Effect of wavelength and beam width on penetration in light-tissue interaction using computational metod. Lasers Med. Sci. 2017, 32, 1909–1918. [Google Scholar] [CrossRef]
- Mayle, K.M.; Dern, K.R.; Wong, V.K.; Sung, S.; Ding, K.; Rodriguez, A.R.; Taylor, Z.; Zhou, Z.H.; Grundfest, W.S.; Deming, T.J.; et al. Polypeptide-based gold nanoshells for photothermal therapy. SLAS Technol. 2017, 22, 18–25. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Waddell, T.G.; Leyden, D.E.; DeBello, M.T. The nature of organosilane to silica-surface bonding. J. Am. Chem. Soc. 1981, 103, 5303–5307. [Google Scholar] [CrossRef]
- Van Blaaderen, A.; Vrij, A. Synthesis and characterization of monodisperse colloidal organo-silica spheres. J. Colloid Interface Sci. 1993, 156, 1–18. [Google Scholar] [CrossRef]
- Duff, D.G.; Baiker, A.; Edwards, P.P. A new hydrosol of gold clusters. 1. formation and particle size variation. Langmuir 1993, 9, 2301–2309. [Google Scholar] [CrossRef]
- Duff, D.G.; Baiker, A.; Gameson, I.; Edwards, P.P. A new hydrosol of gold clusters. 2. a comparison of some different measurement techniques. Langmuir 1993, 9, 2310–2317. [Google Scholar] [CrossRef]
- Westcott, S.L.; Oldenburg, S.J.; Lee, T.R.; Halas, N.J. Formation and adsorption of clusters of gold nanoparticles onto functionalized silica nanoparticle surfaces. Langmuir 1998, 14, 5396–5401. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.S.; Chen, Y.-T.; Srinoi, P.; Marquez, M.D.; Lee, T.R. Hydrogel-Encapsulated Mesoporous Silica-Coated Gold Nanoshells for Smart Drug Delivery. Int. J. Mol. Sci. 2019, 20, 3422. https://doi.org/10.3390/ijms20143422
Kim BS, Chen Y-T, Srinoi P, Marquez MD, Lee TR. Hydrogel-Encapsulated Mesoporous Silica-Coated Gold Nanoshells for Smart Drug Delivery. International Journal of Molecular Sciences. 2019; 20(14):3422. https://doi.org/10.3390/ijms20143422
Chicago/Turabian StyleKim, Bo Sang, Yi-Ting Chen, Pannaree Srinoi, Maria D. Marquez, and T. Randall Lee. 2019. "Hydrogel-Encapsulated Mesoporous Silica-Coated Gold Nanoshells for Smart Drug Delivery" International Journal of Molecular Sciences 20, no. 14: 3422. https://doi.org/10.3390/ijms20143422
APA StyleKim, B. S., Chen, Y.-T., Srinoi, P., Marquez, M. D., & Lee, T. R. (2019). Hydrogel-Encapsulated Mesoporous Silica-Coated Gold Nanoshells for Smart Drug Delivery. International Journal of Molecular Sciences, 20(14), 3422. https://doi.org/10.3390/ijms20143422