Mast Cells in Cardiovascular Disease: From Bench to Bedside


Abstract
1. Introduction
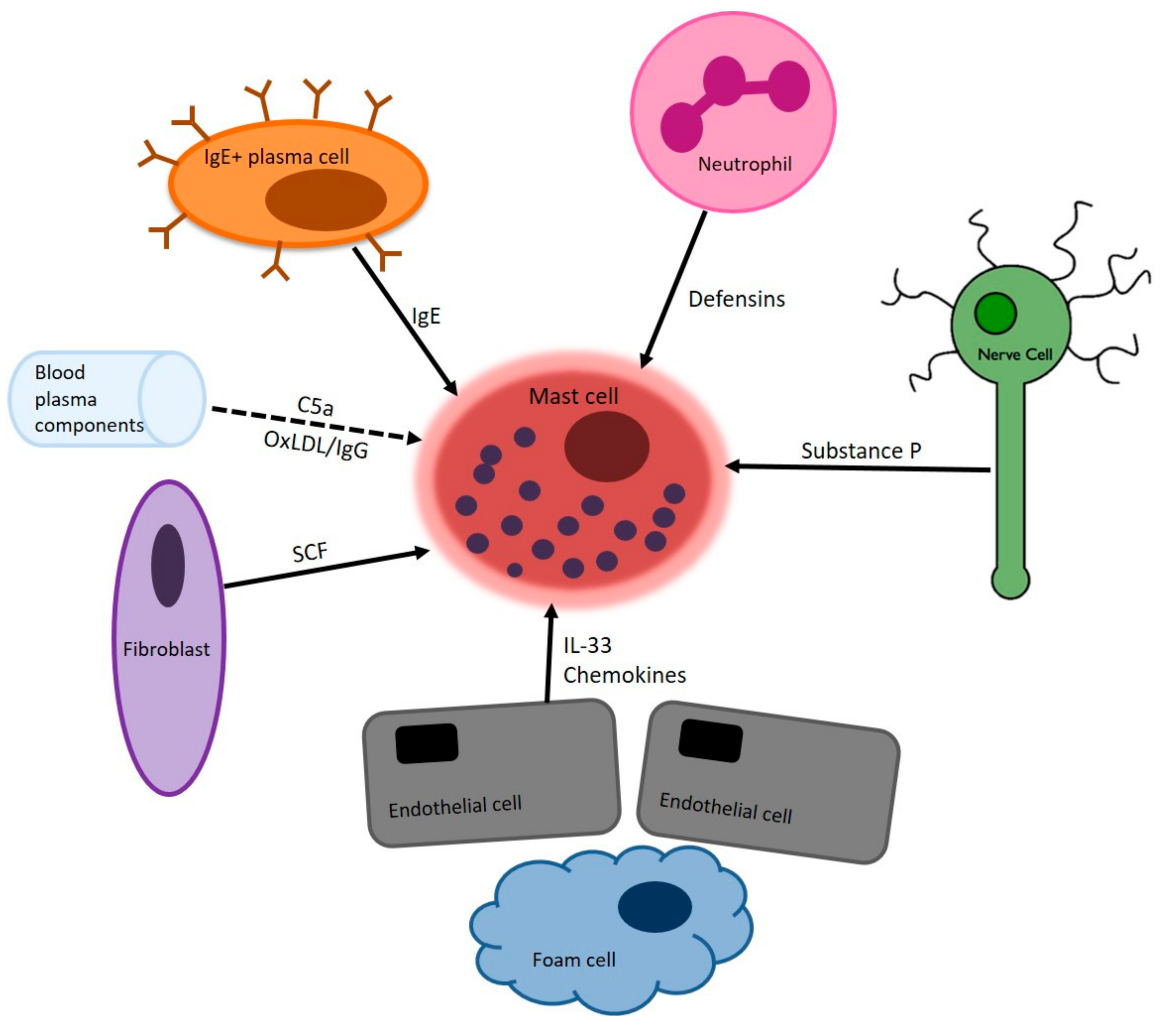
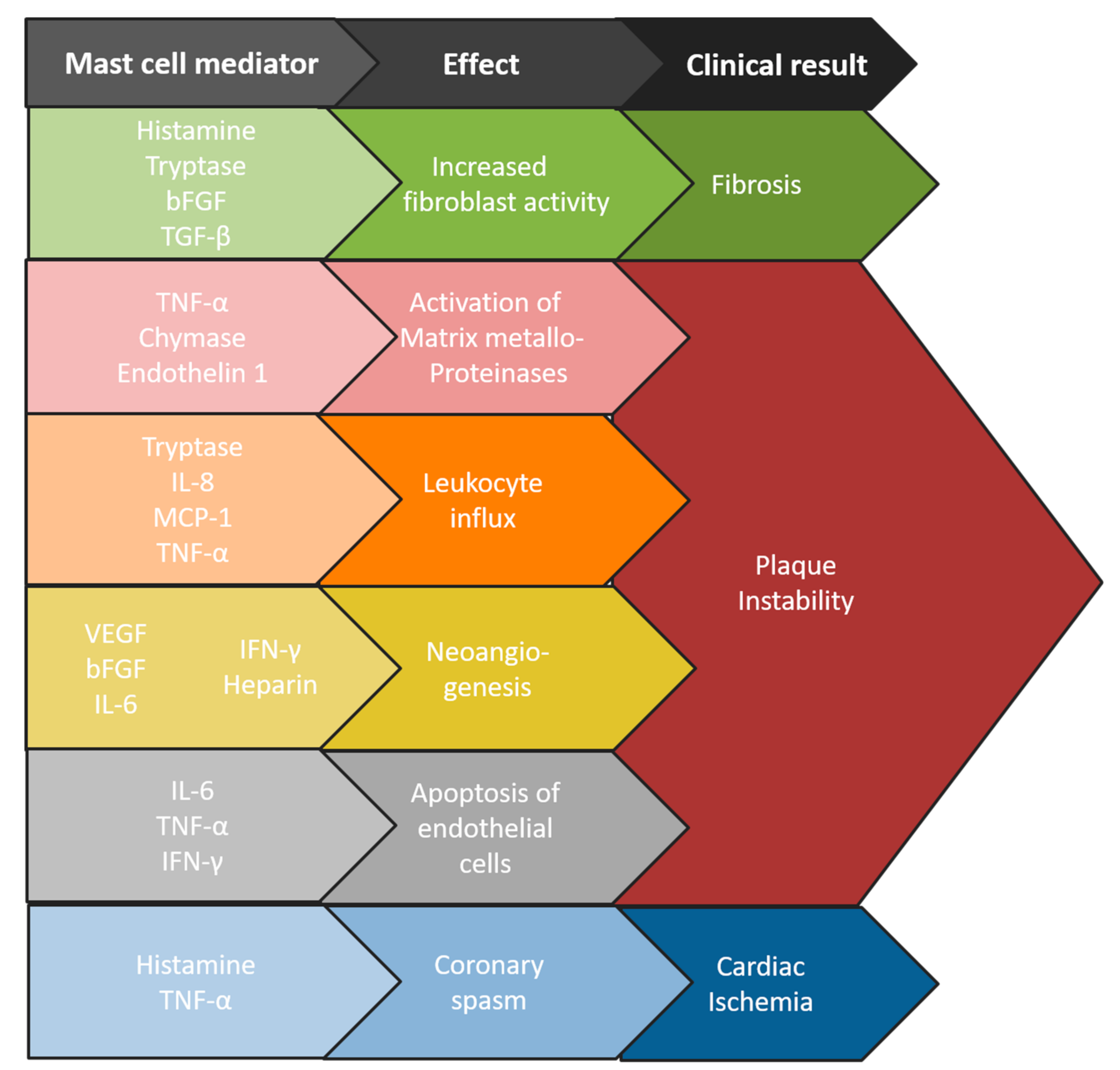
2. Mast Cells in Atherosclerosis
3. Mast Cells in Cardiac Disease
4. Kounis Syndrome
5. Mastocytosis and Cardiovascular Morbidity
6. Allergic Disease and Cardiovascular Morbidity

{kind=link}
{kind=link}
| Cohort | Primary Disease | Comorbidity | Hazard Ratio (95% CI) |
|---|---|---|---|
| Danish population registry [57] | Mastocytosis | Stroke | 1.6 (1.13–2.27) |
| Myocardial infarction | 1.4 (0.9–2.3) | ||
| Dutch case-control study [12] | Mastocytosis | Stroke | 5.0 (0.6–41.3) |
| Coronary heart disease | 2.5 (0.5–12.3) | ||
| Italian population survey [62] | Allergic rhinitis, asthma, or both | Atherosclerosis † | 3.9 (1.3–11.5) |
| Austrian adolescents [62] | Allergic rhinitis, asthma, or both | Atherosclerosis † | 3.0 (1.1–7.9) |
| USA cohort [63] | Allergic rhinitis | Coronary heart disease | 1.40 (1.02–1.92) |
| Wheezing | Coronary heart disease | 2.64 (1.79–3.9) | |
| USA case-matched cohort [64] | Asthma | Coronary heart disease | 1.40 (1.35–1.45) |
| Stroke | 1.20 (1.15–1.25) | ||
| Heart failure | 2.14 (2.06–2.22) | ||
| Polish case-control study [67] | Asthma | Atherosclerotic plaque in LCCA ‡ | 1.2 (0.55–0.91) |
| Atherosclerotic plaque in RCCA ‡ | 0.31 (0.10–0.91) |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huber, M.; Cato, A.C.; Ainooson, G.K.; Freichel, M.; Tsvilovskyy, V.; Jessberger, R.; Riedlinger, E.; Sommerhoff, C.P.; Bischoff, S.C. Regulation of the pleiotropic effects of tissue-resident mast cells. J. Allergy Clin. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Grootens, J.; Ungerstedt, J.S.; Nilsson, G.; Dahlin, J.S. Deciphering the differentiation trajectory from hematopoietic stem cells to mast cells. Blood Adv. 2018, 2, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.; Da Silva, C.A.; Frossard, N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur. J. Pharmacol. 2006, 533, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Caslin, H.L.; Kiwanuka, K.N.; Haque, T.T.; Taruselli, M.T.; Macknight, H.P.; Paranjape, A.; Ryan, J.J. Controlling Mast Cell Activation and Homeostasis: Work Influenced by Bill Paul That Continues Today. Front. Immunol. 2018, 9, 868. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast Cell Mediators: Their Differential Release and the Secretory Pathways Involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P.; Kabesch, M. Current concepts of IgE regulation and impact of genetic determinants. Clin. Exp. Allergy 2012, 42, 852–871. [Google Scholar] [CrossRef]
- Subramanian, H.; Gupta, K.; Ali, H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J. Allergy Clin. Immunol. 2016, 138, 700–710. [Google Scholar] [CrossRef]
- Gaudenzio, N.; Sibilano, R.; Marichal, T.; Starkl, P.; Reber, L.L.; Cenac, N.; McNeil, B.D.; Dong, X.; Hernandez, J.D.; Sagi-Eisenberg, R.; et al. Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Investig. 2016, 126, 3981–3998. [Google Scholar] [CrossRef]
- Wong, G.W.; Zhuo, L.; Kimata, K.; Lam, B.K.; Satoh, N.; Stevens, R.L. Ancient origin of mast cells. Biochem. Biophys. Res. Commun. 2014, 451, 314–318. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Starkl, P.; Marichal, T.; Galli, S.J. IgE and mast cells in host defense against parasites and venoms. Semin. Immunopathol. 2016, 38, 581–603. [Google Scholar] [CrossRef]
- Shi, G.-P.; Bot, I.; Kovanen, P.T. Mast cells in human and experimental cardiometabolic diseases. Nat. Rev. Cardiol. 2015, 12, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Indhirajanti, S.; van Daele, P.L.; Bos, S.; Mulder, M.T.; Bot, I.; van Lennep, J.E.R. Systemic mastocytosis associates with cardiovascular events despite lower plasma lipid levels. Atherosclerosis 2018, 268, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Cairns, A.; Constantinides, P. Mast Cells in Human Atherosclerosis. Science 1954, 120, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, J.O.; Kovanen, P.T. Stimulation of mast cells leads to cholesterol accumulation in macrophages in vitro by a mast cell granule-mediated uptake of low density lipoprotein. Proc. Natl. Acad. Sci. USA 1987, 84, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, P.T. Mast Cell Granule-Mediated Uptake of Low Density Lipoproteins by Macrophages: A Novel Carrier Mechanism Leading to the Formation of Foam Cells. Ann. Med. 1991, 23, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lindstedt, L.K.; Kovanen, P.T. Mast cell-mediated inhibition of reverse cholesterol transport. Arterioscler. Thromb. Vasc. Boil. 1992, 12, 1329–1335. [Google Scholar] [CrossRef]
- Kovanen, P.T.; Kaartinen, M.; Paavonen, T. Infiltrates of Activated Mast Cells at the Site of Coronary Atheromatous Erosion or Rupture in Myocardial Infarction. Circulation 1995, 92, 1084–1088. [Google Scholar] [CrossRef]
- Laine, P.; Kaartinen, M.; Panula, P.; Paavonen, T.; Kovanen, P.T.; Penttilaä, A.; Penttilä, A. Association Between Myocardial Infarction and the Mast Cells in the Adventitia of the Infarct-Related Coronary Artery. Circulation 1999, 99, 361–369. [Google Scholar] [CrossRef]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation 1994, 90, 1669–1678. [Google Scholar] [CrossRef]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Mast cells of two types differing in neutral protease composition in the human aortic intima. Demonstration of tryptase-and tryptase/chymase-containing mast cells in normal intimas, fatty streaks, and the shoulder region of atheromas. Arterioscler. Thromb. Vasc. Boil. 1994, 14, 966–972. [Google Scholar] [CrossRef]
- Willems, S.; Vink, A.; Bot, I.; Quax, P.H.; de Borst, G.J.; de Vries, J.-P.P.; van De Weg, S.M.; Moll, F.L.; Kuiper, J.; Kovanen, P.T.; et al. Mast cells in human carotid atherosclerotic plaques are associated with intraplaque microvessel density and the occurrence of future cardiovascular events. Eur. Hear. J. 2013, 34, 3699–3706. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Mast cells accompany microvessels in human coronary atheromas: Implications for intimal neovascularization and hemorrhage. Atherosclerosis 1996, 123, 123–131. [Google Scholar] [CrossRef]
- Lappalainen, H.; Laine, P.; Sajantila, A.; Kovanen, P.T.; Pentikäinen, M.O. Mast Cells in Neovascularized Human Coronary Plaques Store and Secrete Basic Fibroblast Growth Factor, a Potent Angiogenic Mediator. Arterioscler. Thromb. Vasc. Boil. 2004, 24, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Mayranpaa, M.I.; Trosien, J.A.; Nikkari, S.T.; Kovanen, P.T. Mast cells associate with T-cells and neointimal microvessels in giant cell arteritis. Clin. Exp. Rheumatol. 2008, 26, 63–66. [Google Scholar]
- Kritikou, E.; Depuydt, M.A.C.; de Vries, M.R.; Mulder, K.E.; Govaert, A.M.; Smit, M.D.; van Duijn, J.; Foks, A.C.; Wezel, A.; Smeets, H.J.; et al. Flow Cytometry-Based Characterization of Mast Cells in Human Atherosclerosis. Cells 2019, 8, 334. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sukhova, G.K.; Wolters, P.J.; Yang, M.; Kitamoto, S.; Libby, P.; A Macfarlane, L.; Clair, J.M.-S.; Shi, G.-P. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat. Med. 2007, 13, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Bot, I.; de Jager, S.C.; Zernecke, A.; Lindstedt, K.A.; van Berkel, T.J.; Weber, C.; Biessen, E.A. Perivascular Mast Cells Promote Atherogenesis and Induce Plaque Destabilization in Apolipoprotein E–Deficient Mice. Circulation 2007, 115, 2516–2525. [Google Scholar] [CrossRef]
- Wezel, A.; Lagraauw, H.M.; van Der Velden, D.; de Jager, S.C.; Quax, P.H.; Kuiper, J.; Bot, I. Mast cells mediate neutrophil recruitment during atherosclerotic plaque progression. Atherosclerosis 2015, 241, 289–296. [Google Scholar] [CrossRef]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.-P. Regulation of Endothelial Cell Adhesion Molecule Expression by Mast Cells, Macrophages, and Neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef]
- Heikkila, H.M.; Latti, S.; Leskinen, M.J.; Hakala, J.K.; Kovanen, P.T.; Lindstedt, K.A. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 309–314. [Google Scholar] [CrossRef]
- Leskinen, M.J.; Lindstedt, K.A.; Wang, Y.; Kovanen, P.T. Mast Cell Chymase Induces Smooth Muscle Cell Apoptosis by a Mechanism Involving Fibronectin Degradation and Disruption of Focal Adhesions. Arterioscler. Thromb. Vasc. Boil. 2003, 23, 238–243. [Google Scholar] [CrossRef]
- Leskinen, M.J.; Heikkila, H.M.; Speer, M.Y.; Hakala, J.K.; Laine, M.; Kovanen, P.T.; Lindstedt, K.A. Mast cell chymase induces smooth muscle cell apoptosis by disrupting NF-kappaB-mediated survival signaling. Exp. Cell Res. 2006, 312, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Mayranpaa, M.I.; Heikkila, H.M.; Lindstedt, K.A.; Walls, A.F.; Kovanen, P.T. Desquamation of human coronary artery endothelium by human mast cell proteases: Implications for plaque erosion. Coron. Artery Dis. 2006, 17, 611–621. [Google Scholar] [PubMed]
- Dekker, W.K.D.; Tempel, D.; Bot, I.; Biessen, E.A.; Joosten, L.A.; Netea, M.G.; van Der Meer, J.W.M.; Cheng, C.; Duckers, H.J. Mast Cells Induce Vascular Smooth Muscle Cell Apoptosis via a Toll-Like Receptor 4 Activation Pathway. Arterioscler. Thromb. Vasc. Boil. 2012, 32, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Bot, I.; Bot, M.; van Heiningen, S.H.; van Santbrink, P.J.; Lankhuizen, I.M.; Hartman, P.; Gruener, S.; Hilpert, H.; van Berkel, T.J.; Fingerle, J.; et al. Mast cell chymase inhibition reduces atherosclerotic plaque progression and improves plaque stability in ApoE-/-mice. Cardiovasc. Res. 2011, 89, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Xu, C.; Zhang, H.; Tian, D.; Li, X.; Ning, Y.; Yin, L. Tryptase Promotes Atherosclerotic Plaque Haemorrhage in ApoE-/- Mice. PLoS ONE 2013, 8, e60960. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, M.E.; Oto, A.; Saraclar, Y.; Oram, E.; Oram, A.; Ugurlu, S.; Karamehmetoglu, A.; Karaagaoglu, E. Levels of IgE in the serum of patients with coronary arterial disease. Int. J. Cardiol. 1991, 31, 199–204. [Google Scholar] [CrossRef]
- Kovanen, P.T.; Manttari, M.; Palosuo, T.; Manninen, V.; Aho, K. Prediction of myocardial infarction in dyslipidemic men by elevated levels of immunoglobulin classes A, E, and G, but not M. Arch. Intern. Med. 1998, 158, 1434–1439. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Bot, I.; Kozma, M.O.; Göderle, L.; Perkmann, T.; Hartvigsen, K.; Conrad, D.H.; Kuiper, J.; Mallat, Z.; Binder, C.J.; et al. Increased Plasma IgE Accelerate Atherosclerosis in Secreted IgM Deficiency. Circ. Res. 2017, 120, 78–84. [Google Scholar] [CrossRef]
- Janicki, J.S.; Brower, G.L.; Levick, S.P. The emerging prominence of the cardiac mast cell as a potent mediator of adverse myocardial remodeling. Methods Mol. Biol. 2015, 1220, 121–139. [Google Scholar]
- Frangogiannis, N.G.; Perrard, J.L.; Mendoza, L.H.; Burns, A.R.; Lindsey, M.L.; Ballantyne, C.M.; Michael, L.H.; Smith, C.W.; Entman, M.L. Stem Cell Factor Induction Is Associated with Mast Cell Accumulation after Canine Myocardial Ischemia and Reperfusion. Circulation 1998, 98, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Farwell, K.; Huang, M.; Kempuraj, D.; Donelan, J.; Papaliodis, D.; Vasiadi, M.; Theoharides, T. Mast Cell Deficient W/Wv Mice Have Lower Serum IL-6 and Less Cardiac Tissue Necrosis Than Their Normal Littermates following Myocardial Ischemia-Reperfusion. Int. J. Immunopathol. Pharmacol. 2007, 20, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; Widiapradja, A. Mast Cells: Key Contributors to Cardiac Fibrosis. Int. J. Mol. Sci. 2018, 19, 231. [Google Scholar] [CrossRef] [PubMed]
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 Receptor Antagonists, Left Ventricular Morphology, and Heart Failure Risk: The MESA Study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Shimbori, C.; Upagupta, C.; Bellaye, P.S.; Ayaub, E.A.; Sato, S.; Yanagihara, T.; Zhou, Q.; Ognjanovic, A.; Ask, K.; Gauldie, J.; et al. Mechanical stress-induced mast cell degranulation activates TGF-beta1 signalling pathway in pulmonary fibrosis. Thorax 2019. [Google Scholar] [CrossRef] [PubMed]
- Bradding, P.; Pejler, G. The controversial role of mast cells in fibrosis. Immunol. Rev. 2018, 282, 198–231. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Chen, B.; Zhao, Z.; He, N.; Zeng, Z.; Wu, B.; Fukushima, Y.; Dai, M.; Huang, Q.; Xu, D.; et al. Histamine H2 receptor activation exacerbates myocardial ischemia/reperfusion injury by disturbing mitochondrial and endothelial function. Basic Res. Cardiol. 2013, 108, 342. [Google Scholar] [CrossRef]
- Kounis, N.G.; Zavras, G.M. Histamine-induced coronary artery spasm: The concept of allergic angina. Br. J. Clin. Pract. 1991, 45, 121–128. [Google Scholar]
- Abdelghany, M.; Subedi, R.; Shah, S.; Kozman, H. Kounis syndrome: A review article on epidemiology, diagnostic findings, management and complications of allergic acute coronary syndrome. Int. J. Cardiol. 2017, 232, 1–4. [Google Scholar] [CrossRef]
- González-De-Olano, D.; Matito, A.; Sánchez-López, P.; Sánchez-Muñoz, L.; Morgado, J.; Teodósio, C.; Jara-Acevedo, M.; García-Montero, A.; Orfao, A.; Escribano, L.; et al. Mast cell-related disorders presenting with Kounis syndrome. Int. J. Cardiol. 2012, 161, 56–58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kounis, N.G.; Patsouras, N.; Grapsas, N.; Hahalis, G. Histamine induced coronary artery spasm, fish consumption and Kounis syndrome. Int. J. Cardiol. 2015, 193, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Ghosh, J.; Kapur, R. Mastocytosis: A mutated KIT receptor induced myeloproliferative disorder. Oncotarget 2015, 6, 18250–18264. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A. Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification and management. Am. J. Hematol. 2019, 94, 363–377. [Google Scholar] [CrossRef]
- Brockow, K.; Akin, C.; Huber, M.; Metcalfe, D.D. IL-6 levels predict disease variant and extent of organ involvement in patients with mastocytosis. Clin. Immunol. 2005, 115, 216–223. [Google Scholar] [CrossRef]
- Broesby-Olsen, S.; Farkas, D.K.; Vestergaard, H.; Hermann, A.P.; Møller, M.B.; Mortz, C.G.; Kristensen, T.K.; Bindslev-Jensen, C.; Sørensen, H.T.; Frederiksen, H. Risk of solid cancer, cardiovascular disease, anaphylaxis, osteoporosis and fractures in patients with systemic mastocytosis: A nationwide population-based study. Am. J. Hematol. 2016, 91, 1069–1075. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Atherosclerosis 2016, 252, 207–274. [Google Scholar]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- McFadden, E.R., Jr.; Gilbert, I.A. Asthma. N. Engl. J. Med. 1992, 327, 1928–1937. [Google Scholar] [CrossRef]
- Liu, C.L.; Zhang, J.Y.; Shi, G.P. Interaction between allergic asthma and atherosclerosis. Transl. Res. J. Lab. Clin. Med. 2016, 174, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Knoflach, M.; Kiechl, S.; Mayr, A.; Willeit, J.; Poewe, W.; Wick, G. Allergic Rhinitis, Asthma, and Atherosclerosis in the Bruneck and ARMY Studies. Arch. Intern. Med. 2005, 165, 2521–2526. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Purushottam, B.; Chae, Y.K.; Chebrolu, L.; Amanullah, A. Relation Between Common Allergic Symptoms and Coronary Heart Disease Among NHANES III Participants. Am. J. Cardiol. 2010, 106, 984–987. [Google Scholar] [CrossRef] [PubMed]
- Tolstykh, I.V.; Miller, M.K.; Sobel, E.; Iribarren, C.; Eisner, M.D. Adult Asthma and Risk of Coronary Heart Disease, Cerebrovascular Disease, and Heart Failure: A Prospective Study of 2 Matched Cohorts. Am. J. Epidemiol. 2012, 176, 1014–1024. [Google Scholar]
- Evelein, A.M.; Visseren, F.L.; van der Ent, C.K.; Grobbee, D.E.; Uiterwaal, C.S. Allergies are associated with arterial changes in young children. Eur. J. Prev. Cardiol. 2015, 22, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, J.; Magnussen, C.G.; Sinaiko, A.; Woo, J.; Urbina, E.; Jacobs, D.R., Jr.; Steinberger, J.; Prineas, R.; Sabin, M.A.; Burns, T.; et al. Childhood Age and Associations Between Childhood Metabolic Syndrome and Adult Risk for Metabolic Syndrome, Type 2 Diabetes Mellitus and Carotid Intima Media Thickness: The International Childhood Cardiovascular Cohort Consortium. J. Am. Heart Assoc. 2017, 6, e005632. [Google Scholar] [CrossRef] [PubMed]
- Podgórski, M.; Kupczyk, M.; Grzelak, P.; Bocheńska-Marciniak, M.; Polguj, M.; Kuna, P.; Stefańczyk, L. Inhaled Corticosteroids in Asthma: Promoting or Protecting Against Atherosclerosis? Med. Sci. Monit. 2017, 23, 5337–5344. [Google Scholar] [CrossRef]
- Potaczek, D.P. Links between allergy and cardiovascular or hemostatic system. Int. J. Cardiol. 2014, 170, 278–285. [Google Scholar] [CrossRef]
- Naqvi, T.Z.; Lee, M.-S. Carotid Intima-Media Thickness and Plaque in Cardiovascular Risk Assessment. JACC Cardiovasc. Imaging 2014, 7, 1025–1038. [Google Scholar] [CrossRef]
- Pimenta, E.; Wolley, M.; Stowasser, M. Adverse Cardiovascular Outcomes of Corticosteroid Excess. Endocrinology 2012, 153, 5137–5142. [Google Scholar] [CrossRef]
- Kantor, R.; Kim, A.; Thyssen, J.P.; Silverberg, J.I. Association of atopic dermatitis with smoking: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2016, 75, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Silverberg, J.I. Association of atopic dermatitis with being overweight and obese: A systematic review and metaanalysis. J. Am. Acad. Dermatol. 2015, 72, 606–616.e4. [Google Scholar] [CrossRef] [PubMed]
- Miethe, S.; Guarino, M.; Alhamdan, F.; Simon, H.-U.; Renz, H.; Dufour, J.-F.; Potaczek, D.P.; Garn, H. The effects of obesity on asthma: Immunometabolic links. Pol. Arch. Intern. Med. 2018, 128, 469–477. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermans, M.A.W.; Roeters van Lennep, J.E.; van Daele, P.L.A.; Bot, I. Mast Cells in Cardiovascular Disease: From Bench to Bedside. Int. J. Mol. Sci. 2019, 20, 3395. https://doi.org/10.3390/ijms20143395
Hermans MAW, Roeters van Lennep JE, van Daele PLA, Bot I. Mast Cells in Cardiovascular Disease: From Bench to Bedside. International Journal of Molecular Sciences. 2019; 20(14):3395. https://doi.org/10.3390/ijms20143395
Chicago/Turabian StyleHermans, M. A. W., J. E. Roeters van Lennep, P. L. A. van Daele, and I. Bot. 2019. "Mast Cells in Cardiovascular Disease: From Bench to Bedside" International Journal of Molecular Sciences 20, no. 14: 3395. https://doi.org/10.3390/ijms20143395
APA StyleHermans, M. A. W., Roeters van Lennep, J. E., van Daele, P. L. A., & Bot, I. (2019). Mast Cells in Cardiovascular Disease: From Bench to Bedside. International Journal of Molecular Sciences, 20(14), 3395. https://doi.org/10.3390/ijms20143395