Epileptic Encephalopathy In A Patient With A Novel Variant In The Kv7.2 S2 Transmembrane Segment: Clinical, Genetic, and Functional Features

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Genetic and Clinical Description of the Proband
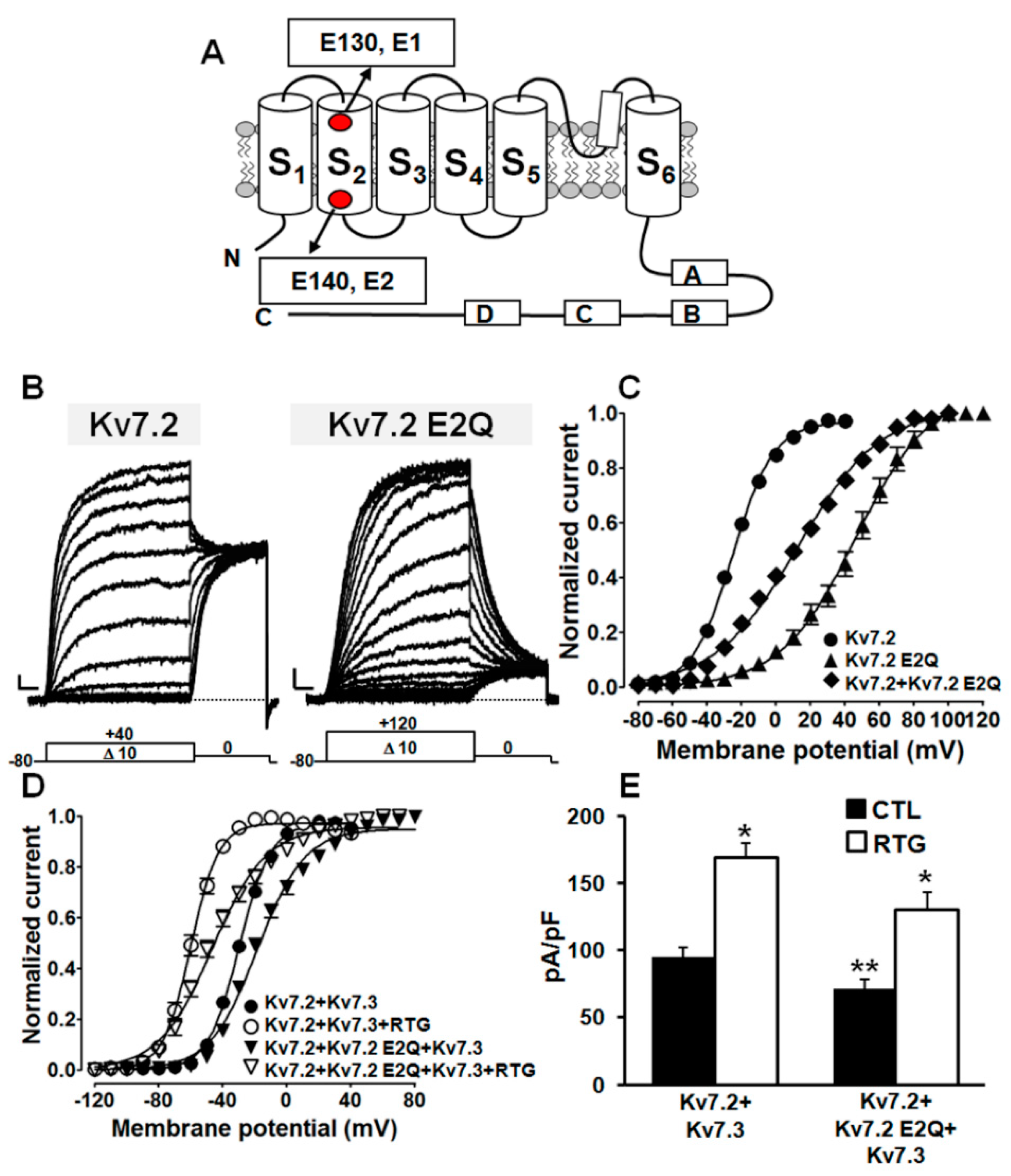
2.2. The E140Q Substitution Prompts Loss-of-Function Effects on Kv7.2 Channels
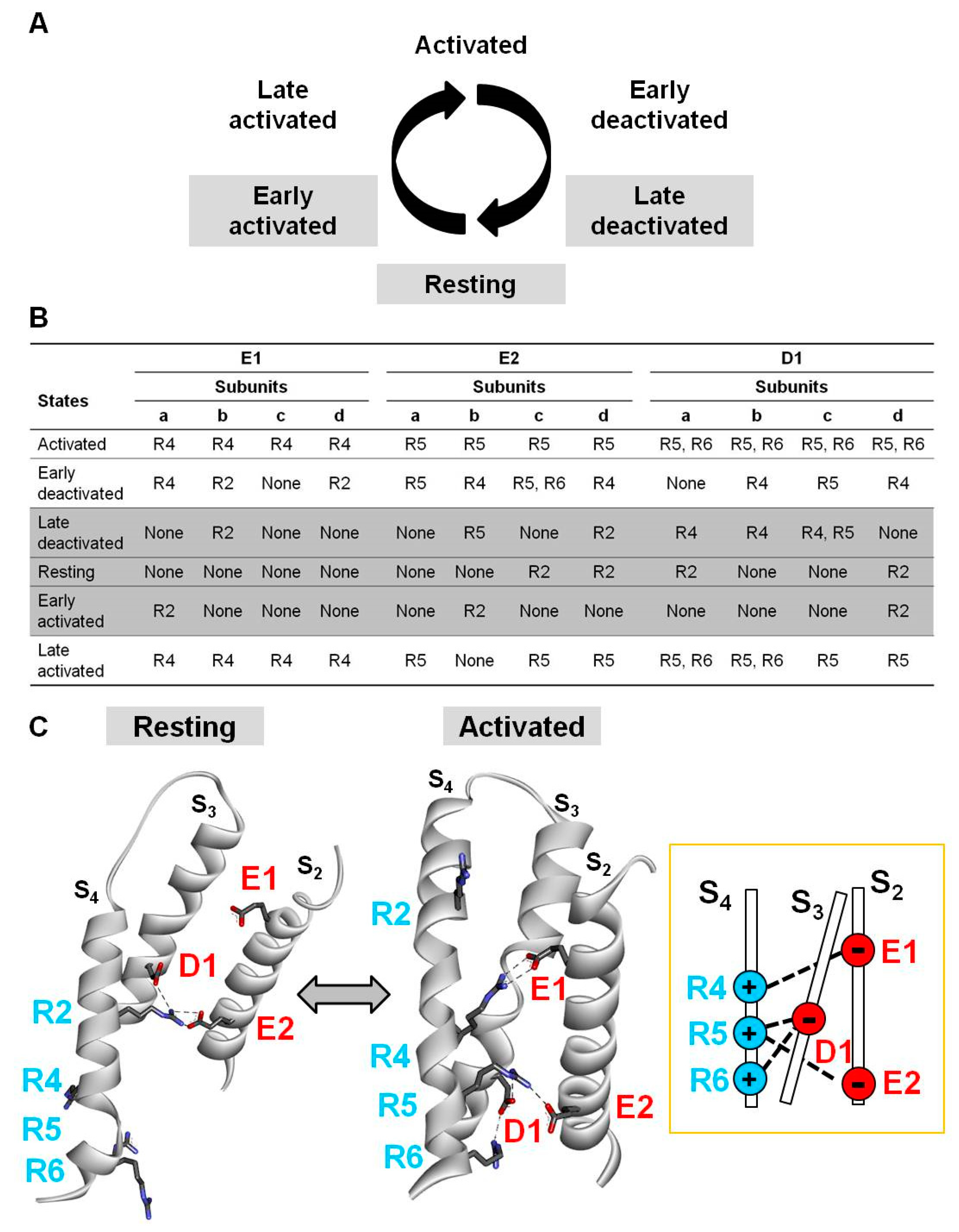
2.3. Electrostatic Interactions in Distinct Kv7.2 VSD States Revealed by Homology Modelling
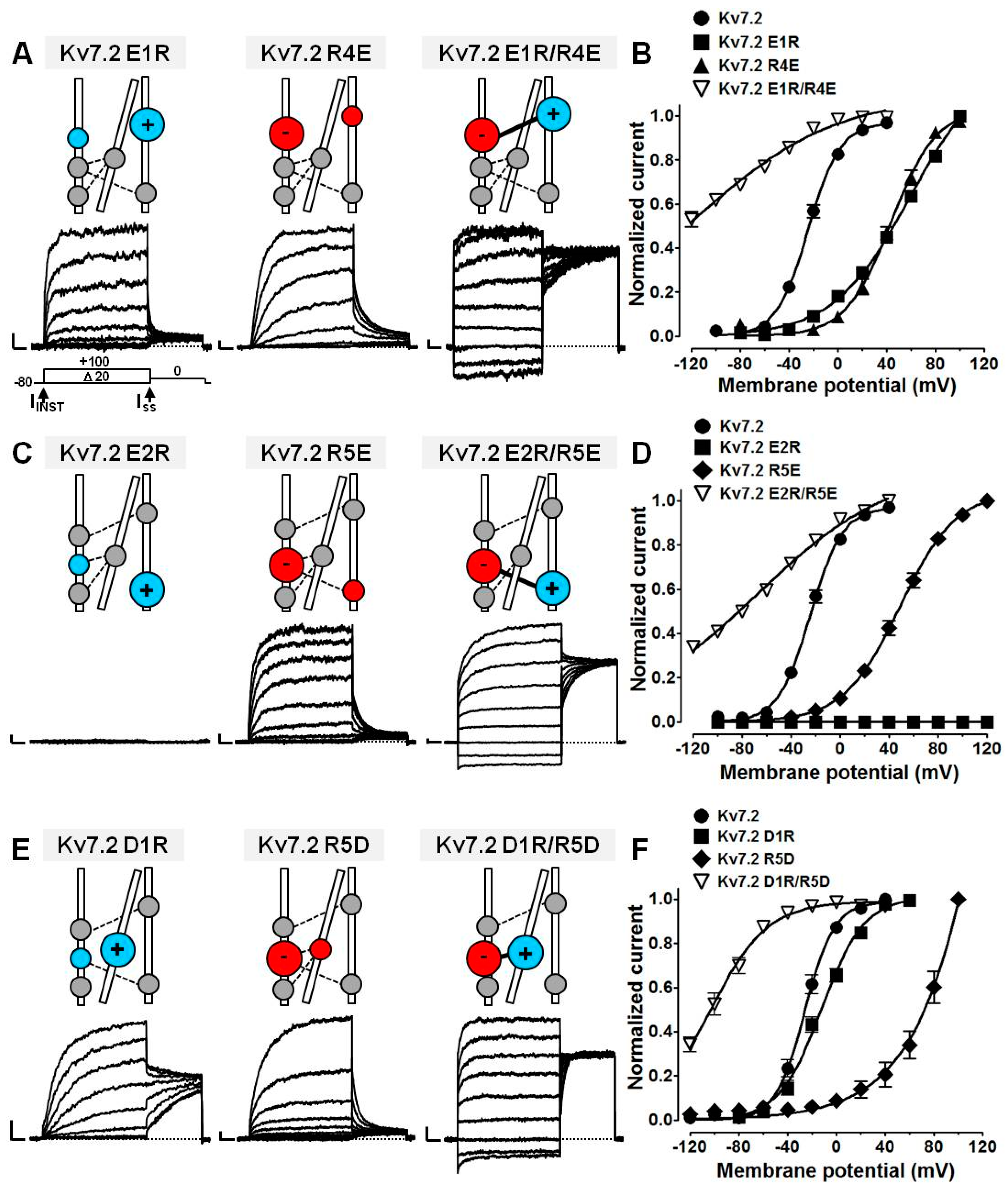
2.4. Probing E1/R4, E2/R5, D1/R5, and D1/R6 Interactions Using Coupled Charge Reversal Strategy
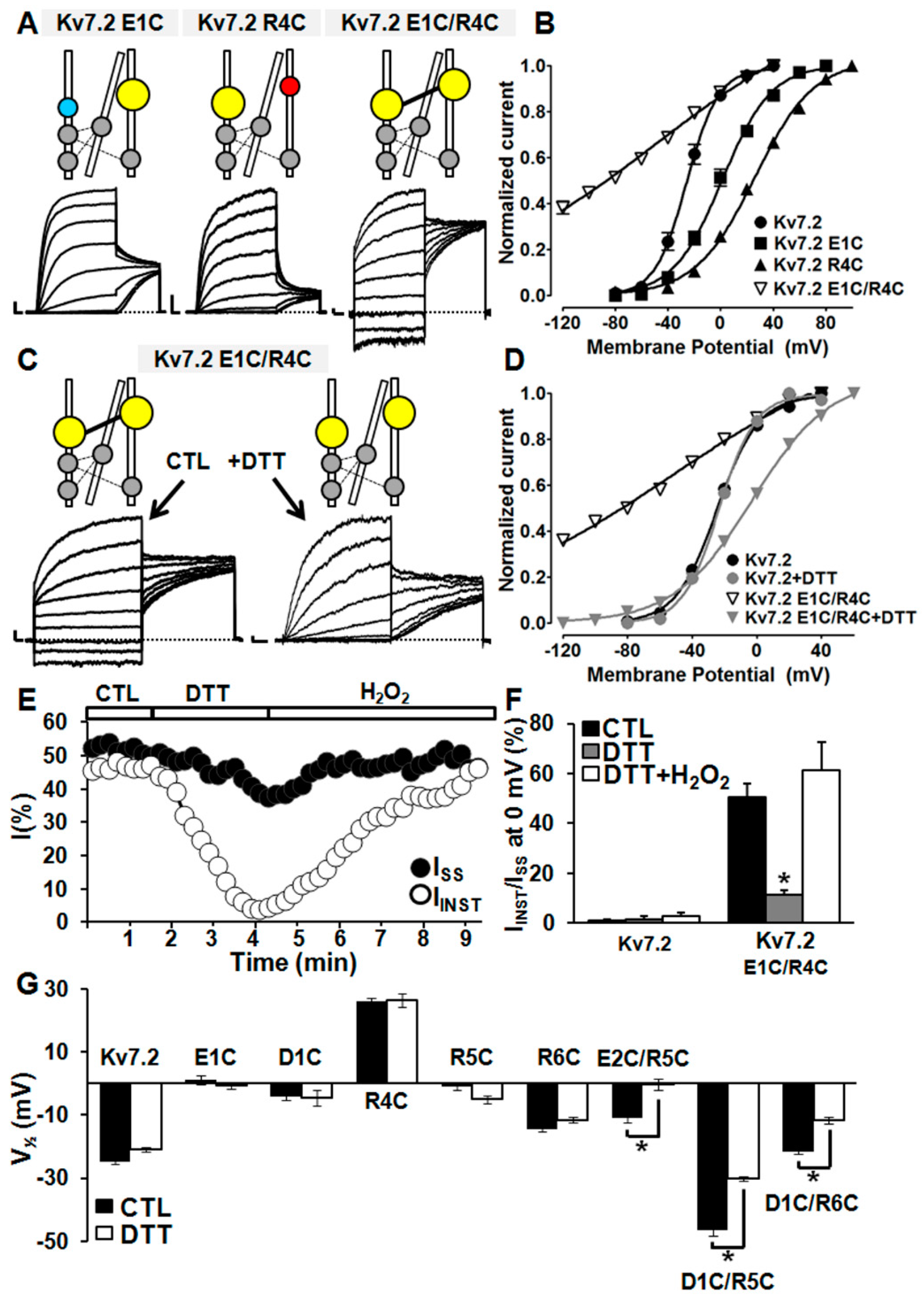
2.5. Probing E1/R4, E2/R5, D1/R5, and D1/R6 Interactions Using Cysteine Substitutions in Disulfide Trapping Experiments
3. Materials and Methods
3.1. Genetic Testing
3.2. Mutagenesis
3.3. Cell Cultures and Transfections
3.4. Cell Surface Biotinylation and Western Blot
3.5. Whole-Cell Electrophysiology
3.6. Multistate Structural Modelling
3.7. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooper, E.C.; Harrington, E.; Jan, Y.N.; Jan, L.Y. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 2001, 21, 9529–9540. [Google Scholar] [CrossRef] [PubMed]
- Soldovieri, M.V.; Miceli, F.; Taglialatela, M. Driving With No Brakes: Molecular Pathophysiology of Kv7 Potassium Channels. Physiology 2011, 26, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Schwake, M.; Athanasiadu, D.; Beimgraben, C.; Blanz, J.; Beck, C.; Jentsch, T.J.; Saftig, P.; Friedrich, T. Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J. Neurosci. 2006, 26, 3757–3766. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.J.; Clark, K.A.; Holton, J.M.; Minor, D.L. Structural Insight into KCNQ (Kv7) Channel Assembly and Channelopathy. Neuron 2007, 53, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohács, T.; Lopes, C.M.B.; Jin, T.; Logothetis, D.E. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef]
- Soldovieri, M.V.; Ambrosino, P.; Mosca, I.; De Maria, M.; Moretto, E.; Miceli, F.; Alaimo, A.; Iraci, N.; Manocchio, L.; Medoro, A.; et al. Early-onset epileptic encephalopathy caused by a reduced sensitivity of Kv7.2 potassium channels to phosphatidylinositol 4,5-bisphosphate. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yus-Nájera, E.; Santana-Castro, I.; Villarroel, A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 2002, 277, 28545–28553. [Google Scholar] [CrossRef]
- Ambrosino, P.; Alaimo, A.; Bartollino, S.; Manocchio, L.; De Maria, M.; Mosca, I.; Gomis-Perez, C.; Alberdi, A.; Scambia, G.; Lesca, G.; et al. Epilepsy-causing mutations in Kv7.2 C-terminus affect binding and functional modulation by calmodulin. Biochim. Biophys. Acta—Mol. Basis Dis. 2015, 1852, 1856–1866. [Google Scholar] [CrossRef]
- Gomis-Perez, C.; Soldovieri, M.V.; Malo, C.; Ambrosino, P.; Taglialatela, M.; Areso, P.; Villarroel, A. Differential regulation of PI(4,5)P2 sensitivity of kv7.2 and kv7.3 channels by calmodulin. Front. Mol. Neurosci. 2017, 10, 117. [Google Scholar] [CrossRef]
- Regev, N.; Degani-Katzav, N.; Korngreen, A.; Etzioni, A.; Siloni, S.; Alaimo, A.; Chikvashvili, D.; Villarroel, A.; Attali, B.; Lotan, I. Selective interaction of syntaxin 1A with KCNQ2: Possible implications for specific modulation of presynaptic activity. PLoS ONE 2009, 4, e6586. [Google Scholar] [CrossRef]
- Soldovieri, M.V.; Boutry-Kryza, N.; Milh, M.; Doummar, D.; Heron, B.; Bourel, E.; Ambrosino, P.; Miceli, F.; De Maria, M.; Dorison, N.; et al. Novel KCNQ2 and KCNQ3 mutations in a large cohort of families with benign neonatal epilepsy: First evidence for an altered channel regulation by syntaxin-1A. Hum. Mutat. 2014, 35, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, N.; Zhang, J.S.; Omaki, M.; Takeuchi, T.; Yokoyama, S.; Wanaverbecq, N.; Langeberg, L.K.; Yoneda, Y.; Scott, J.D.; Brown, D.A.; et al. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 2003, 6, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z. A Common Ankyrin-G-Based Mechanism Retains KCNQ and NaV Channels at Electrically Active Domains of the Axon. J. Neurosci. 2006, 26, 2599–2613. [Google Scholar] [CrossRef] [PubMed]
- Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; Barrese, V.; Migliore, M.; Cilio, M.R.; Taglialatela, M. Genotype–phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of Kv7.2 potassium channel subunits. Proc. Natl. Acad. Sci. USA 2013, 110, 4386–4391. [Google Scholar] [CrossRef] [PubMed]
- Orhan, G.; Bock, M.; Schepers, D.; Ilina, E.I.; Reichel, S.N.; Loffler, H.; Jezutkovic, N.; Weckhuysen, S.; Mandelstam, S.; Suls, A.; et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 2014, 75, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef]
- Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; De Maria, M.; Migliore, M.; Migliore, R.; Taglialatela, M. Early-Onset Epileptic Encephalopathy Caused by Gain-of-Function Mutations in the Voltage Sensor of Kv7.2 and Kv7.3 Potassium Channel Subunits. J. Neurosci. 2015, 35, 3782–3793. [Google Scholar] [CrossRef]
- Millichap, J.J.; Miceli, F.; De Maria, M.; Keator, C.; Joshi, N.; Tran, B.; Soldovieri, M.V.; Ambrosino, P.; Shashi, V.; Mikati, M.A.; et al. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 2017, 58, e10–e15. [Google Scholar] [CrossRef]
- Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; Manocchio, L.; Mosca, I.; Taglialatela, M. Pharmacological Targeting of Neuronal Kv7.2/3 Channels: A Focus on Chemotypes and Receptor Sites. Curr. Med. Chem. 2018, 25, 2637–2660. [Google Scholar] [CrossRef]
- Ihara, Y.; Tomonoh, Y.; Deshimaru, M.; Zhang, B.; Uchida, T.; Ishii, A.; Hirose, S. Retigabine, a Kv7.2/Kv7.3-channel opener, attenuates drug-induced seizures in knock-in mice harboring Kcnq2 mutations. PLoS ONE 2016, 11, e0150095. [Google Scholar] [CrossRef] [PubMed]
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Joshi, N.; Levisohn, P.M.; Marsh, E.; Nangia, S.; et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.F.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Ivanovic, V.; Hendrickx, R.; Van Coster, R.; Hjalgrim, H.; Møller, R.S.; Grønborg, S.; Schoonjans, A.S.; Ceulemans, B.; Heavin, S.B.; et al. Extending the KCNQ2 encephalopathy spectrum: Clinical and neuroimaging findings in 17 patients. Neurology 2013, 81, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Podesta, B.; Thibert, R.L.; Shapiro, K.A.; Guerrini, R.; et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015, 56, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.S.; Barnes, P.D.; Clegg, N.J.; Stashinko, E.E. Septopreoptic holoprosencephaly: A mild subtype associated with midline craniofacial anomalies. Am. J. Neuroradiol. 2010, 31, 1596–1601. [Google Scholar] [CrossRef]
- Dubourg, C.; Carre, W.; Hamdi-Roze, H.; Mouden, C.; Roume, J.; Abdelmajid, B.; Amram, D.; Baumann, C.; Chassaing, N.; Coubes, C.; et al. Mutational Spectrum in Holoprosencephaly Shows That FGF is a New Major Signaling Pathway. Hum. Mutat. 2016, 37, 1329–1339. [Google Scholar] [CrossRef]
- Roessler, E.; El-Jaick, K.B.; Dubourg, C.; Velez, J.I.; Solomon, B.D.; Pineda-Alvarez, D.E.; Lacbawan, F.; Zhou, N.; Ouspenskaia, M.; Paulussen, A.; et al. The mutational spectrum of holoprosencephaly-associated changes within the SHH gene in humans predicts loss-of-function through either key structural alterations of the ligand or its altered synthesis. Hum. Mutat. 2009, 30, E921–E935. [Google Scholar] [CrossRef]
- Lenkov, D.N.; Volnova, A.B.; Pope, A.R.D.; Tsytsarev, V. Advantages and limitations of brain imaging methods in the research of absence epilepsy in humans and animal models. J. Neurosci. Methods 2013, 212, 195–202. [Google Scholar] [CrossRef]
- Stewart, A.P.; Gómez-Posada, J.C.; McGeorge, J.; Rouhani, M.J.; Villarroel, A.; Murrell-Lagnado, R.D.; Edwardson, J.M. The Kv7.2/Kv7.3 heterotetramer assembles with a random subunit arrangement. J. Biol. Chem. 2012, 287, 11870–11877. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature 1991, 350, 232–235. [Google Scholar] [CrossRef]
- Minassian, N.A.; Lin, M.-C.A.; Papazian, D.M. Altered Kv3.3 channel gating in early-onset spinocerebellar ataxia type 13. J. Physiol. 2012, 590, 1599–1614. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Tinel, N.; Lauritzen, I.; Chouabe, C.; Lazdunski, M.; Borsotto, M.; Antipolis, S. The KCNQ2 potassium channel splice varinats functional and develpmental expression. FEBS Lett. 1998, 438, 171–176. [Google Scholar] [CrossRef]
- Hadley, J.K.; Passmore, G.M.; Tatulian, L.; Al-Qatari, M.; Ye, F.; Wickenden, A.D.; Brown, D.A. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J. Neurosci. 2003, 23, 5012–5019. [Google Scholar] [CrossRef]
- Kanaumi, T.; Takashima, S.; Iwasaki, H.; Itoh, M.; Mitsudome, A.; Hirose, S. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: Possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 2008, 30, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Pérez, C.; Urrutia, J.; Marcé-Grau, A.; Malo, C.; López-Laso, E.; Felipe-Rucián, A.; Raspall-Chaure, M.; Macaya, A.; Villarroel, A. Homomeric Kv7.2 current suppression is a common feature in KCNQ2 epileptic encephalopathy. Epilepsia 2019, 60, 139–148. [Google Scholar] [CrossRef]
- Long, S.B.; Tao, X.; Campbell, E.B.; Mackinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450. [Google Scholar] [CrossRef]
- Jensen, M.Ø.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of voltage gating in potassium channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef]
- Meisel, E.; Dvir, M.; Haitin, Y.; Giladi, M.; Peretz, A.; Attali, B. KCNQ1 channels do not undergo concerted but sequential gating transitions in both the absence and the presence of KCNE1 protein. J. Biol. Chem. 2012, 287, 34212–34224. [Google Scholar] [CrossRef] [PubMed]
- Miceli, F.; Soldovieri, M.V.; Hernandez, C.C.; Shapiro, M.S.; Annunziato, L.; Taglialatela, M. Gating consequences of charge neutralization of arginine residues in the S4 segment of Kv7.2, an epilepsy-linked K+ channel subunit. Biophys. J. 2008, 95, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Gourgy-Hacohen, O.; Kornilov, P.; Pittel, I.; Peretz, A.; Attali, B.; Paas, Y. Capturing distinct KCNQ2 channel resting states by metal ion bridges in the voltage-sensor domain. J. Gen. Physiol. 2014, 144, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, P.; del Giudice, E.M.; Coppola, G.; Pascotto, A.; Annunziato, L.; Taglialatela, M. Benign familial neonatal convulsions caused by altered gating of KCNQ2/KCNQ3 potassium channels. J. Neurosci. 2002, 22, RC199. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Delaloye, K.; Zaydman, M.A.; Nekouzadeh, A.; Rudy, Y.; Cui, J. State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J. Gen. Physiol. 2010, 135, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.; Eldstrom, J.; Murray, C.I.; Thompson, E.; Fedida, D. IKs ion-channel pore conductance can result from individual voltage sensor movements. Proc. Natl. Acad. Sci. USA 2019, 116, 7879–7888. [Google Scholar] [CrossRef] [PubMed]
- Gamper, N.; Zaika, O.; Li, Y.; Martin, P.; Hernandez, C.C.; Perez, M.R.; Wang, A.Y.C.; Jaffe, D.B.; Shapiro, M.S. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006, 25, 4996–5004. [Google Scholar] [CrossRef]
- Miceli, F.; Soldovieri, M.V.; Lugli, L.; Bellini, G.; Ambrosino, P.; Migliore, M.; del Giudice, E.M.; Ferrari, F.; Pascotto, A.; Taglialatela, M. Neutralization of a unique, negatively-charged residue in the voltage sensor of KV7.2 subunits in a sporadic case of benign familial neonatal seizures. Neurobiol. Dis. 2009, 34, 501–510. [Google Scholar] [CrossRef]
- Miceli, F.; Vargas, E.; Bezanilla, F.; Taglialatela, M. Gating currents from K v7 channels carrying neuronal hyperexcitability mutations in the voltage-sensing domain. Biophys. J. 2012, 102, 1372–1382. [Google Scholar] [CrossRef]
- Soldovieri, M.V.; Cilio, M.R.; Miceli, F.; Bellini, G.; Miraglia del Giudice, E.; Castaldo, P.; Hernandez, C.C.; Shapiro, M.S.; Pascotto, A.; Annunziato, L.; et al. Atypical Gating Of M-Type Potassium Channels Conferred by Mutations in Uncharged Residues in the S4 Region of KCNQ2 Causing Benign Familial Neonatal Convulsions. J. Neurosci. 2007, 27, 4919–4928. [Google Scholar] [CrossRef]
- Hortigüela, M.; Fernández-Marmiesse, A.; Cantarín, V.; Gouveia, S.; García-Peñas, J.J.; Fons, C.; Armstrong, J.; Barrios, D.; Díaz-Flores, F.; Tirado, P.; et al. Clinical and genetic features of 13 Spanish patients with KCNQ2 mutations. J. Hum. Genet. 2017, 62, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Sands, T.T.; Balestri, M.; Bellini, G.; Mulkey, S.B.; Danhaive, O.; Bakken, E.H.; Taglialatela, M.; Oldham, M.S.; Vigevano, F.; Holmes, G.L.; et al. Rapid and safe response to low-dose carbamazepine in neonatal epilepsy. Epilepsia 2016, 57, 2019–2030. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Channel | n | Current Density (pA/pF) # | V1/2 (mV) | k (mV/e-Fold) |
|---|---|---|---|---|
| Kv7.2 | 44–108 | 33.2 ± 2.0 (0 mV) | −25.0 ± 0.4 | 11.7 ± 0.3 |
| Kv7.2 E2Q | 12 | 19.6 ± 4.5 (+80 mV) * | 46.1 ± 2.2 * | 21.8 ± 1.3 * |
| Kv7.2 E2Q+Kv7.2 | 13 | 39.8 ± 4.0 (+80 mV) | 12.4 ± 1.0 * | 24.0 ± 0.7 * |
| Kv7.3 | 28 | 12.8 ± 1.2 (0 mV) | −38.6 ± 0.4 | 6.3 ± 0.4 |
| Kv7.2 E2Q+Kv7.3 | 18 | 97.2 ± 7.8 (+80 mV) ** | 14.2 ± 1.0 ** | 22.0 ± 0.7 ** |
| Kv7.2+Kv7.3 | 25 | 165.2 ± 8.8 (0 mV) | −29.7 ± 0.4 | 9.8 ± 0.3 |
| Kv7.2+Kv7.2 E2Q+Kv7.3 | 21 | 119.8 ± 11.7 (+40 mV) ** | −18.4 ± 0.7 ** | 14.8 ± 0.6 ** |
| Kv7.2 E1R | 7 | 29.0 ± 4.1 (+100 mV) | 59.5 ± 3.4 * | 31.7 ± 1.5 * |
| Kv7.2 E2R | 23 | 0.8 ± 0.1 (0 mV) * | - | - |
| Kv7.2 D1R | 12–14 | 13.2 ± 2.2 (+40 mV) * | −11.9 ± 1.5 * | 17.5 ± 1.2 * |
| Kv7.2 R4E | 12–13 | 31.8 ± 6.5 (+100 mV) | 43.8 ± 1.9 * | 18.7 ± 1.4 * |
| Kv7.2 R5E | 16 | 73.7 ± 7.8 (+120 mV) * | 49.1 ± 1.7 * | 23.1 ± 1.2 * |
| Kv7.2 R5D | 8 | 26.9 ± 7.1 (+100mV) | ≈200 * | 39.0 ± 7.5 * |
| Kv7.2 R6D | 7 | 0.2 ± 0.2 (0 mV) * | - | - |
| Kv7.2 E1R/R4E | 27–30 | 62.2 ± 6.8 (0 mV) * | - | - |
| Kv7.2 E1R+Kv7.2 R4E | 5 | 56.3 ± 3.4 (+100 mV) * | 37.7 ± 1.8 * | 22.8 ± 1.2 * |
| Kv7.2 E2R/R5E | 13 | 30.4 ± 5.1 (0 mV) | - | - |
| Kv7.2 E2R+Kv7.2 R5E | 5–6 | 50.0 ± 6.2 (+120 mV) * | 53.7 ± 1.7 * | 24.1 ± 1.1 * |
| Kv7.2 D1R/R5D | 16 | 54.3 ± 11.7 (+0 mV) * | - | - |
| Kv7.2 D1R+Kv7.2 R5D | 9–13 | 30.2 ± 4.4 (+100 mV) | 51.9 ± 5.2 * | 34.4 ± 2.4 * |
| Kv7.2 D1R/R6D | 9 | 0.7 ± 0.2 (0 mV) * | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soldovieri, M.V.; Ambrosino, P.; Mosca, I.; Miceli, F.; Franco, C.; Canzoniero, L.M.T.; Kline-Fath, B.; Cooper, E.C.; Venkatesan, C.; Taglialatela, M. Epileptic Encephalopathy In A Patient With A Novel Variant In The Kv7.2 S2 Transmembrane Segment: Clinical, Genetic, and Functional Features. Int. J. Mol. Sci. 2019, 20, 3382. https://doi.org/10.3390/ijms20143382
Soldovieri MV, Ambrosino P, Mosca I, Miceli F, Franco C, Canzoniero LMT, Kline-Fath B, Cooper EC, Venkatesan C, Taglialatela M. Epileptic Encephalopathy In A Patient With A Novel Variant In The Kv7.2 S2 Transmembrane Segment: Clinical, Genetic, and Functional Features. International Journal of Molecular Sciences. 2019; 20(14):3382. https://doi.org/10.3390/ijms20143382
Chicago/Turabian StyleSoldovieri, Maria Virginia, Paolo Ambrosino, Ilaria Mosca, Francesco Miceli, Cristina Franco, Lorella Maria Teresa Canzoniero, Beth Kline-Fath, Edward C. Cooper, Charu Venkatesan, and Maurizio Taglialatela. 2019. "Epileptic Encephalopathy In A Patient With A Novel Variant In The Kv7.2 S2 Transmembrane Segment: Clinical, Genetic, and Functional Features" International Journal of Molecular Sciences 20, no. 14: 3382. https://doi.org/10.3390/ijms20143382
APA StyleSoldovieri, M. V., Ambrosino, P., Mosca, I., Miceli, F., Franco, C., Canzoniero, L. M. T., Kline-Fath, B., Cooper, E. C., Venkatesan, C., & Taglialatela, M. (2019). Epileptic Encephalopathy In A Patient With A Novel Variant In The Kv7.2 S2 Transmembrane Segment: Clinical, Genetic, and Functional Features. International Journal of Molecular Sciences, 20(14), 3382. https://doi.org/10.3390/ijms20143382