Kinase Inhibitors with Antiepileptic Properties Identified with a Novel in Vitro Screening Platform


Abstract
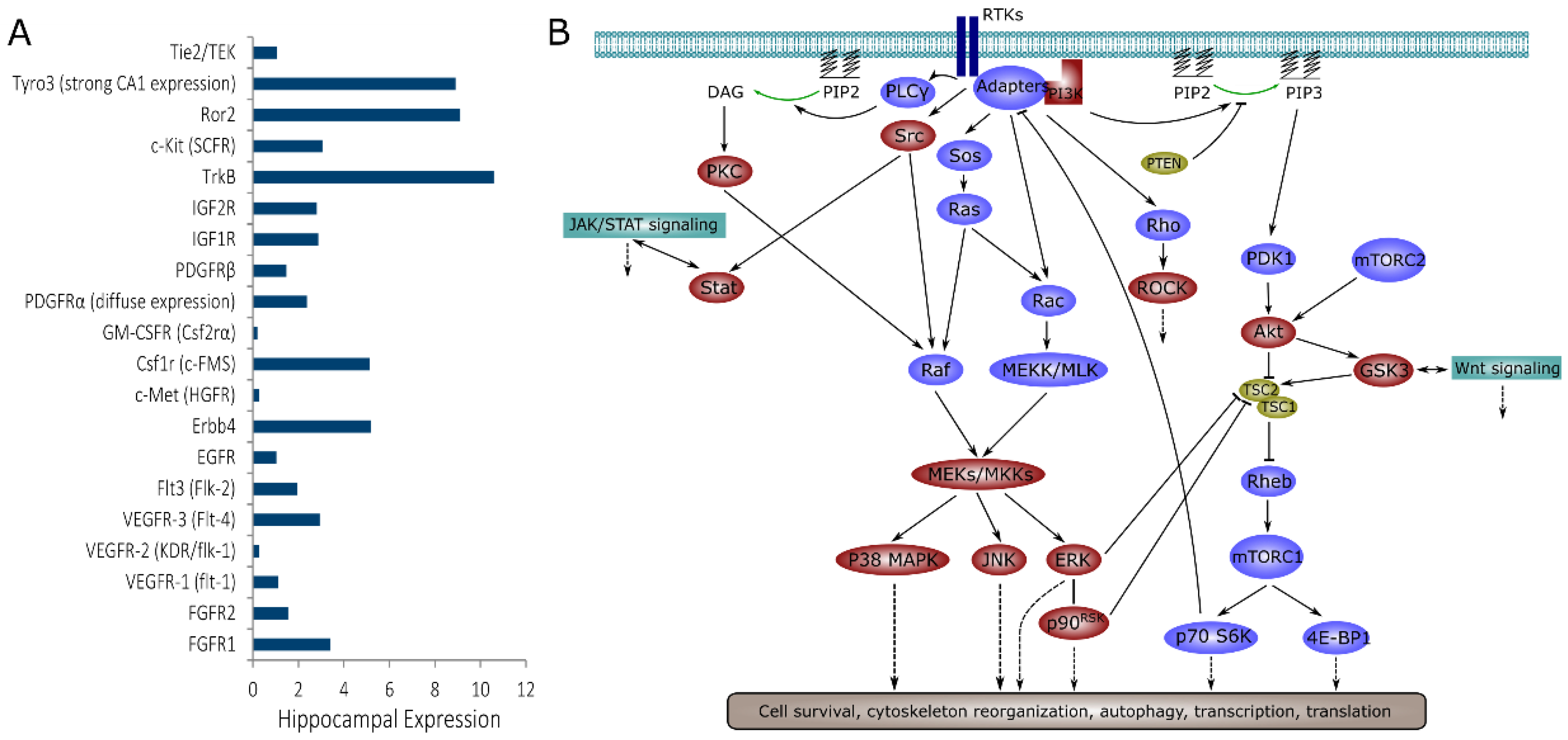
1. Introduction
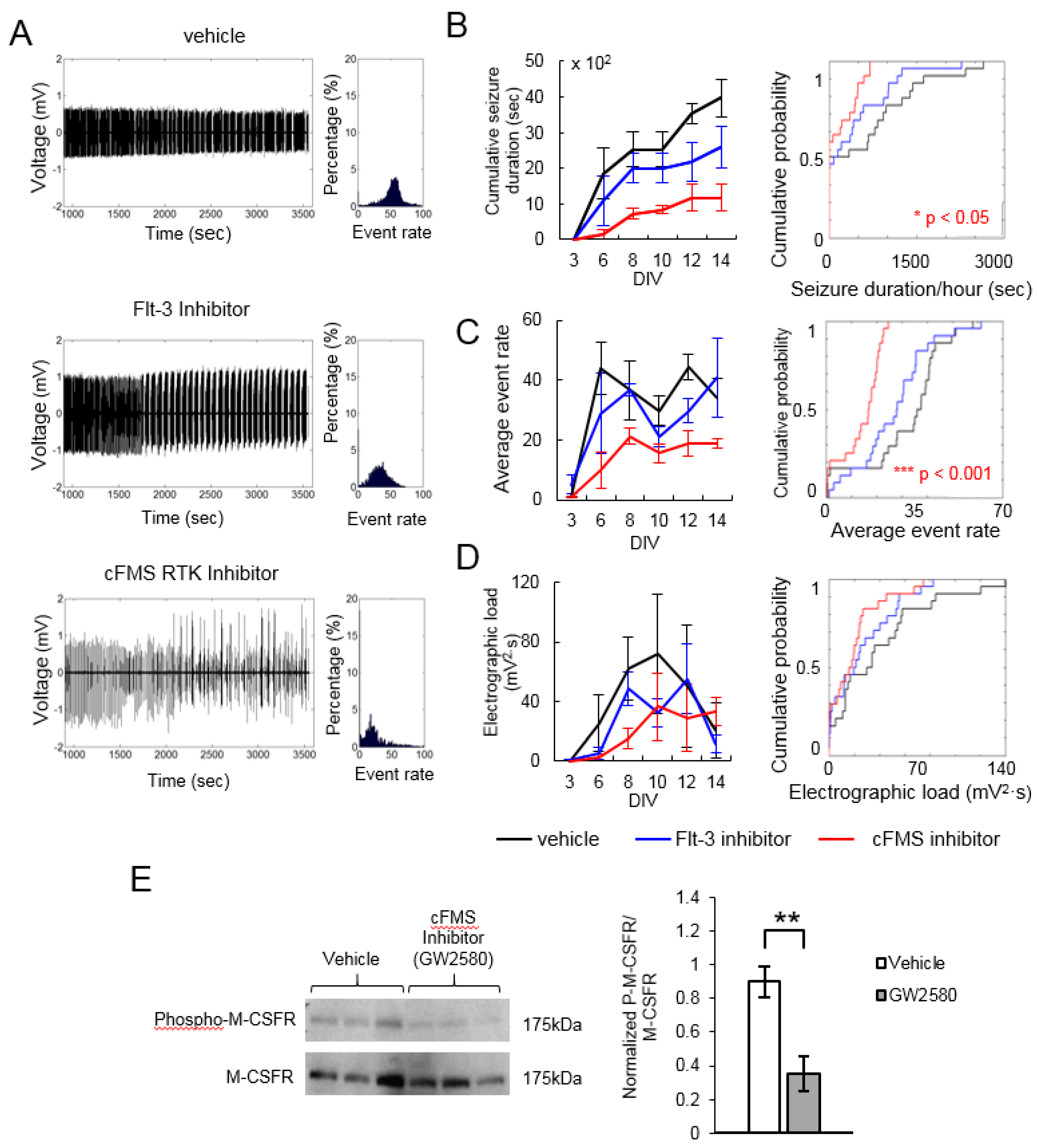
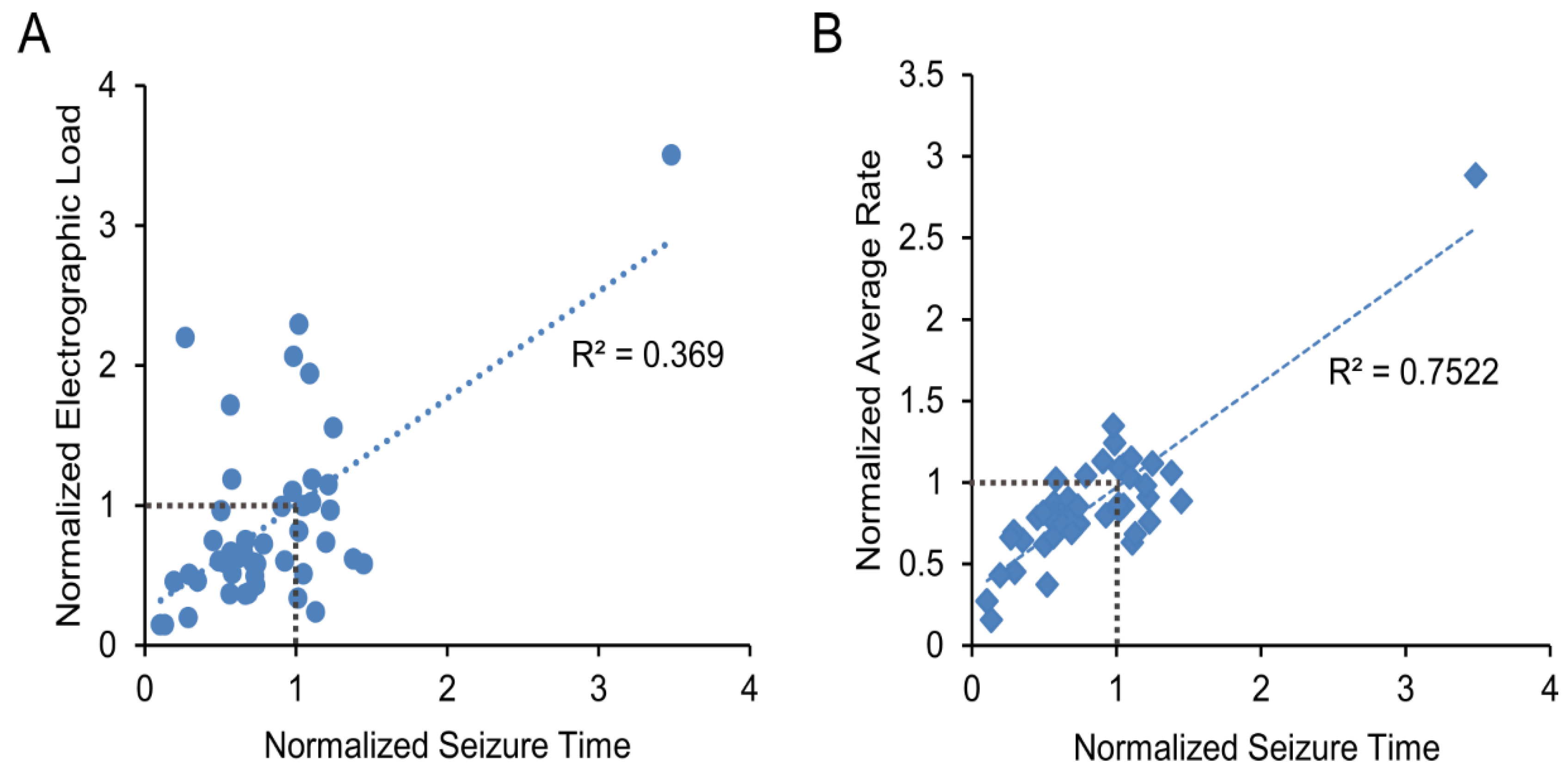
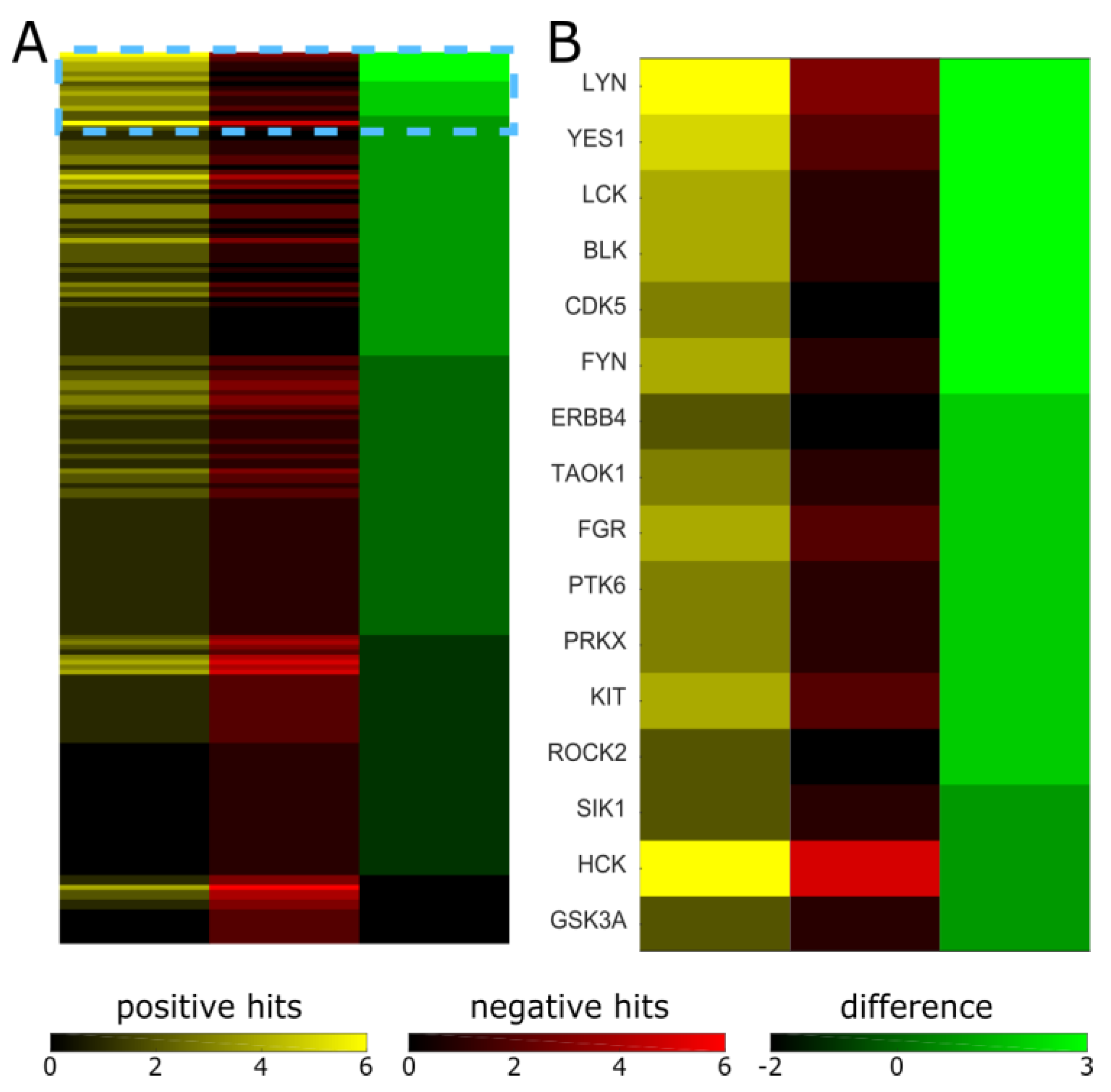
2. Results
3. Discussion
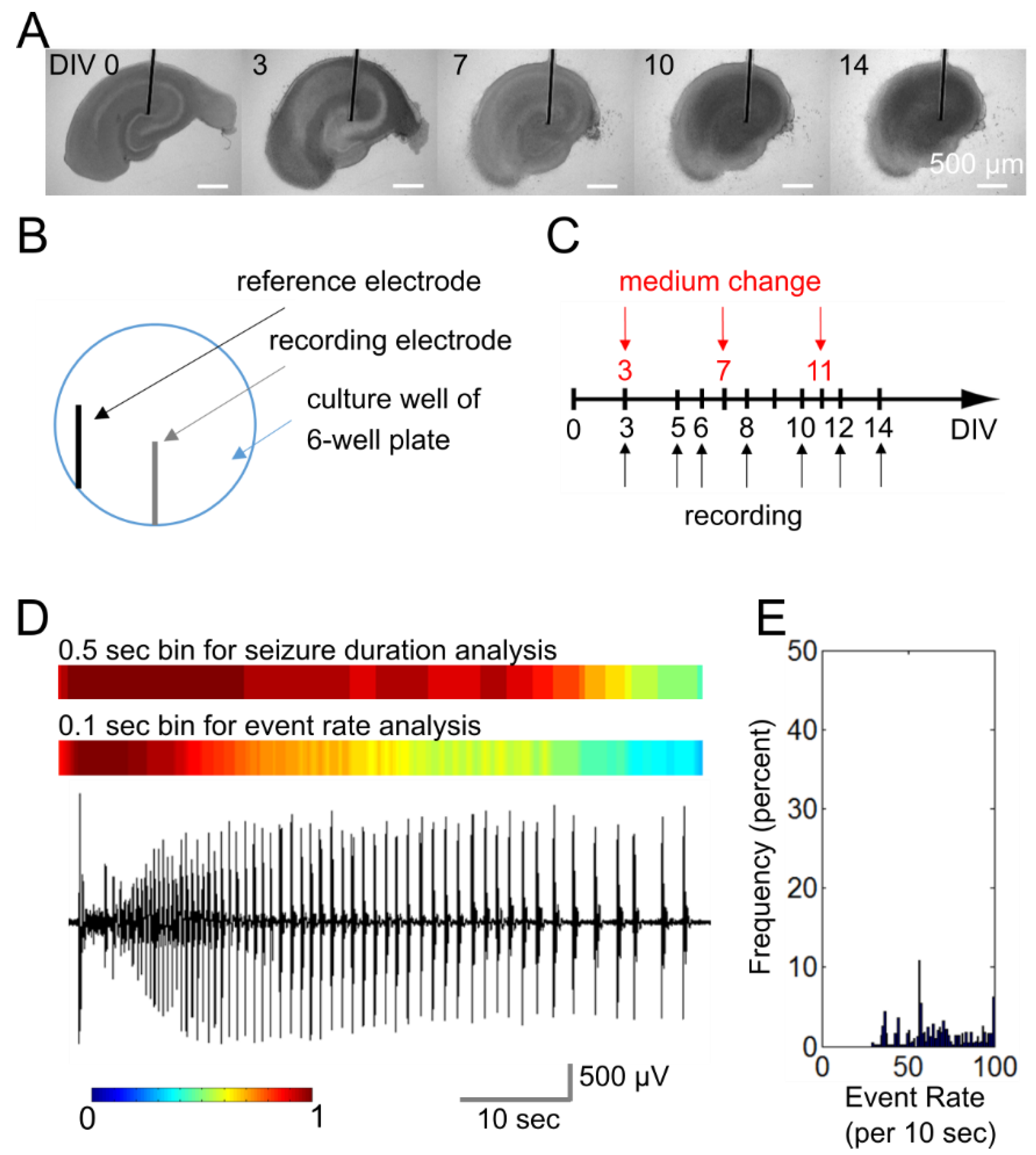
4. Materials and Methods
4.1. Microwire-Integrated Culture Plate
4.2. Organotypic Hippocampal Slice Cultures
4.3. Electrophysiology and Data Analysis
4.4. Determination of Maximum Non-Toxic Inhibitor Concentration
4.5. Drug Application
4.6. Western Blots and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MEA | Multiple Electrode Array |
| RTK | Receptor Tyrosine Kinase |
| DIV | Days in Vitro |
| LDH | Lactate Dehydrogenase |
References
- Pitkänen, A.; Lukasiuk, K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011, 10, 173–186. [Google Scholar] [CrossRef]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Grabenstatter, H.L.; Russek, S.J.; Brooks-Kayal, A.R. Molecular pathways controlling inhibitory receptor expression. Epilepsia 2012, 53, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Lukasiuk, K. Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. EB 2009, 14, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Amato, S.; Liu, X.; Zheng, B.; Cantley, L.; Rakic, P.; Man, H.-Y. AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science 2011, 332, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.B.; Liu, X.; Pradoldej, S.; Hao, C.; An, J.; Yepes, M.; Luo, H.R.; Ye, K. Phosphoinositide 3-kinase enhancer regulates neuronal dendritogenesis and survival in neocortex. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 8083–8092. [Google Scholar] [CrossRef]
- Cuesto, G.; Enriquez-Barreto, L.; Caramés, C.; Cantarero, M.; Gasull, X.; Sandi, C.; Ferrús, A.; Acebes, Á.; Morales, M. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 2721–2733. [Google Scholar] [CrossRef]
- Oliva, A.A.; Atkins, C.M.; Copenagle, L.; Banker, G.A. Activated c-Jun N-terminal kinase is required for axon formation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 9462–9470. [Google Scholar] [CrossRef]
- Berdichevsky, Y.; Dryer, A.M.; Saponjian, Y.; Mahoney, M.M.; Pimentel, C.A.; Lucini, C.A.; Usenovic, M.; Staley, K.J. PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 9056–9067. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.-H.; Rensing, N.R.; Wong, M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 6964–6972. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Ingram, E.A.; Wen, X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 8259–8269. [Google Scholar] [CrossRef]
- Grabenstatter, H.L.; Del Angel, Y.C.; Carlsen, J.; Wempe, M.F.; White, A.M.; Cogswell, M.; Russek, S.J.; Brooks-Kayal, A.R. The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol. Dis. 2014, 62, 73–85. [Google Scholar] [CrossRef]
- Scharfman, H.E. Brain-derived neurotrophic factor and epilepsy—A missing link? Epilepsy Curr. 2005, 5, 83–88. [Google Scholar] [CrossRef]
- Liu, G.; Gu, B.; He, X.-P.; Joshi, R.B.; Wackerle, H.D.; Rodriguiz, R.M.; Wetsel, W.C.; McNamara, J.O. Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron 2013, 79, 31–38. [Google Scholar] [CrossRef]
- Aungst, S.; England, P.M.; Thompson, S.M. Critical role of trkB receptors in reactive axonal sprouting and hyperexcitability after axonal injury. J. Neurophysiol. 2013, 109, 813–824. [Google Scholar] [CrossRef]
- Dinocourt, C.; Gallagher, S.E.; Thompson, S.M. Injury-induced axonal sprouting in the hippocampus is initiated by activation of trkB receptors. Eur. J. Neurosci. 2006, 24, 1857–1866. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Martin, K.J.; Arthur, J.S.C. Selective kinase inhibitors as tools for neuroscience research. Neuropharmacology 2012, 63, 1227–1237. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Gao, Y.; Davies, S.P.; Augustin, M.; Woodward, A.; Patel, U.A.; Kovelman, R.; Harvey, K.J. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem. J. 2013, 451, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, H.; Johns, T.G.; Neufeld, A.H. Epidermal growth factor receptor activation: An upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 7532–7540. [Google Scholar] [CrossRef] [PubMed]
- Leadbeater, W.E.; Gonzalez, A.-M.; Logaras, N.; Berry, M.; Turnbull, J.E.; Logan, A. Intracellular trafficking in neurones and glia of fibroblast growth factor-2, fibroblast growth factor receptor 1 and heparan sulphate proteoglycans in the injured adult rat cerebral cortex. J. Neurochem. 2006, 96, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, T.; Nagayama, M.; Kohara, S.; Kamiguchi, H.; Shibuya, M.; Katoh, Y.; Itoh, J.; Shinohara, Y. Post-ischemic delayed expression of hepatocyte growth factor and c-Met in mouse brain following focal cerebral ischemia. Brain Res. 2004, 999, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Schäbitz, W.-R.; Krüger, C.; Pitzer, C.; Weber, D.; Laage, R.; Gassler, N.; Aronowski, J.; Mier, W.; Kirsch, F.; Dittgen, T.; et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2008, 28, 29–43. [Google Scholar] [CrossRef]
- Tokita, Y.; Keino, H.; Matsui, F.; Aono, S.; Ishiguro, H.; Higashiyama, S.; Oohira, A. Regulation of neuregulin expression in the injured rat brain and cultured astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 1257–1264. [Google Scholar] [CrossRef]
- Sköld, M.K.; von Gertten, C.; Sandberg-Nordqvist, A.-C.; Mathiesen, T.; Holmin, S. VEGF and VEGF receptor expression after experimental brain contusion in rat. J. Neurotrauma 2005, 22, 353–367. [Google Scholar] [CrossRef]
- Helmy, A.; Carpenter, K.L.H.; Menon, D.K.; Pickard, J.D.; Hutchinson, P.J.A. The cytokine response to human traumatic brain injury: Temporal profiles and evidence for cerebral parenchymal production. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2011, 31, 658–670. [Google Scholar] [CrossRef]
- Castañeda-Cabral, J.L.; Beas-Zárate, C.; Rocha-Arrieta, L.L.; Orozco-Suárez, S.A.; Alonso-Vanegas, M.; Guevara-Guzmán, R.; Ureña-Guerrero, M.E. Increased protein expression of VEGF-A, VEGF-B, VEGF-C and their receptors in the temporal neocortex of pharmacoresistant temporal lobe epilepsy patients. J. Neuroimmunol. 2018, 328, 68–72. [Google Scholar] [CrossRef]
- Song, Y.; Pimentel, C.; Walters, K.; Boller, L.; Ghiasvand, S.; Liu, J.; Staley, K.J.; Berdichevsky, Y. Neuroprotective levels of IGF-1 exacerbate epileptogenesis after brain injury. Sci. Rep. 2016, 6, 32095. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Caron, E.; Ghosh, S.; Matsuoka, Y.; Ashton-Beaucage, D.; Therrien, M.; Lemieux, S.; Perreault, C.; Roux, P.P.; Kitano, H. A comprehensive map of the mTOR signaling network. Mol. Syst. Biol. 2010, 6, 453. [Google Scholar] [CrossRef]
- van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Lim, K.-C.; Crino, P.B. Focal malformations of cortical development: New vistas for molecular pathogenesis. Neuroscience 2013, 252, 262–276. [Google Scholar] [CrossRef]
- Jastrzebski, K.; Hannan, K.M.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors Chur Switz. 2007, 25, 209–226. [Google Scholar] [CrossRef]
- Cho, C.H. Frontier of epilepsy research-mTOR signaling pathway. Exp. Mol. Med. 2011, 43, 231–274. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Alemán, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Chang, S.-S.; Hsu, J.L.; Hung, M.-C. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene 2014, 33, 1073–1081. [Google Scholar] [CrossRef]
- Liu, J.; Sternberg, A.R.; Ghiasvand, S.; Berdichevsky, Y. Epilepsy-on-a-chip System for Antiepileptic Drug Discovery. IEEE Trans. Biomed. Eng. 2018, 66, 1231–1241. [Google Scholar] [CrossRef]
- Patch, R.J.; Baumann, C.A.; Liu, J.; Gibbs, A.C.; Ott, H.; Lattanze, J.; Player, M.R. Identification of 2-acylaminothiophene-3-carboxamides as potent inhibitors of FLT3. Bioorg. Med. Chem. Lett. 2006, 16, 3282–3286. [Google Scholar] [CrossRef]
- Conway, J.G.; McDonald, B.; Parham, J.; Keith, B.; Rusnak, D.W.; Shaw, E.; Jansen, M.; Lin, P.; Payne, A.; Crosby, R.M.; et al. Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proc. Natl. Acad. Sci. USA. 2005, 102, 16078–16083. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Kumar, A.; Jaggi, A.S.; Singh, N. Pharmacology of Src family kinases and therapeutic implications of their modulators. Fundam. Clin. Pharmacol. 2015, 29, 115–130. [Google Scholar] [CrossRef]
- Sharma, S.; Carlson, S.; Puttachary, S.; Sarkar, S.; Showman, L.; Putra, M.; Kanthasamy, A.G.; Thippeswamy, T. Role of the Fyn-PKCδ signaling in SE-induced neuroinflammation and epileptogenesis in experimental models of temporal lobe epilepsy. Neurobiol. Dis. 2018, 110, 102–121. [Google Scholar] [CrossRef]
- Berdichevsky, Y.; Saponjian, Y.; Park, K.-I.; Roach, B.; Pouliot, W.; Lu, K.; Swiercz, W.; Dudek, F.E.; Staley, K.J. Staged anticonvulsant screening for chronic epilepsy. Ann. Clin. Transl. Neurol. 2016, 3, 908–923. [Google Scholar] [CrossRef]
- Berdichevsky, Y.; Dzhala, V.; Mail, M.; Staley, K.J. Interictal spikes, seizures and ictal cell death are not necessary for post-traumatic epileptogenesis in vitro. Neurobiol. Dis. 2012, 45, 774–785. [Google Scholar] [CrossRef]
- Wuarin, J.P.; Dudek, F.E. Electrographic seizures and new recurrent excitatory circuits in the dentate gyrus of hippocampal slices from kainate-treated epileptic rats. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 4438–4448. [Google Scholar] [CrossRef]
- Khazipov, R.; Khalilov, I.; Tyzio, R.; Morozova, E.; Ben-Ari, Y.; Holmes, G.L. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur. J. Neurosci. 2004, 19, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Dzhala, V.I.; Talos, D.M.; Sdrulla, D.A.; Brumback, A.C.; Mathews, G.C.; Benke, T.A.; Delpire, E.; Jensen, F.E.; Staley, K.J. NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 2005, 11, 1205–1213. [Google Scholar] [CrossRef]
- Jirsa, V.K.; Stacey, W.C.; Quilichini, P.P.; Ivanov, A.I.; Bernard, C. On the nature of seizure dynamics. Brain, J. Neurol. 2014, 137, 2210–2230. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Conc. (μM) | CAS | Cumulative Seizure Duration | Average Event Rate | Electrographic Load | |||
|---|---|---|---|---|---|---|---|---|
| Norm. | p | Norm. | p | Norm. | p | |||
| Rho Kinase Inhibitor III, Rockout | 10 | 7272-84-6 | 0.569 | 0.3 | 0.873 | 0.576 | 1.713 | 0.49 |
| Aurora Kinase Inhibitor II | 0.5 | 331770-21-9 | 0.577 | 0.023 | 0.748 | 0.023 | 1.185 | 0.497 |
| GSK3, GSK3β inhibitor, Bisindolylmaleimide I | 0.5 | 133052-90-1 | 0.572 | 0.135 | 0.803 | 0.135 | 0.663 | 0.275 |
| Flt-3 Inhibitor | 2 | 301305-73-7 | 0.656 | 0.387 | 0.856 | 0.051 | 0.664 | 0.622 |
| cFMS Receptor Tyrosine Kinase Inhibitor | 2 | 870483-87-7 | 0.297 | 0.021 | 0.45 | 0.001 | 0.505 | 0.051 |
| EGFR/ErbB-2 Inhibitor | 2 | 179248-61-4 | 0.35 | 0.088 | 0.645 | 0.169 | 0.458 | 0.49 |
| GTP-14564, cFMS, c-kit, Flt3 inhibitor | 5 | 34823-86-4 | 1.049 | 0.49 | 1.101 | 0.72 | 0.996 | 0.169 |
| Aurora Kinase Inhibitor II | 1 | 331770-21-9 | 0.742 | 0.6 | 0.747 | 0.89 | 0.58 | 0.72 |
| PDGF Receptor Tyrosine Kinase Inhibitor III | 2 | 205254-94-0 | 0.505 | 0.003 | 0.616 | 0.001 | 0.959 | 0.917 |
| VEGF Receptor Tyrosine Kinase Inhibitor II | 5 | 269390-69-4 | 0.668 | 0.207 | 0.787 | 0.029 | 0.362 | 0.021 |
| VEGFR Tyrosine Kinase Inhibitor IV (Tivozanib, KRN951) | 0.3 | 475108-18-0 | 1.23 | 0.367 | 0.759 | 0.148 | 0.964 | 0.001 |
| PKC, PKCβ, Bisindolylmaleimide IV | 10 | 119139-23-0 | 0.541 | 0.109 | 0.789 | 0.109 | 0.587 | 0.216 |
| EGFR Inhibitor, BPIQ-I | 10 | 174709-30-9 | 0.667 | 0.216 | 0.898 | 0.216 | 0.748 | 0.387 |
| Src Kinase Inhibitor I (Src-I1) | 5 | 179248-59-0 | 0.453 | 0.008 | 0.782 | 0.001 | 0.748 | 0.387 |
| PKC, PKCβ, Chelerythrine Chloride | 5 | 3895-92-9 | 0.927 | 0.861 | 0.798 | 0.387 | 0.599 | 0.109 |
| SL0101, p90 RSK inhibitor | 5 | 77307-50-7 | 1.05 | 0.987 | 0.858 | 0.051 | 0.507 | 0.003 |
| GSK-3b, GSK-3β Inhibitor XI | 10 | 626604-39-5 | 1.104 | 0.987 | 1.148 | 0.861 | 1.018 | 0.987 |
| Ki20227, cFMS Inhibitor | 5 | 623142-96-1 | 1.45 | 0.166 | 0.884 | 0.144 | 0.581 | 0.131 |
| GW2580, cFMS Receptor Tyrosine Kinase Inhibitor | 5 | 870483-87-7 | 0.105 | 0.216 | 0.273 | 0.001 | 0.146 | 0.001 |
| SB203580, p38 MAPK Inhibitor | 2.65 | 152121-47-6 | 1.248 | 0.861 | 1.116 | 0.622 | 1.554 | 0.109 |
| JAK Inhibitor VI | 2 | 856436-16-3 | 1.11 | 0.622 | 0.631 | 0.003 | 1.184 | 0.861 |
| JAK Inhibitor I (Pyridone 6) | 2 | 457081-03-7 | 1.217 | 0.72 | 0.91 | 0.3 | 1.147 | 0.3 |
| CHIR99021, GSK3β Inhibitor | 1 | 252917-06-9 | 0.493 | 0.088 | 0.813 | 0.6 | 0.601 | 0.088 |
| EGFR Inhibitor | 0.1 | 879127-07-8 | 0.788 | 0.72 | 1.04 | 0.003 | 0.724 | 0.088 |
| Rho Kinase Inhibitor IV | 10 | 913844-45-8 | 0.29 | 0.088 | 0.695 | 0.008 | 0.198 | 3.4E-4 |
| JAK Inhibitor I | 1 | 457081-03-7 | 0.716 | 0.3 | 0.728 | 0.088 | 0.589 | 0.088 |
| Akt Inhibitor VIII, Isozyme-Selective, Akti-1/2 | 0.5 | 612847-09-3 | 0.731 | 0.72 | 0.744 | 0.088 | 0.491 | 0.088 |
| Gö 6976, PKC, PKCβ | 0.1 | 136194-77-9 | 0.522 | 0.088 | 0.373 | 1.8E-6 | 0.594 | 0.003 |
| Akt Inhibitor V, Triciribine | 0.5 | 35943-35-2 | 0.563 | 0.169 | 0.667 | 0.019 | 0.368 | 0.042 |
| FR180204, MAPK (ERK1/2) | 10 | 865362-74-9 | 1.2 | 0.711 | 0.979 | 0.994 | 0.736 | 0.162 |
| PKCbII/EGFR Inhibitor (CGP 53353) | 10 | 145915-60-2 | 0.271 | 0.003 | 0.66 | 0.008 | 2.197 | 2.7E-4 |
| IRAK-1/4 Inhibitor | 10 | 509093-47-4 | 0.987 | 0.861 | 1.243 | 0.387 | 2.062 | 0.021 |
| PI3Kγ Inhibitor (AS 605240) | 10 | 648450-29-7 | 0.979 | 0.995 | 1.348 | 0.169 | 1.099 | 0.169 |
| JNK VIII inhibitor | 10 | 894804-07-0 | 3.485 | 0.008 | 2.883 | 3.9E-7 | 3.502 | 0.001 |
| Y-27632, ROCK Inhibitor | 10 | 146986-50-7 | 1.02 | 1 | 0.832 | 0.49 | 0.813 | 0.49 |
| PD 0325901, MEK Inhibitor | 10 | 391210-10-9 | 0.135 | 2.8E-5 | 0.156 | 8.0E-8 | 0.144 | 3.4E-4 |
| CP-690550, Tofacitinib, JAK3 Inhibitor | 10 | 540737-29-9 | 0.626 | 0.088 | 0.746 | 0.042 | 0.628 | 0.72 |
| BIRB 796, Doramapimod, p38 MAPK inhibitor | 10 | 285983-48-4 | 0.907 | 0.042 | 1.13 | 0.169 | 0.992 | 0.019 |
| BI-D1870, RSK1/2/3/4 Inhibitor | 10 | 501437-28-1 | 0.194 | 3.4E-4 | 0.432 | 1.0E-4 | 0.454 | 0.008 |
| AZD0530, Saracatinib, Src Inhibitor | 2 | 379231-04-6 | 0.689 | 0.169 | 0.694 | 0.169 | 0.37 | 0.008 |
| JNK Inhibitor IX | 0.2 | 312917-14-9 | 1.131 | 0.188 | 0.682 | 0.023 | 0.237 | 3.7E-5 |
| PI-103, PI3K | 0.2 | 371935-74-9 | 1.094 | 0.088 | 1.034 | 0.49 | 1.939 | 0.088 |
| CyclotraxinB, TRK Inhibitor | 10 | 1203586-72-4 | 0.735 | 0.184 | 0.853 | 0.22 | 0.428 | 0.24 |
| GNF5837, TRK Inhibitor | 2 | 1033769-28-6 | 0.582 | 0.003 | 1.016 | 0.169 | 0.512 | 0.72 |
| PKR Inhibitor | 0.05 | 608512-97-6 | 1.016 | 0.917 | 0.864 | 0.72 | 0.334 | 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Schenker, M.; Ghiasvand, S.; Berdichevsky, Y. Kinase Inhibitors with Antiepileptic Properties Identified with a Novel in Vitro Screening Platform. Int. J. Mol. Sci. 2019, 20, 2502. https://doi.org/10.3390/ijms20102502
Liu J, Schenker M, Ghiasvand S, Berdichevsky Y. Kinase Inhibitors with Antiepileptic Properties Identified with a Novel in Vitro Screening Platform. International Journal of Molecular Sciences. 2019; 20(10):2502. https://doi.org/10.3390/ijms20102502
Chicago/Turabian StyleLiu, Jing, Madison Schenker, Shabnam Ghiasvand, and Yevgeny Berdichevsky. 2019. "Kinase Inhibitors with Antiepileptic Properties Identified with a Novel in Vitro Screening Platform" International Journal of Molecular Sciences 20, no. 10: 2502. https://doi.org/10.3390/ijms20102502
APA StyleLiu, J., Schenker, M., Ghiasvand, S., & Berdichevsky, Y. (2019). Kinase Inhibitors with Antiepileptic Properties Identified with a Novel in Vitro Screening Platform. International Journal of Molecular Sciences, 20(10), 2502. https://doi.org/10.3390/ijms20102502