The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism



Abstract
1. Introduction
2. GBA Variants, GD, PD, and How They Interplay
3. Other Diseases of Sphingolipid Metabolism Determining or Predisposing Individuals to Neurodegenerative Disorders
4. Specific Diseases of Ganglioside Biosynthesis Determining Neurodegenerative Disorders
4.1. GM2 Synthase Deficiency (B4GALNT1-CDG), HSP26, and Other HSPs
4.2. GM3 Synthase Deficiency (ST3GAL5-CDG)
4.3. ST3GAL3-CDG, Non-Syndromic Intellectual Disability and West Syndrome
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDG | Congenital disorders of glycosylation |
| ER | Endoplasmic reticulum |
| GalCer | Galactosylceramide |
| GD | Gaucher disease |
| GlcCer | Glucosylceramide |
| GlcSph | Glucosylsphingosine |
| GlcNAc | N-acetylglucosamine |
| GSL | Glycosphingolipid |
| HSP | Hereditary spastic paraplegia |
| LacCer | Lactosylceramide |
| PD | Parkinson disease |
| ROS | Reactive oxygen species |
| SCARB2 | Scavenger receptor class B member 2/lysosomal integral protein 2 (LIMP2) |
| SM | Sphingomyelin |
| S1P | Sphingosine-1-phosphate |
References
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, Y. The sphingomyelin synthase family: proteins, diseases, and inhibitors. Biol. Chem. 2017, 398, 1319–1325. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nemoto-Sasaki, Y.; Matsumoto, N.; Hama, K.; Tanikawa, T.; Oka, S.; Saeki, T.; Kumasaka, T.; Koizumi, T.; Arai, S.; et al. Complex formation of sphingomyelin synthase 1 with glucosylceramide synthase increases sphingomyelin and decreases glucosylceramide levels. J. Biol. Chem. 2018, 293, 17505–17522. [Google Scholar] [CrossRef]
- Nickel, W.; Brugger, B.; Wieland, F.T. Protein and lipid sorting between the endoplasmic reticulum and the Golgi complex. Seminars Cell Dev. Biol. 1998, 9, 493–501. [Google Scholar] [CrossRef]
- Russo, D.; Capolupo, L.; Loomba, J.S.; Sticco, L.; D’Angelo, G. Glycosphingolipid metabolism in cell fate specification. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Bejaoui, K.; Wu, C.; Scheffler, M.D.; Haan, G.; Ashby, P.; Wu, L.; de Jong, P.; Brown, R.H., Jr. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat. Genet. 2001, 27, 261–262. [Google Scholar] [CrossRef]
- Wilson, E.R.; Kugathasan, U.; Abramov, A.Y.; Clark, A.J.; Bennett, D.L.H.; Reilly, M.M.; Greensmith, L.; Kalmar, B. Hereditary sensory neuropathy type 1-associated deoxysphingolipids cause neurotoxicity, acute calcium handling abnormalities and mitochondrial dysfunction in vitro. Neu. Dis. 2018, 117, 1–14. [Google Scholar] [CrossRef]
- Boyden, L.M.; Vincent, N.G.; Zhou, J.; Hu, R.; Craiglow, B.G.; Bayliss, S.J.; Rosman, I.S.; Lucky, A.W.; Diaz, L.A.; Goldsmith, L.A.; et al. Mutations in KDSR Cause Recessive Progressive Symmetric Erythrokeratoderma. Am.J. Hum. Genet. 2017, 100, 978–984. [Google Scholar] [CrossRef]
- Takeichi, T.; Torrelo, A.; Lee, J.Y.W.; Ohno, Y.; Lozano, M.L.; Kihara, A.; Liu, L.; Yasuda, Y.; Ishikawa, J.; Murase, T.; et al. Biallelic Mutations in KDSR Disrupt Ceramide Synthesis and Result in a Spectrum of Keratinization Disorders Associated with Thrombocytopenia. J. Investig. Dermatol. 2017, 137, 2344–2353. [Google Scholar] [CrossRef]
- Bariana, T.K.; Labarque, V.; Heremans, J.; Thys, C.; De Reys, M.; Greene, D.; Jenkins, B.; Grassi, L.; Seyres, D.; Burden, F.; et al. Sphingolipid dysregulation due to lack of functional KDSR impairs proplatelet formation causing thrombocytopenia. Haematologica 2019, 104, 1036–1045. [Google Scholar] [CrossRef]
- Vanni, N.; Fruscione, F.; Ferlazzo, E.; Striano, P.; Robbiano, A.; Traverso, M.; Sander, T.; Falace, A.; Gazzerro, E.; Bramanti, P.; et al. Impairment of ceramide synthesis causes a novel progressive myoclonus epilepsy. Ann. Neurol. 2014, 76, 206–212. [Google Scholar] [CrossRef]
- Godeiro Junior, C.O.; Vale, T.C.; Afonso, C.O.M.; Kok, F.; Pedroso, J.L.; Barsottini, O.G. Progressive Myoclonic Epilepsy Type 8 Due to CERS1 Deficiency: A Novel Mutation with Prominent Ataxia. Mov. Disord. Clin. Pract. 2018, 5, 330–332. [Google Scholar] [CrossRef]
- Eckl, K.M.; Tidhar, R.; Thiele, H.; Oji, V.; Hausser, I.; Brodesser, S.; Preil, M.L.; Onal-Akan, A.; Stock, F.; Muller, D.; et al. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J. Investig. Dermatol. 2013, 133, 2202–2211. [Google Scholar] [CrossRef]
- Mosbech, M.B.; Olsen, A.S.; Neess, D.; Ben-David, O.; Klitten, L.L.; Larsen, J.; Sabers, A.; Vissing, J.; Nielsen, J.E.; Hasholt, L.; et al. Reduced ceramide synthase 2 activity causes progressive myoclonic epilepsy. Ann. Clin. Trans. Neurol. 2014, 1, 88–98. [Google Scholar] [CrossRef]
- Wegner, M.S.; Schiffmann, S.; Parnham, M.J.; Geisslinger, G.; Grosch, S. The enigma of ceramide synthase regulation in mammalian cells. Prog. Lipid Res. 2016, 63, 93–119. [Google Scholar] [CrossRef]
- Skolova, B.; Kovacik, A.; Tesar, O.; Opalka, L.; Vavrova, K. Phytosphingosine, sphingosine and dihydrosphingosine ceramides in model skin lipid membranes: permeability and biophysics. Biochim. Biophys. Acta Biomembr. 2017, 1859, 824–834. [Google Scholar] [CrossRef]
- Pant, D.C.; Dorboz, I.; Schluter, A.; Fourcade, S.; Launay, N.; Joya, J.; Aguilera-Albesa, S.; Yoldi, M.E.; Casasnovas, C.; Willis, M.J.; et al. Loss of the sphingolipid desaturase DEGS1 causes hypomyelinating leukodystrophy. J. Clin. Investig. 2019, 129, 1240–1256. [Google Scholar] [CrossRef]
- Karsai, G.; Kraft, F.; Haag, N.; Korenke, G.C.; Hanisch, B.; Othman, A.; Suriyanarayanan, S.; Steiner, R.; Knopp, C.; Mull, M.; et al. DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans. J. Clin. Investig. 2019, 129, 1229–1239. [Google Scholar] [CrossRef]
- Bienias, K.; Fiedorowicz, A.; Sadowska, A.; Prokopiuk, S.; Car, H. Regulation of sphingomyelin metabolism. Pharmaco. Rep. 2016, 68, 570–581. [Google Scholar] [CrossRef]
- Pekkinen, M.; Terhal, P.A.; Botto, L.D.; Henning, P.; Makitie, R.E.; Roschger, P.; Jain, A.; Kol, M.; Kjellberg, M.A.; Paschalis, E.P.; et al. Osteoporosis and skeletal dysplasia caused by pathogenic variants in SGMS2. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Monies, D.; Anabrees, J.; Ibrahim, N.; Elbardisy, H.; Abouelhoda, M.; Meyer, B.F.; Alkuraya, F.S. Identification of a novel lethal form of autosomal recessive ichthyosis caused by UDP-glucose ceramide glucosyltransferase deficiency. Clin. Genet. 2018, 93, 1252–1253. [Google Scholar] [CrossRef]
- Trinchera, M.; Parini, R.; Indellicato, R.; Domenighini, R.; dall’Olio, F. Diseases of ganglioside biosynthesis: An expanding group of congenital disorders of glycosylation. Mol. Genet. Metab. 2018, 124, 230–237. [Google Scholar] [CrossRef]
- Ng, B.G.; Freeze, H.H. Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet. 2018, 34, 466–476. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8. [Google Scholar] [CrossRef]
- Woeste, M.A.; Wachten, D. The Enigmatic Role of GBA2 in Controlling Locomotor Function. Frontiers in Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef]
- Edvardson, S.; Yi, J.K.; Jalas, C.; Xu, R.; Webb, B.D.; Snider, J.; Fedick, A.; Kleinman, E.; Treff, N.R.; Mao, C.; et al. Deficiency of the alkaline ceramidase ACER3 manifests in early childhood by progressive leukodystrophy. J. Med. Genet. 2016, 53, 389–396. [Google Scholar] [CrossRef]
- Dunn, T.M.; Tifft, C.J.; Proia, R.L. A perilous path: the inborn errors of sphingolipid metabolism. J. Lipid Res. 2019, 60, 475–483. [Google Scholar] [CrossRef]
- Alfonso, P.; Navascues, J.; Navarro, S.; Medina, P.; Bolado-Carrancio, A.; Andreu, V.; Irun, P.; Rodriguez-Rey, J.C.; Pocovi, M.; Espana, F.; et al. Characterization of variants in the glucosylceramide synthase gene and their association with type 1 Gaucher disease severity. Hum. Mutat. 2013, 34, 1396–1403. [Google Scholar] [CrossRef]
- Mullin, S.; Hughes, D.; Mehta, A.; Schapira, A.H.V. Neurological effects of glucocerebrosidase gene mutations. Eur. J. Neurol. 2019, 26, 388–e29. [Google Scholar] [CrossRef]
- de Souza, P.V.S.; de Rezende Pinto, W.B.V.; de Rezende Batistella, G.N.; Bortholin, T.; Oliveira, A.S.B. Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum 2017, 16, 525–551. [Google Scholar] [CrossRef]
- Edvardson, S.; Baumann, A.M.; Muhlenhoff, M.; Stephan, O.; Kuss, A.W.; Shaag, A.; He, L.; Zenvirt, S.; Tanzi, R.; Gerardy-Schahn, R.; et al. West syndrome caused by ST3Gal-III deficiency. Epilepsia 2013, 54, e24–e27. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- Scott-Hewitt, N.J.; Folts, C.J.; Noble, M.D. Heterozygous carriers of galactocerebrosidase mutations that cause Krabbe disease have impaired microglial function and defective repair of myelin damage. Neural Regen. Res. 2018, 13, 393–401. [Google Scholar] [CrossRef]
- Berger, J.R. Misdiagnosis of multiple sclerosis in a female heterozygote with Fabry’s disease. Multiple sclerosis and related disorders 2019, 30, 45–47. [Google Scholar] [CrossRef]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the structural biology of Gaucher disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef]
- Erickson, A.H.; Ginns, E.I.; Barranger, J.A. Biosynthesis of the lysosomal enzyme glucocerebrosidase. J. Biol. Chem. 1985, 260, 14319–14324. [Google Scholar]
- Berg-Fussman, A.; Grace, M.E.; Ioannou, Y.; Grabowski, G.A. Human acid beta-glucosidase. N-glycosylation site occupancy and the effect of glycosylation on enzymatic activity. J. Biol. Chem. 1993, 268, 14861–14866. [Google Scholar]
- Liou, B.; Haffey, W.D.; Greis, K.D.; Grabowski, G.A. The LIMP-2/SCARB2 binding motif on acid beta-glucosidase: basic and applied implications for Gaucher disease and associated neurodegenerative diseases. J. Biol. Chem. 2014, 289, 30063–30074. [Google Scholar] [CrossRef]
- Zunke, F.; Andresen, L.; Wesseler, S.; Groth, J.; Arnold, P.; Rothaug, M.; Mazzulli, J.R.; Krainc, D.; Blanz, J.; Saftig, P.; et al. Characterization of the complex formed by beta-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc. Natl. Acad. Sci. USA 2016, 113, 3791–3796. [Google Scholar] [CrossRef]
- Martin, E.; Schule, R.; Smets, K.; Rastetter, A.; Boukhris, A.; Loureiro, J.L.; Gonzalez, M.A.; Mundwiller, E.; Deconinck, T.; Wessner, M.; et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 238–244. [Google Scholar] [CrossRef]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; McKay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Beavan, M.; McNeill, A.; Proukakis, C.; Hughes, D.A.; Mehta, A.; Schapira, A.H. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neuro. 2015, 72, 201–208. [Google Scholar] [CrossRef]
- Gatto, E.M.; Da Prat, G.; Etcheverry, J.L.; Drelichman, G.; Cesarini, M. Parkinsonisms and Glucocerebrosidase Deficiency: A Comprehensive Review for Molecular and Cellular Mechanism of Glucocerebrosidase Deficiency. Brain Sci. 2019, 9. [Google Scholar] [CrossRef]
- O’Regan, G.; deSouza, R.M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase Mutations in Parkinson Disease. J. Parkinson’s Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef]
- Ysselstein, D.; Shulman, J.M.; Krainc, D. Emerging links between pediatric lysosomal storage diseases and adult parkinsonism. Mov. Disord. 2019, 34, 614–624. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200. [Google Scholar] [CrossRef]
- Gegg, M.E.; Burke, D.; Heales, S.J.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease. Mov. Disord. 2016, 31, 1610–1618. [Google Scholar] [CrossRef]
- Fussi, N.; Hollerhage, M.; Chakroun, T.; Nykanen, N.P.; Rosler, T.W.; Koeglsperger, T.; Wurst, W.; Behrends, C.; Hoglinger, G.U. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018, 9, 757. [Google Scholar] [CrossRef]
- Ikuno, M.; Yamakado, H.; Akiyama, H.; Parajuli, L.K.; Taguchi, K.; Hara, J.; Uemura, N.; Hatanaka, Y.; Higaki, K.; Ohno, K.; et al. GBA haploinsufficiency accelerates alpha-synuclein pathology with altered lipid metabolism in a prodromal model of Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1894–1904. [Google Scholar] [CrossRef]
- Gundner, A.L.; Duran-Pacheco, G.; Zimmermann, S.; Ruf, I.; Moors, T.; Baumann, K.; Jagasia, R.; van de Berg, W.D.J.; Kremer, T. Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol. Dis. 2019, 121, 205–213. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef]
- Kim, M.J.; Jeon, S.; Burbulla, L.F.; Krainc, D. Acid ceramidase inhibition ameliorates alpha-synuclein accumulation upon loss of GBA1 function. Hum. Mol. Genet. 2018, 27, 1972–1988. [Google Scholar] [CrossRef]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef]
- Thomas, R.E.; Vincow, E.S.; Merrihew, G.E.; MacCoss, M.J.; Davis, M.Y.; Pallanck, L.J. Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles. PLoS Genet. 2018, 14, e1007694. [Google Scholar] [CrossRef]
- Huang, Y.; Deng, L.; Zhong, Y.; Yi, M. The Association between E326K of GBA and the Risk of Parkinson’s Disease. Parkinson’s Dis. 2018, 2018, 1048084. [Google Scholar] [CrossRef]
- Schmitz, M.; Alfalah, M.; Aerts, J.M.; Naim, H.Y.; Zimmer, K.P. Impaired trafficking of mutants of lysosomal glucocerebrosidase in Gaucher’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 2310–2320. [Google Scholar] [CrossRef]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef]
- Papadopoulos, V.E.; Nikolopoulou, G.; Antoniadou, I.; Karachaliou, A.; Arianoglou, G.; Emmanouilidou, E.; Sardi, S.P.; Stefanis, L.; Vekrellis, K. Modulation of beta-glucocerebrosidase increases alpha-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum. Mol. Genet. 2018, 27, 1696–1710. [Google Scholar] [CrossRef]
- Thomas, R.; Kermode, A.R. Enzyme enhancement therapeutics for lysosomal storage diseases: Current status and perspective. Mol. Genet. Metab. 2019, 126, 83–97. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8, 448. [Google Scholar] [CrossRef]
- Davidson, B.A.; Hassan, S.; Garcia, E.J.; Tayebi, N.; Sidransky, E. Exploring genetic modifiers of Gaucher disease: The next horizon. Hum. Mutat. 2018, 39, 1739–1751. [Google Scholar] [CrossRef]
- Campbell, P.; Morris, H.; Schapira, A. Chaperone-mediated autophagy as a therapeutic target for Parkinson disease. Expert. Opin. Ther. Targets 2018, 22, 823–832. [Google Scholar] [CrossRef]
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Mallett, V.; Vanderperre, B.; Tavassoly, O.; Dauvilliers, Y.; Wu, R.Y.J.; Ruskey, J.A.; Leblond, C.S.; Ambalavanan, A.; Laurent, S.B.; et al. SMPD1 mutations, activity, and alpha-synuclein accumulation in Parkinson’s disease. Mov. Disord. 2019, 34, 526–535. [Google Scholar] [CrossRef]
- Wheeler, S.; Haberkant, P.; Bhardwaj, M.; Tongue, P.; Ferraz, M.J.; Halter, D.; Sprong, H.; Schmid, R.; Aerts, J.; Sullo, N.; et al. Cytosolic glucosylceramide regulates endolysosomal function in Niemann-Pick type C disease. Neurobiol. Dis. 2019, 127, 242–252. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Barmherzig, R.; Bullivant, G.; Cordeiro, D.; Sinasac, D.S.; Blaser, S.; Mercimek-Mahmutoglu, S. A New Patient With Intermediate Severe Salla Disease With Hypomyelination: A Literature Review for Salla Disease. Pediatr. Neurol. 2017, 74, 87–91. [Google Scholar] [CrossRef]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 4. [Google Scholar] [CrossRef]
- Wise, A.H.; Yang, A.; Naik, H.; Stauffer, C.; Zeid, N.; Liong, C.; Balwani, M.; Desnick, R.J.; Alcalay, R.N. Parkinson’s disease prevalence in Fabry disease: A survey study. Mol. Genet. Metab. reports 2018, 14, 27–30. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Wolf, P.; Levy, O.A.; Kang, U.J.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.H.; Vanegas, N.; Shah, H.; et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Gene. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Scott-Hewitt, N.J.; Folts, C.J.; Hogestyn, J.M.; Piester, G.; Mayer-Proschel, M.; Noble, M.D. Heterozygote galactocerebrosidase (GALC) mutants have reduced remyelination and impaired myelin debris clearance following demyelinating injury. Hum. Mol. Genet. 2017, 26, 2825–2837. [Google Scholar] [CrossRef]
- Marshall, M.S.; Bongarzone, E.R. Beyond Krabbe’s disease: The potential contribution of galactosylceramidase deficiency to neuronal vulnerability in late-onset synucleinopathies. J. Neurosci. Res. 2016, 94, 1328–1332. [Google Scholar] [CrossRef]
- Folts, C.J.; Scott-Hewitt, N.; Proschel, C.; Mayer-Proschel, M.; Noble, M. Lysosomal Re-acidification Prevents Lysosphingolipid-Induced Lysosomal Impairment and Cellular Toxicity. PLoS Biol. 2016, 14, e1002583. [Google Scholar] [CrossRef]
- Suzuki, K.; Yamaguchi, A.; Yamanaka, S.; Kanzaki, S.; Kawashima, M.; Togo, T.; Katsuse, O.; Koumitsu, N.; Aoki, N.; Iseki, E.; et al. Accumulated alpha-synuclein affects the progression of GM2 gangliosidoses. Exp. Neurol. 2016, 284, 38–49. [Google Scholar] [CrossRef]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Perkowska, K. Parkinsonism in Inherited Metabolic Disorders: Key Considerations and Major Features. Front. Neurol. 2018, 9, 857. [Google Scholar] [CrossRef]
- Gaspar, R.; Pallbo, J.; Weininger, U.; Linse, S.; Sparr, E. Ganglioside lipids accelerate alpha-synuclein amyloid formation. Biochim. Biophys. Acta Proteins proteomics 2018. [Google Scholar] [CrossRef]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef]
- Schneider, J.S. Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson’s disease. PloS ONE 2018, 13, e0199189. [Google Scholar] [CrossRef]
- Chan, R.B.; Perotte, A.J.; Zhou, B.; Liong, C.; Shorr, E.J.; Marder, K.S.; Kang, U.J.; Waters, C.H.; Levy, O.A.; Xu, Y.; et al. Elevated GM3 plasma concentration in idiopathic Parkinson’s disease: A lipidomic analysis. PloS ONE 2017, 12, e0172348. [Google Scholar] [CrossRef]
- Lutz, M.S.; Jaskiewicz, E.; Darling, D.S.; Furukawa, K.; Young, W.W., Jr. Cloned beta 1,4 N-acetylgalactosaminyltransferase synthesizes GA2 as well as gangliosides GM2 and GD2. GM3 synthesis has priority over GA2 synthesis for utilization of lactosylceramide substrate in vivo. J. Biol. Chem. 1994, 269, 29227–29231. [Google Scholar]
- Hidari, J.K.; Ichikawa, S.; Furukawa, K.; Yamasaki, M.; Hirabayashi, Y. beta 1-4N-acetylgalactosaminyltransferase can synthesize both asialoglycosphingolipid GM2 and glycosphingolipid GM2 in vitro and in vivo: isolation and characterization of a beta 1-4N-acetylgalactosaminyltransferase cDNA clone from rat ascites hepatoma cell line AH7974F. Biochem. J. 1994, 303, 957–965. [Google Scholar] [CrossRef]
- Boukhris, A.; Schule, R.; Loureiro, J.L.; Lourenco, C.M.; Mundwiller, E.; Gonzalez, M.A.; Charles, P.; Gauthier, J.; Rekik, I.; Acosta Lebrigio, R.F.; et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 93, 118–123. [Google Scholar] [CrossRef]
- Harlalka, G.V.; Lehman, A.; Chioza, B.; Baple, E.L.; Maroofian, R.; Cross, H.; Sreekantan-Nair, A.; Priestman, D.A.; Al-Turki, S.; McEntagart, M.E.; et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013, 136, 3618–3624. [Google Scholar] [CrossRef]
- Wakil, S.M.; Monies, D.M.; Ramzan, K.; Hagos, S.; Bastaki, L.; Meyer, B.F.; Bohlega, S. Novel B4GALNT1 mutations in a complicated form of hereditary spastic paraplegia. Clin. Genet. 2014, 86, 500–501. [Google Scholar] [CrossRef]
- Bhuiyan, R.H.; Ohmi, Y.; Ohkawa, Y.; Zhang, P.; Takano, M.; Hashimoto, N.; Okajima, T.; Furukawa, K. Loss of Enzyme Activity in Mutated B4GALNT1 Gene Products in Patients with Hereditary Spastic Paraplegia Results in Relatively Mild Neurological Disorders: Similarity with Phenotypes of B4galnt1 Knockout Mice. Neuroscience 2019, 397, 94–106. [Google Scholar] [CrossRef]
- Sheikh, K.A.; Sun, J.; Liu, Y.; Kawai, H.; Crawford, T.O.; Proia, R.L.; Griffin, J.W.; Schnaar, R.L. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc. Natl. Acad. Sci. USA 1999, 96, 7532–7537. [Google Scholar] [CrossRef]
- Takamiya, K.; Yamamoto, A.; Furukawa, K.; Yamashiro, S.; Shin, M.; Okada, M.; Fukumoto, S.; Haraguchi, M.; Takeda, N.; Fujimura, K.; et al. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc. Natl. Acad. Sci. USA 1996, 93, 10662–10667. [Google Scholar] [CrossRef]
- Dick, K.J.; Eckhardt, M.; Paisan-Ruiz, C.; Alshehhi, A.A.; Proukakis, C.; Sibtain, N.A.; Maier, H.; Sharifi, R.; Patton, M.A.; Bashir, W.; et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum. Mutat. 2010, 31, E1251–E1260. [Google Scholar] [CrossRef]
- Hama, H. Fatty acid 2-Hydroxylation in mammalian sphingolipid biology. Biochim. Biophys. Acta 2010, 1801, 405–414. [Google Scholar] [CrossRef]
- Boutry, M.; Branchu, J.; Lustremant, C.; Pujol, C.; Pernelle, J.; Matusiak, R.; Seyer, A.; Poirel, M.; Chu-Van, E.; Pierga, A.; et al. Inhibition of Lysosome Membrane Recycling Causes Accumulation of Gangliosides that Contribute to Neurodegeneration. Cell Rep. 2018, 23, 3813–3826. [Google Scholar] [CrossRef]
- Sandhoff, R.; Schulze, H.; Sandhoff, K. Ganglioside Metabolism in Health and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 1–62. [Google Scholar] [CrossRef]
- Indellicato, R.; Parini, R.; Domenighini, R.; Malagolini, N.; Iascone, M.; Gasperini, S.; Masera, N.; dall’Olio, F.; Trinchera, M. Total loss of GM3 synthase activity by a normally processed enzyme in a novel variant and in all ST3GAL5 variants reported to cause a distinct congenital disorder of glycosylation. Glycobiology 2019, 29, 229–241. [Google Scholar] [CrossRef]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A.; et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef]
- Boccuto, L.; Aoki, K.; Flanagan-Steet, H.; Chen, C.F.; Fan, X.; Bartel, F.; Petukh, M.; Pittman, A.; Saul, R.; Chaubey, A.; et al. A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum. Mol. Genet. 2014, 23, 418–433. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Song, J.; Choi, M.; Chae, J.H. GM3 synthase deficiency due to ST3GAL5 variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype. Am. J. Med. Geneti. Part A 2016, 170, 2200–2205. [Google Scholar] [CrossRef]
- Bowser, L.E.; Young, M.; Wenger, O.K.; Ammous, Z.; Brigatti, K.W.; Carson, V.J.; Moser, T.; Deline, J.; Aoki, K.; Morlet, T.; et al. Recessive GM3 synthase deficiency: Natural history, biochemistry, and therapeutic frontier. Mol. Genet. Metab. 2019. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohmi, Y.; Ji, S.; Zhang, P.; Bhuiyan, R.H.; Ohkawa, Y.; Tajima, O.; Hashimoto, N. Glycolipids: Essential regulator of neuro-inflammation, metabolism and gliomagenesis. Biochim. Biophys. Acta General subjects 2017, 1861, 2479–2484. [Google Scholar] [CrossRef]
- Yamashita, T.; Hashiramoto, A.; Haluzik, M.; Mizukami, H.; Beck, S.; Norton, A.; Kono, M.; Tsuji, S.; Daniotti, J.L.; Werth, N.; et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA 2003, 100, 3445–3449. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Go, S.; Takasaki, K.; Kakazu, Y.; Ohashi, M.; Nagafuku, M.; Kabayama, K.; Sekimoto, J.; Suzuki, S.; Takaiwa, K.; et al. Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc. Natl. Acad. Sci. USA 2009, 106, 9483–9488. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Harduin-Lepers, A.; Magalhaes, A.; Machado, E.; Mendes, N.; Costa, L.T.; Matthiesen, R.; Almeida, R.; Costa, J.; Reis, C.A. Differential expression of alpha-2,3-sialyltransferases and alpha-1,3/4-fucosyltransferases regulates the levels of sialyl Lewis a and sialyl Lewis x in gastrointestinal carcinoma cells. The international journal of biochemistry & cell biology 2010, 42, 80–89. [Google Scholar] [CrossRef]
- Kitagawa, H.; Paulson, J.C. Cloning and expression of human Gal beta 1,3(4)GlcNAc alpha 2,3-sialyltransferase. Biochem Bioph Res Commun. 1993, 194, 375–382. [Google Scholar] [CrossRef]
- Kono, M.; Ohyama, Y.; Lee, Y.C.; Hamamoto, T.; Kojima, N.; Tsuji, S. Mouse beta-galactoside alpha 2,3-sialyltransferases: comparison of in vitro substrate specificities and tissue specific expression. Glycobiology 1997, 7, 469–479. [Google Scholar] [CrossRef]
- Sturgill, E.R.; Aoki, K.; Lopez, P.H.; Colacurcio, D.; Vajn, K.; Lorenzini, I.; Majic, S.; Yang, W.H.; Heffer, M.; Tiemeyer, M.; et al. Biosynthesis of the major brain gangliosides GD1a and GT1b. Glycobiology 2012, 22, 1289–1301. [Google Scholar] [CrossRef]
- Aronica, A.; Avagliano, L.; Caretti, A.; Tosi, D.; Bulfamante, G.P.; Trinchera, M. Unexpected distribution of CA19.9 and other type 1 chain Lewis antigens in normal and cancer tissues of colon and pancreas: Importance of the detection method and role of glycosyltransferase regulation. Biochim. Biophys. Acta General subjects 2017, 1861, 3210–3220. [Google Scholar] [CrossRef]
- Zulueta, A.; Caretti, A.; Signorelli, P.; Dall’olio, F.; Trinchera, M. Transcriptional control of the B3GALT5 gene by a retroviral promoter and methylation of distant regulatory elements. FASEB J. 2014, 28, 946–955. [Google Scholar] [CrossRef]
- Mare, L.; Caretti, A.; Albertini, R.; Trinchera, M. CA19.9 antigen circulating in the serum of colon cancer patients: where is it from? Int. J. Biochem. Cell Biol. 2013, 45, 792–797. [Google Scholar] [CrossRef]
- Hu, H.; Eggers, K.; Chen, W.; Garshasbi, M.; Motazacker, M.M.; Wrogemann, K.; Kahrizi, K.; Tzschach, A.; Hosseini, M.; Bahman, I.; et al. ST3GAL3 mutations impair the development of higher cognitive functions. Am. J. Hum. Genet. 2011, 89, 407–414. [Google Scholar] [CrossRef]
- van Diepen, L.; Buettner, F.F.R.; Hoffmann, D.; Thiesler, C.T.; von Bohlen Und Halbach, O.; von Bohlen Und Halbach, V.; Jensen, L.R.; Steinemann, D.; Edvardson, S.; Elpeleg, O.; et al. A patient-specific induced pluripotent stem cell model for West syndrome caused by ST3GAL3 deficiency. Eur.J. Hum. Geneti. 2018, 26, 1773–1783. [Google Scholar] [CrossRef]
- Ellies, L.G.; Sperandio, M.; Underhill, G.H.; Yousif, J.; Smith, M.; Priatel, J.J.; Kansas, G.S.; Ley, K.; Marth, J.D. Sialyltransferase specificity in selectin ligand formation. Blood 2002, 100, 3618–3625. [Google Scholar] [CrossRef]
- Kiwamoto, T.; Brummet, M.E.; Wu, F.; Motari, M.G.; Smith, D.F.; Schnaar, R.L.; Zhu, Z.; Bochner, B.S. Mice deficient in the St3gal3 gene product alpha2,3 sialyltransferase (ST3Gal-III) exhibit enhanced allergic eosinophilic airway inflammation. J. Allergy Clin. Immunol. 2014, 133, 240–247. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- van Echten-Deckert, G.; Alam, S. Sphingolipid metabolism―An ambiguous regulator of autophagy in the brain. Biological chemistry 2018, 399, 837–850. [Google Scholar] [CrossRef]
- Qiang, L.; Piermarini, E.; Baas, P.W. New Hypothesis for the Etiology of SPAST-based Hereditary Spastic Paraplegia. Cytoskeleton 2019. [Google Scholar] [CrossRef]
- Fragaki, K.; Ait-El-Mkadem, S.; Chaussenot, A.; Gire, C.; Mengual, R.; Bonesso, L.; Beneteau, M.; Ricci, J.E.; Desquiret-Dumas, V.; Procaccio, V.; et al. Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency. Eur. J. Hum. Genet. 2013, 21, 528–534. [Google Scholar] [CrossRef]
- Kiebish, M.A.; Han, X.; Cheng, H.; Lunceford, A.; Clarke, C.F.; Moon, H.; Chuang, J.H.; Seyfried, T.N. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J. Neurochem. 2008, 106, 299–312. [Google Scholar] [CrossRef]
- Saffari, A.; Kolker, S.; Hoffmann, G.F.; Ebrahimi-Fakhari, D. Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 2017, 40, 631–640. [Google Scholar] [CrossRef]
- Sahu, S.K.; Hannun, Y.A.; Yao, N. Emergence of membrane sphingolipids as a potential therapeutic target. Biochimie 2019, 158, 257–264. [Google Scholar] [CrossRef]
- Cui, Y.; Hettinghouse, A.; Liu, C.J. Progranulin: A conductor of receptors orchestra, a chaperone of lysosomal enzymes and a therapeutic target for multiple diseases. Cytokine Growth Factor Rev. 2019, 45, 53–64. [Google Scholar] [CrossRef]
- Zielonka, M.; Garbade, S.F.; Kölker, S.; Hoffmann, G.F.; Ries, M. A cross-sectional quantitative analysis of the natural history of Farber disease: an ultra-orphan condition with rheumatologic and neurological cardinal disease features. Genetet. Med. 2018, 20, 524–530. [Google Scholar] [CrossRef]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases; roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. [Google Scholar] [CrossRef]
- Li, F.; Xu, R.; Low, B.E.; Lin, CL.; Garcia-Barros, M.; Schrandt, J.; Mileva, I.; Snider, A.; Luo, C.K.; Jiang, X.C.; et al. Alkaline ceramidase 2 is essential for the homeostasis of plasma sphingoid bases and their phosphates. FASEB J. 2018, 32, 3058–3069. [Google Scholar] [CrossRef]
- Hatoum, D.; Haddadi, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: challenges for SphK as an oncotarget. Oncotarget 2017, 8, 36898–36929. [Google Scholar] [CrossRef]
- Lovric, S.; Goncalves, S.; Gee, H.Y.; Oskouian, B.; Srinivas, H.; Choi, W.I.; Shril, S.; Ashraf, S.; Tan, W.; Rao, J.; et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Investig. 2017, 127, 912–928. [Google Scholar] [CrossRef]
- Oliveira, J.P.; Ferreira, S. Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype- phenotype correlations. Appl. Clin. Genet. 2019, 12, 35–50. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Enzyme | Hugo Symbol | Subcellular Site | Disease | Main Clinical Features | Biochemical Features | Ref |
|---|---|---|---|---|---|---|
| Acid ceramidase | ASAH1 | Lysosome | Farber disease and spinal muscular atrophy with progressive myoclonic epilepsy | Typical spectrum disease varying from the classic triad of subcutaneous nodules, joint contractures, and hoarse voice to moderate or severe forms involving hematopoietic, gastrointestinal, respiratory, and neurologic symptoms, including seizures; developmental delay and death in the early childhood. | The same Y137C mutation provided very mild phenotype in a patient and severe neurologic phenotype in another. Two SNPs are associated with schizophrenia. Residual activity >5% is associated with survival. Candidate risk factor for Parkinson´s disease (PD). | [29,73,127] |
| Neutral ceramidase | ASAH2 | Plasma membrane | None reported | Main expression in the small intestine and colon, probable role in digestion. | [128] | |
| Alkaline ceramidases | ACER1 | Endoplasmic reticulum (ER) | Main expression in the skin. | [129] | ||
| ACER2 | Golgi apparatus | |||||
| ACER3 | ER and Golgi apparatus | Progressive leukodystrophy | Developmental regression at 6–13 months, starting with peripheral neuropathy and leading to severe dysmorphic facial feature and psychomotor impairment, requiring mechanical ventilation. | Plasma accumulation of ceramides, dihydroceramides, glucosylceramide (GlcCer), and lactosylceramide (LacCer). Increased blood lactate levels. | [30] | |
| Sphingosine kinases | SPHK1 | Plasma membrane (main) | None reported | [130] | ||
| SPHK2 | ER (main) | |||||
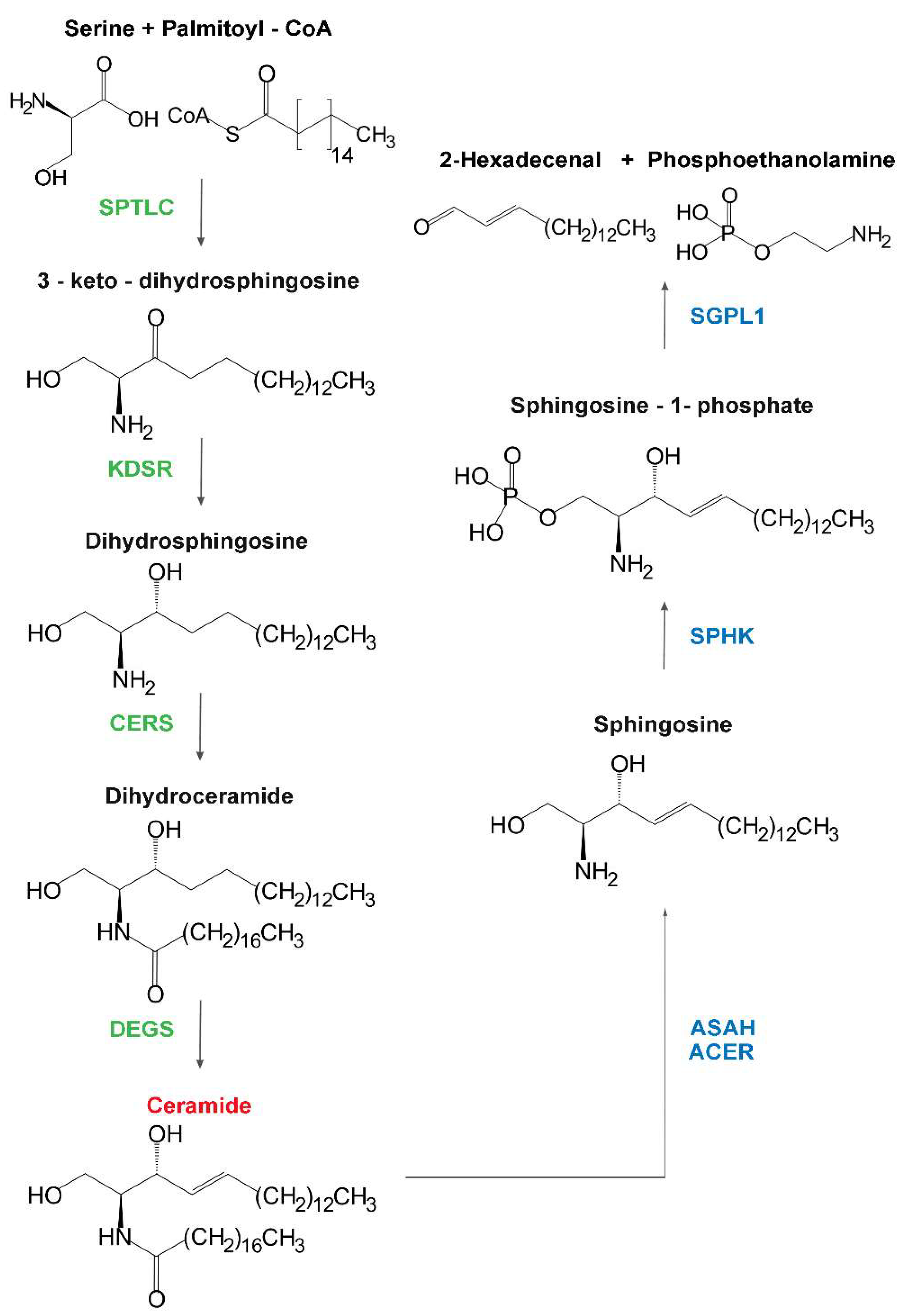
| Sphingosine lyase | SGPL1 | ER | Syndromic steroid-resistant nephrotic syndrome | Steroid-resistant nephrotic syndrome with facultative ichthyosis, adrenal insufficiency, immunodeficiency, and neurological defects. | Reduced activity and protein mislocalization are frequent between mutations. Ceramides are elevated in the conditioned culture medium of patient fibroblasts. | [131] |
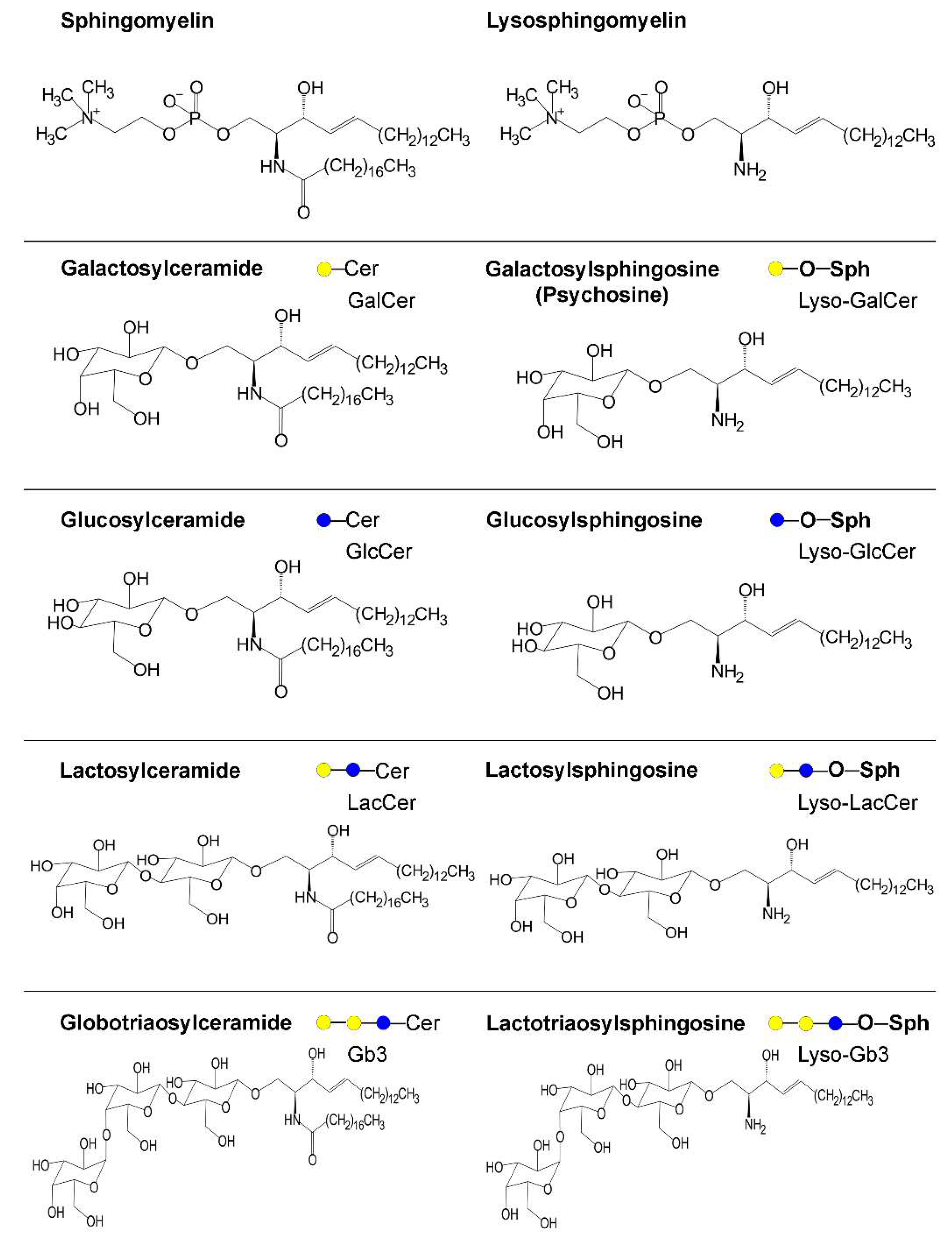
| Galacto-cerebrosidase | GALC | Lysosome | Krabbe disease | Infantile onset (within 6 months). Increased irritability, spasticity, developmental delay along with unexplained fever, blindness, and deafness. Severe motor and mental deterioration. | Poor genotype–phenotype relationship. Galactosyl-sphingosine (psychosine) accumulates, affecting endolysosomal transport and pH. Aggregated forms of α-synuclein reported. | [37,49,79,91] |
| Gluco-cerebrosidases | GBA | Lysosome | Gaucher disease | Type 1 disease classically includes inflammatory signs in visceral organs that appear in adulthood; types 2 and 3 are instead neuronopathic, with early onset and progression at different rates. Recently proposed to be a spectrum disease. Main genetic risk factor for PD even in heterozygous carriers (see details in the text). | Poor genotype–phenotype relationship. GlcCer and glucosylsphingosine accumulate, affecting vesicle traffic and autophagy including mitophagy, which are also impaired by altered proteostasis. Strong evidence that glucocerebrosidase (GBA) variants affect α-synuclein accumulation (see details in the text). | [27,33,50,54,57] |
| GBA2 | Microsomes | HSP46/Cerebellar ataxia with late-onset spasticity | Early onset of motor impairment with mental retardation, cataract, and hypogonadism in males. MRI: cerebellar and corpus callosum atrophy. | Loss of enzymatic activity in almost all known mutations. Inhibition of activity in fibroblasts from Niemann–Pick patients restores endolysosomal pH. | [28,44,72] | |
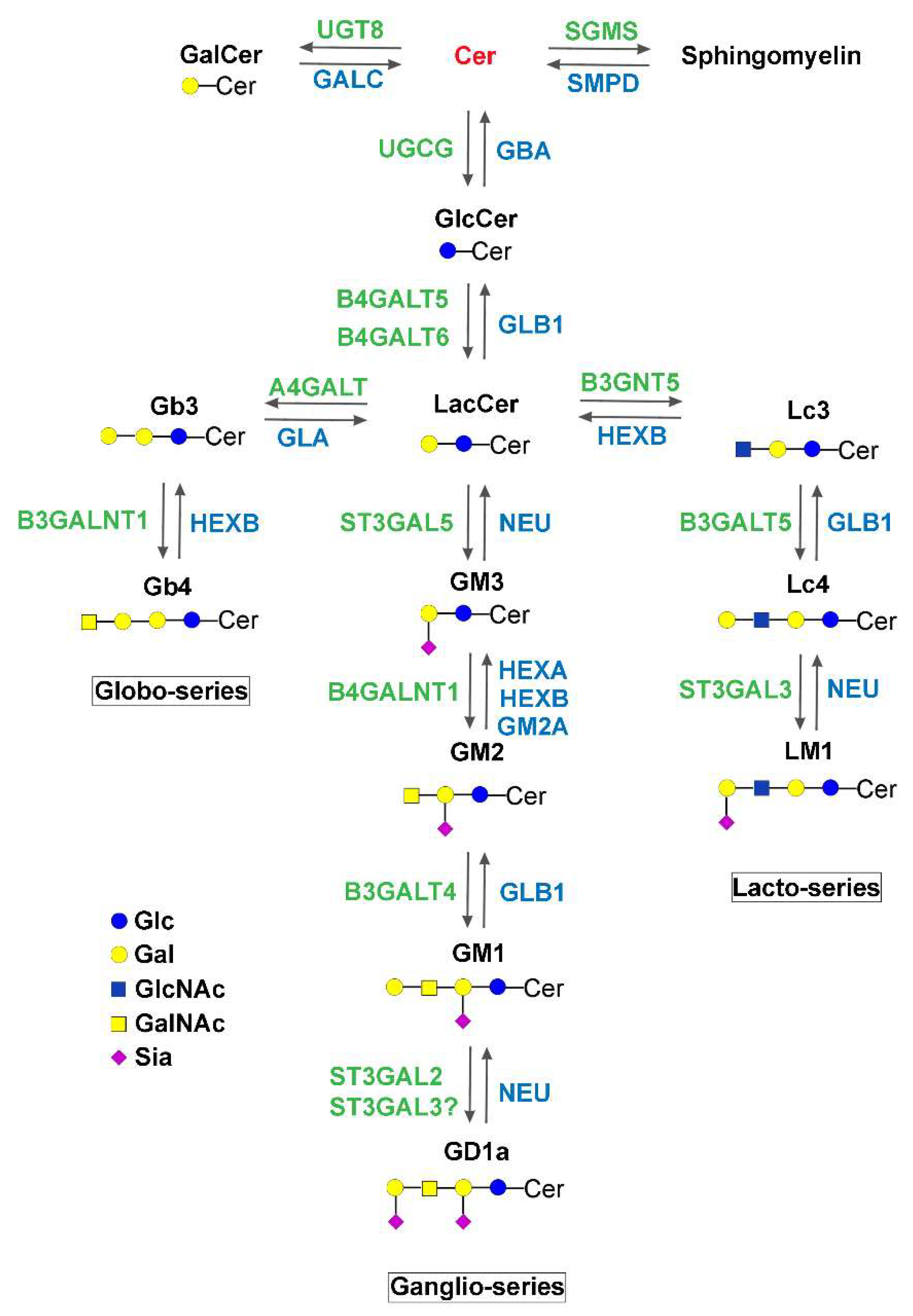
| β-galactosidase | GLB1 | Lysosome | GM1 gangliosidosis | Infantile form: early onset and rapid progressive psychomotor deterioration, skeletal abnormalities, visceromegaly, and death. Juvenile and adult phenotypes characterized by slowly progressive neurological degeneration and mild skeletal changes. | Poor genotype–phenotype correlation. GM1, LacCer, and lactosylsphingosine accumulate causing impairment of endolysosomal transport and pH, autophagy, and mitochondrial function). ER stress detected. | [81,99] |
| Hexosaminidase A | HEXA | Lysosome | Tay–Sachs disease | Infantile form: early onset of neurodevelopmental dysfunctions, hypotonia and eye movement abnormalities. Progression includes dysphagia, seizures, macrocephaly, and death until age 3.5 years. Juvenile onset includes ataxia, dysarthria, dysphagia, progressive hypotonia, seizures, and death until 15 years of age. | [99] | |
| Hexosaminidase B | HEXB | Lysosome | Sandhoff disease | Juvenile onset form: reduced attention, weakness, hypotonia, and progressive psychomotor impairment. Adult-onset form: milder phenotype due to residual enzymatic activity. Muscle weakness and motor symptoms. | GM2, asialo-GM2, and globoside accumulate. Deposits of α-synuclein reported. | [49,99] |
| GM2 activator | GM2A | Lysosome | GM2 gangliosidosis | Similar to Tay–Sachs disease. | [99] | |
| α-galactosidase | GLA | Lysosome | Fabry disease | X-linked recessive, phenotypes from healthy to severe in women, severe to fatal in men. Various organs potentially involved, including peripheral and central nervous system. Cardiovascular involvement is frequent and at high risk for stroke and arrhythmias. | Globotriaosylceramide and globotriaosylsphingosine accumulate. Activity of respiratory chain enzymes reduced, protein trafficking and sorting altered, autophagy-lysosome pathway dysregulated. Impaired α-synuclein degradation. | [38,76,132] |
| Acid sphingomyelinase | SMPD1 | Lysosome and secretory | Niemann–Pick disease types A and B | The gene is paternally imprinted. Type A: acute, early onset with failure to thrive and hepatosplenomegaly. Rapid and progressive neurodegenerative course, hypothonia and death until age of 3 years. Cherry-red spot in the macula. Type B: chronic, no neurologic signs. Hepato-splenomegaly and signs of liver failure. Impaired pulmonary function. High levels of serum triglycerides and LDL-cholesterol, low levels of HDL-cholesterol. Reddish brown or cherry red spot in the macula. SMPD1 variants are confirmed risk factor for PD. | Good genotype-phenotype correlation. Sphingomyelin and lysosphingomyelin (sphingosine-phosphocoline) accumulate. Increased levels of cholesterol, GlcCer, LacCer, and gangliosides. Decreased activity levels led to α-synuclein accumulation. | [22,36] |
| Neutral sphingomyelinases | SMPD2 | Plasma membrane | None reported | [22] | ||
| SMPD3 | ER, Golgi apparatus, and nucleus | |||||
| SMPD4 | ER and Golgi apparatus | |||||
| SMPD5 | Mitochondria and ER |
| Enzyme | Hugo Symbol | Disease | Inheritance | Main Clinical Features | Biochemical Features | Ref. |
|---|---|---|---|---|---|---|
| Serine palmitoyl transferases | SPTLC1 | Hereditary sensory neuropathy (Type 1) | Autosomal dominant | Onset of sensory impairment spanning the second to fifth decades, frequent motor impairment and burning pain episodes; distal to proximal progression. Mutations of either one or two subunits determine identical clinical phenotypes. | Alanine and glycine used instead of serine producing deoxysphinganine and deoxyceramide, which have mitochondrial toxicity in vitro. | [9,10] |
| SPTLC2 | ||||||
| SPTLC3 | None reported | |||||
| 3-keto-dihydro-sphingosine reductase | KDSR | Erythrokeratoderma or ichtyosis with anemia and thrombocytopenia | Autosomal recessive | No neurologic signs. Heterogeneous skin and hematologic symptoms; spontaneous remission with age in some cases. | Retinoic acid therapy effective, probably stimulating salvage pathway from sphingosine. | [11,12,13] |
| Dihydro-ceramide synthases | CERS1 | Myoclonus epilepsy | Autosomal recessive | Ataxia at the age of one year, delay in development, generalized tonic–clonic seizures, action myoclonus with onset between 6 and 16 years of age. Cognitive deterioration up to dementia.Magnetic resonance imaging: brainstem atrophy. | [14,15] | |
| CERS2 | Progressive myoclonus epilepsy | 27 kb heterozygous deletion | Tonic–clonic seizures prevented by valproic acid, learning disability, progressive myoclonic epilepsy, moderate intellectually disability with dysarthria and ataxia. | [17] | ||
| CERS3 | Congenital ichthyosis | Autosomal recessive | No neurologic signs. Congenital ichthyosis characterized by collodion membranes at birth, generalized scaling of the skin, and mild erythroderma. | Specific loss of ceramides with acyl chains from C26 up to C34 in keratinocytes. | [16] | |
| CERS4 | None reported | [18] | ||||
| CERS5 | ||||||
| CERS6 | ||||||
| Dihydro-ceramide desaturases | DEGS1 | Hypomyelinating Leukodystrophy | Autosomal recessive | Onset at 0.5–24 months. Failure to thrive, developmental delay, epilepsy, neurogenic muscular atrophy, severe motor arrest, microcephaly, dystonia and severe spasticity. MRI: hypomyelination, thin white matter, progressive thalamic and cerebellar atrophy. | Presence of Δ14-cis sphingolipids; inhibition of CERS ameliorates phenotype in zebrafish model, and reactive oxygen species (ROS) levels in patient fibroblasts. | [20,21] |
| DEGS2 | None reported | Relevant in stratum corneum. | Involved in the metabolism of sphingolipid containing 4-hydroxysphingosine (phytosphingosine). | [19] | ||
| SM synthase related protein | SAMD8 | None reported | Involved in the synthesis of ceramide phosphoethanolamine. | [22] | ||
| GalCer synthase | UGT8 | None reported |
| Enzyme | Hugo Symbol | Disease | Inheritance | Main Clinical Features | Biochemical Features | Ref. |
|---|---|---|---|---|---|---|
| Sphingomyelin synthases | SGMS1 | None reported | [22] | |||
| SGMS2 (plasma membrane resident) | Osteoporosis with skeletal dysplasia | Autosomal dominant | Minor neurologic signs detectable in some cases. Childhood onset osteoporosis with or without cranial sclerosis, neonatal fractures, short stature, and spondylometaphyseal dysplasia. | Variants are frequently mislocalized or retained in the ER; catalytic activity maintained by some variants. | [23] | |
| Glucosylceramide synthase | UGCG | Congenital ichthyosis | Autosomal recessive | Normal growth parameters at birth, but covered with a collodion membrane; death at age 2 weeks because of severe hypernatremic anuric renal failure. | Phenotype similar to that of the keratinocyte-conditional KO mouse. | [24] |
| UDP-Gal: GlcCer β1,4-galactosyltransferase | B4GALT6 | None reported | Synthesizes lactosylceramide. | |||
| UDP-Gal: lactosylceramideα-1,4-galactosyl-transferase | A4GALT | Synthesizes globotriosyl ceramide. | ||||
| UDP-GlcNAc: lactosylceramide β-1,4-GlcNAc transferase | B3GNT5 | Synthesizes lacto-N-triosyl ceramide. | ||||
| GM3 synthase | ST3GAL5 | ST3GAL5-CDG | Autosomal recessive | Normal at birth, early onset severe neurological signs. Failure to thrive, regression, severe hearing, visual, motor, and cognitive impairment (see details in the text). | Mitochondrial defects in patients. Globosides accumulate in human fibroblasts. | [25,100,101,102,103,104] |
| GM2/GD2/GA2 synthase | B4GALNT1 | Hereditary spastic paraplegia 26 (B4GALNT1-CDG) | Autosomal recessive | Late onset motor impairment of the legs accompanied by mild to moderate cognitive impairment, sometimes associated with psychiatric illness and/or non-neurological symptoms (see details in the text). | GM3 and GD3 accumulate in vivo and in vitro models. | [90,91,92] |
| UDP-Gal: GM2/GD2/GA2 β1,3-galactosyltransferase | B3GALT4 | None reported | Synthesizes gangliosides GM1, GD1a, and GD1b. | |||
| CMP-Sial: GlcNAcβ1,3(4) sialyltransferase | ST3GAL3 | Non syndromic autosomal recessive intellectual disability/West syndrome | Autosomal recessive | Only intellectual disability reported when diagnosed in adults, West syndrome when diagnosed in early childhood (see details in the text). | ER retention frequent in variants, enzyme activity maintained in one variant. | [35,115,116] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indellicato, R.; Trinchera, M. The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. Int. J. Mol. Sci. 2019, 20, 3304. https://doi.org/10.3390/ijms20133304
Indellicato R, Trinchera M. The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. International Journal of Molecular Sciences. 2019; 20(13):3304. https://doi.org/10.3390/ijms20133304
Chicago/Turabian StyleIndellicato, Rossella, and Marco Trinchera. 2019. "The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism" International Journal of Molecular Sciences 20, no. 13: 3304. https://doi.org/10.3390/ijms20133304
APA StyleIndellicato, R., & Trinchera, M. (2019). The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. International Journal of Molecular Sciences, 20(13), 3304. https://doi.org/10.3390/ijms20133304