NMR Fragment-Based Screening against Tandem RNA Recognition Motifs of TDP-43

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
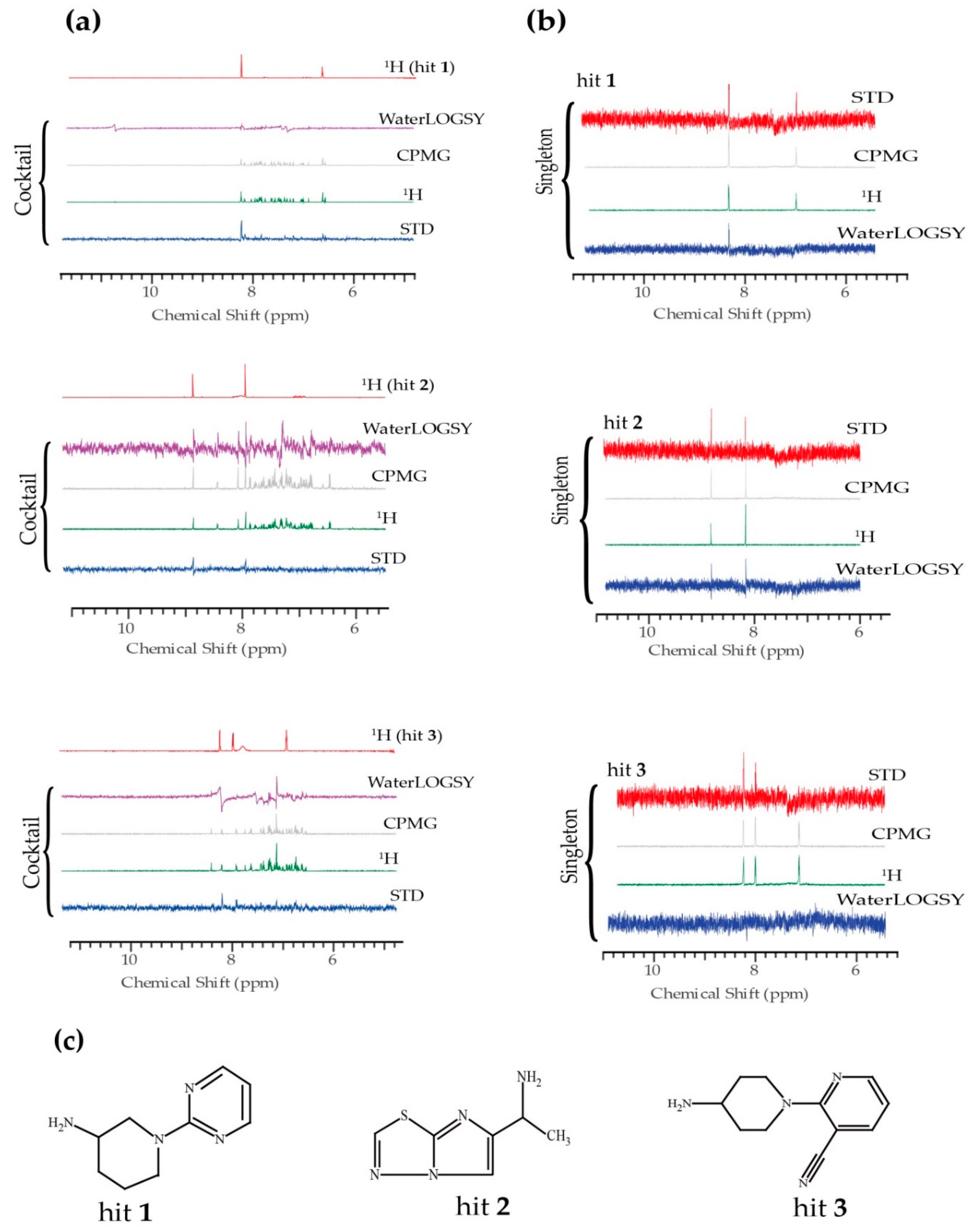
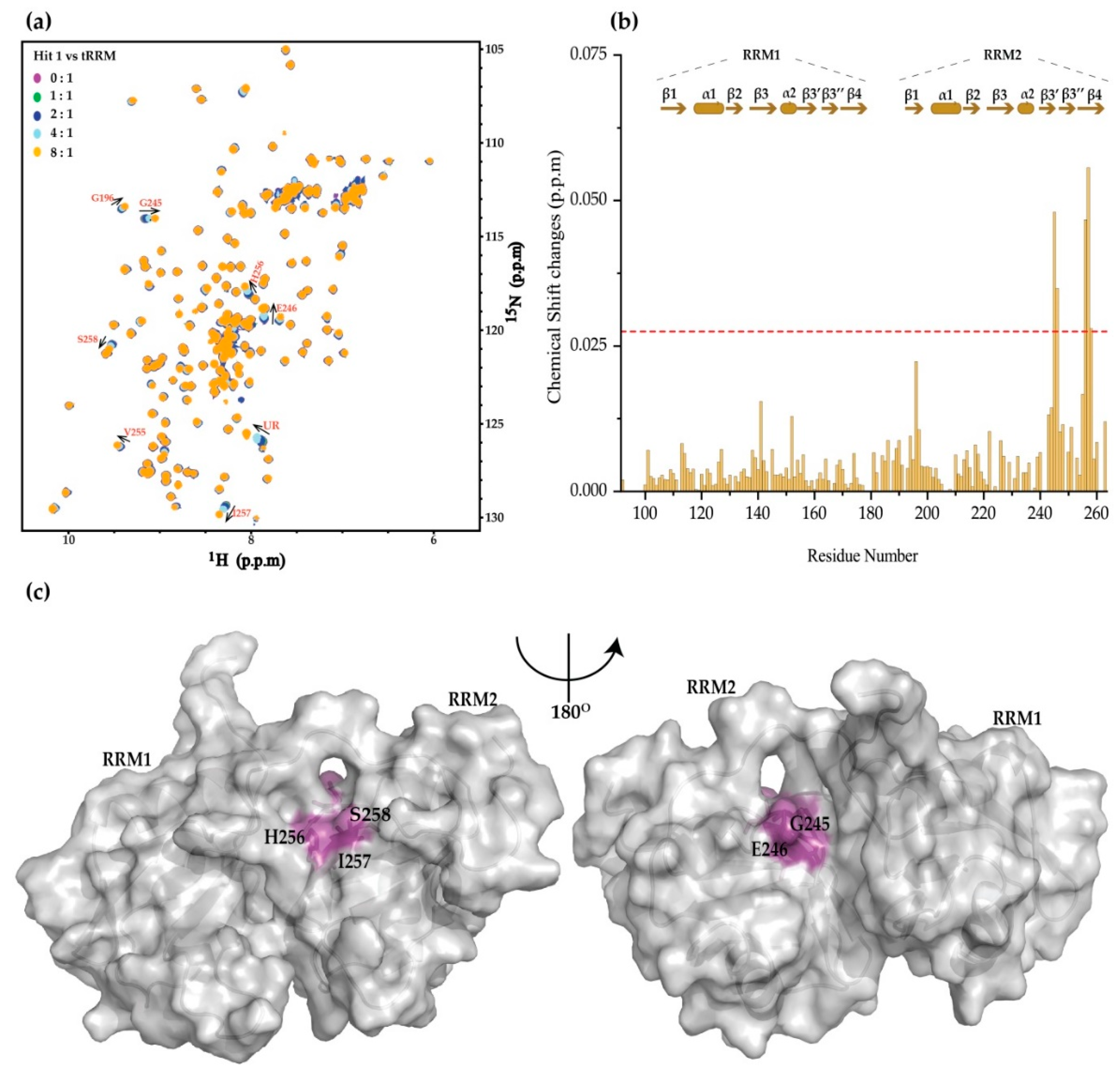
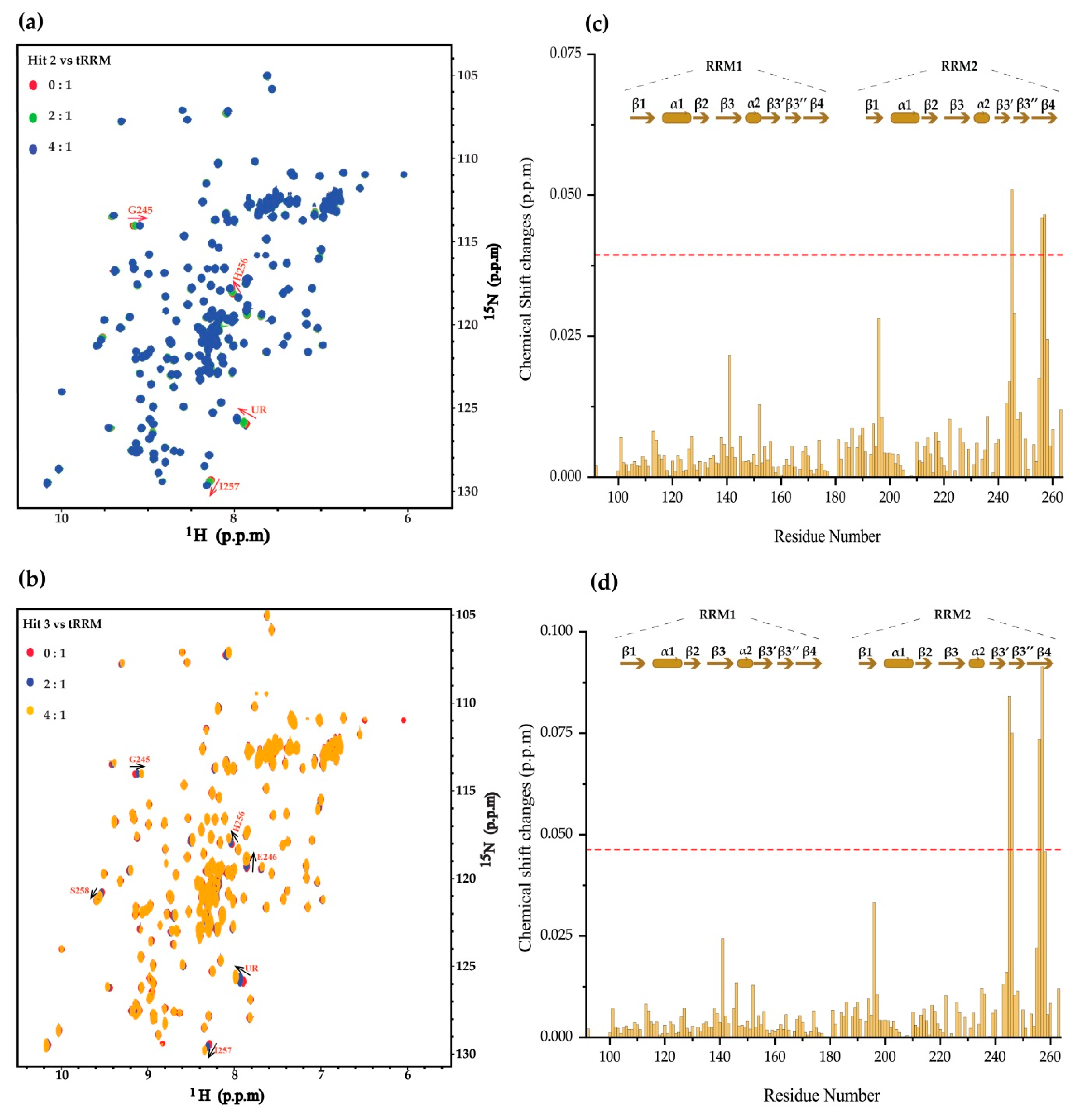
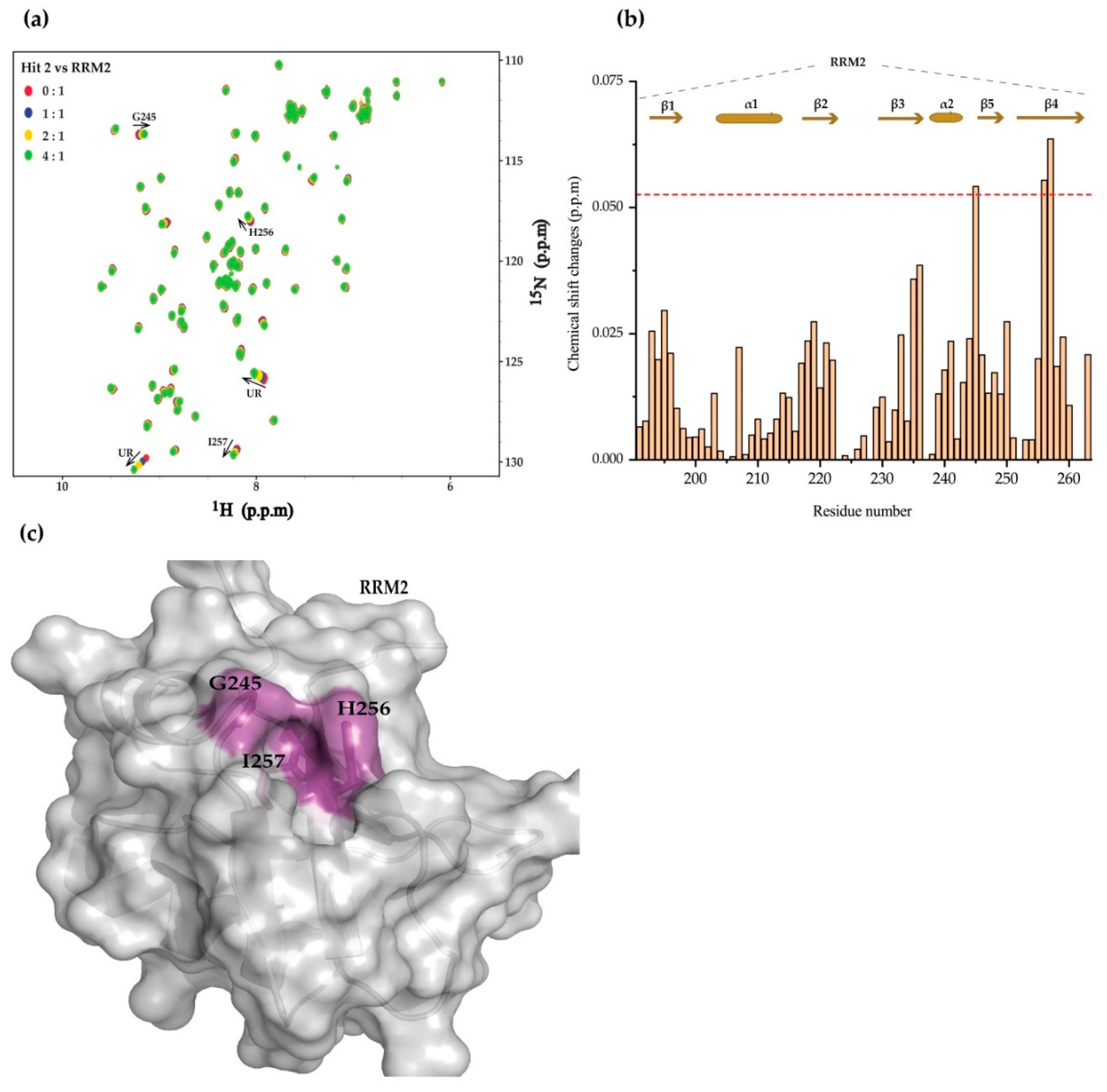
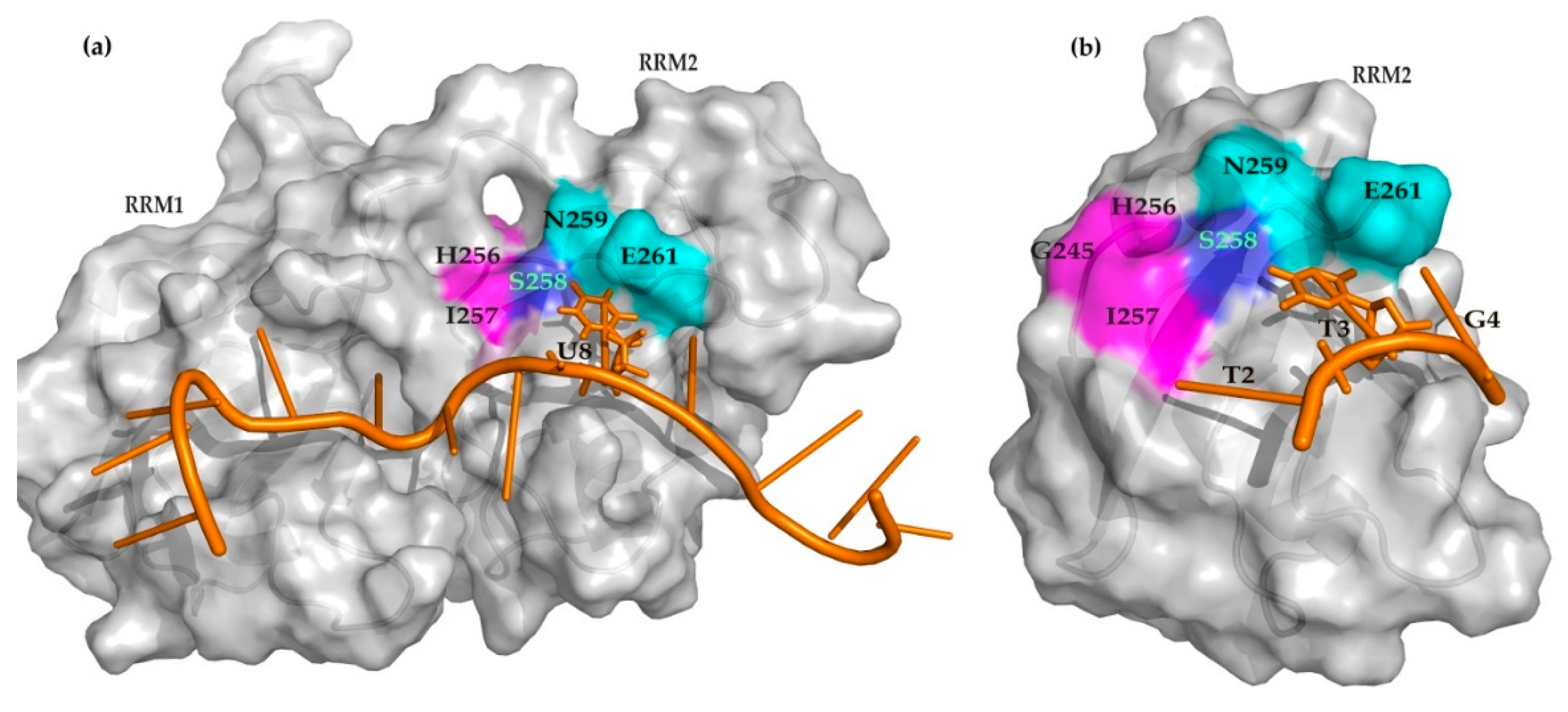
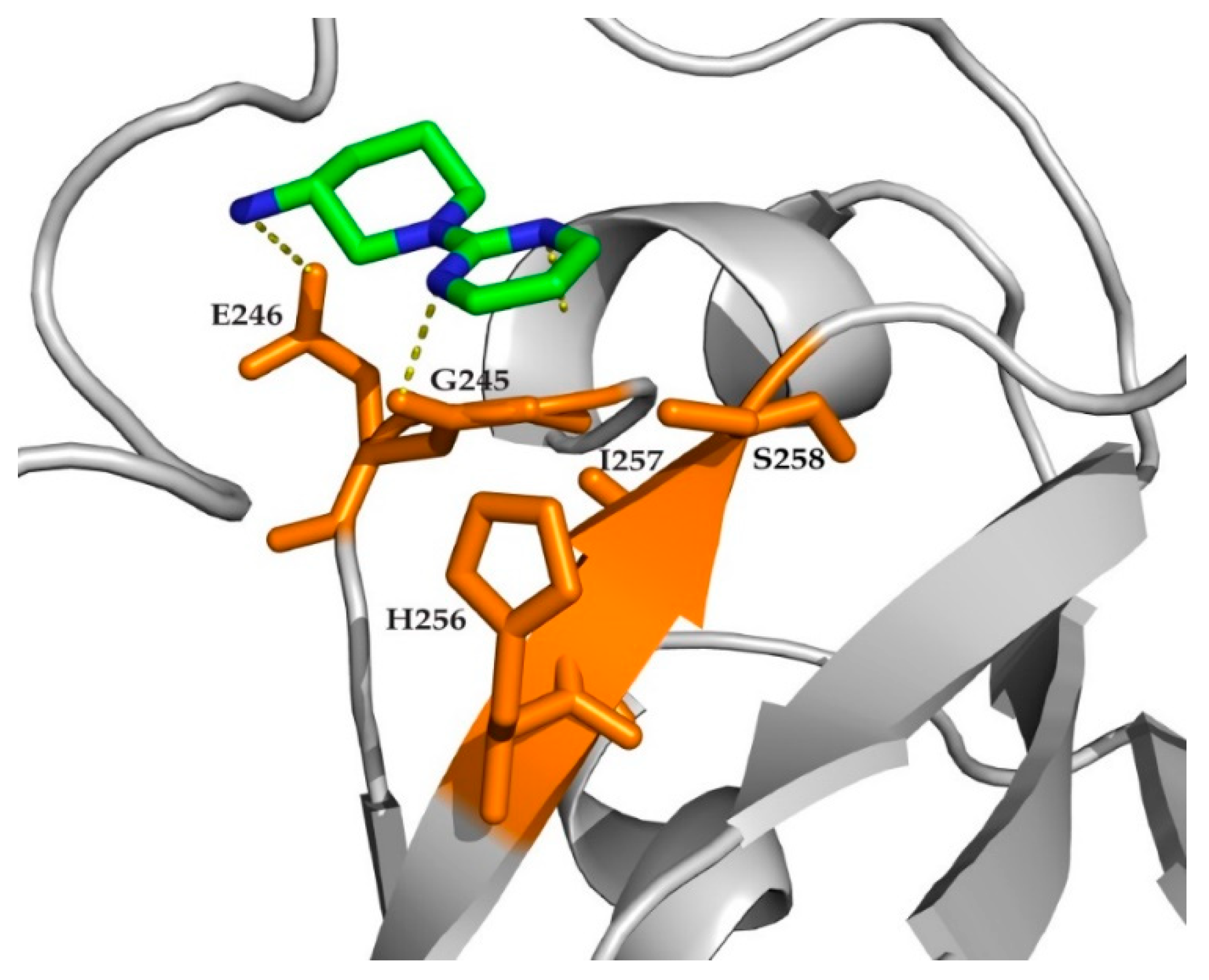
2. Results
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Protein Purification
4.2. NMR Fragment-Based Screening
4.3. NMR Chemical Shift Perturbation
4.4. Molecular Docking
4.5. Linewidth Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| ATG7 | Autophagy-related protein 7 |
| BMRB | Biological Magnetic Resonance Data Bank |
| CPMG | Carr–Purcell–Meiboom–Gill |
| CSPs | Chemical shift perturbations |
| FBS | Fragment-based screening |
| FTLD | Frontotemporal lobar degeneration |
| HADDOCK | High Ambiguity Driven biomolecular DOCKing |
| HDAC6 | Histone deacetylase 6 |
| hNFL | human low molecular weight neurofilament |
| HSQC | Heteronuclear single-quantum coherence |
| ITC | Isothermal titration calorimetry |
| NMR | Nuclear Magnetic Resonance |
| RNP | Ribonucleoproteins |
| RRM | RNA Recognition Motifs |
| SGs | Stress granules |
| SNHG12 | Small nucleolar RNA host gene 12 |
| SPR | Surface Plasmon Resonance |
| STD | Saturation Transfer difference |
| TDP-43 | Transactive response DNA-binding Protein 43 |
| TNBC | Triple-negative breast cancer |
| TRIM16 | Tripartite motif-containing protein 16 |
| WaterLOGSY | Water ligand observed via gradient spectroscopy |
References
- Venter, J.C. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Cienikova, Z.; Clery, A.; Allain, F.H. One, Two, Three, Four! How Multiple RRMs Read the Genome Sequence. Methods Enzym. 2015, 558, 235–278. [Google Scholar] [CrossRef]
- Clery, A.; Blatter, M.; Allain, F.H. RNA recognition motifs: Boring? Not quite. Curr. Opin. Struct. Biol. 2008, 18, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Kielkopf, C.L.; Lucke, S.; Green, M.R. U2AF homology motifs: Protein recognition in the RRM world. Genes Dev. 2004, 18, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef] [PubMed]
- Mercado, P.A.; Ayala, Y.M.; Romano, M.; Buratti, E.; Baralle, F.E. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005, 33, 6000–6010. [Google Scholar] [CrossRef] [PubMed]
- Banks, G.T.; Kuta, A.; Isaacs, A.M.; Fisher, E.M. TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm. Genome 2008, 19, 299–305. [Google Scholar] [CrossRef]
- Strong, M.J.; Volkening, K.; Hammond, R.; Yang, W.C.; Strong, W.; Leystra-Lantz, C.; Shoesmith, C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 2007, 35, 320–327. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Misteli, T.; Baralle, F.E. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. USA 2008, 105, 3785–3789. [Google Scholar] [CrossRef]
- Weskamp, K.; Barmada, S.J. TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 2018, 1693, 67–74. [Google Scholar] [CrossRef]
- Wilson, C.A.; Dugger, B.; Dickson, D.W.; Wang, D.S. TDP-43 in aging and Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2011, 4, 147–155. [Google Scholar] [PubMed]
- Sun, M.; Yamashita, T.; Shang, J.; Liu, N.; Deguchi, K.; Liu, W.; Ikeda, Y.; Feng, J.; Abe, K. Acceleration of TDP43 and FUS/TLS protein expressions in the preconditioned hippocampus following repeated transient ischemia. J. Neurosci. Res. 2014, 92, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014, 127, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2012, 287, 23079–23094. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef]
- Chen, Y.; Cohen, T.J. Aggregation of the nucleic acid–binding protein TDP-43 °Ccurs via distinct routes that are coordinated with stress granule formation. J. Biol. Chem. 2019. [Google Scholar]
- Ash, P.E.; Zhang, Y.J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef]
- Li, Y.; Ray, P.; Rao, E.J.; Shi, C.; Guo, W.; Chen, X.; Woodruff, E.A., 3rd; Fushimi, K.; Wu, J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 3169–3174. [Google Scholar] [CrossRef]
- Voigt, A.; Herholz, D.; Fiesel, F.C.; Kaur, K.; Muller, D.; Karsten, P.; Weber, S.S.; Kahle, P.J.; Marquardt, T.; Schulz, J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 2010, 5, e12247. [Google Scholar] [CrossRef]
- Bhandare, V.V.; Ramaswamy, A. The proteinopathy of D169G and K263E mutants at the RNA Recognition Motif (RRM) domain of tar DNA-binding protein (tdp43) causing neurological disorders: A computational study. J. Biomol. Struct. Dyn. 2018, 36, 1075–1093. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Chen, S.B.; Guo, D.J.; Pan, S.Y.; Yu, Z.L. Proteomic identification of differentially expressed proteins in curcumin-treated MCF-7 cells. Phytomedicine 2011, 18, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.Y.; Tan, O.; Liu, B.; Trahair, T.; Liu, T.; Haber, M.; Norris, M.D.; Marshall, G.M.; Cheung, B.B. High TDP43 expression is required for TRIM16-induced inhibition of cancer cell growth and correlated with good prognosis of neuroblastoma and breast cancer patients. Cancer Lett. 2016, 374, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.N.; Zhu, J.Y.; Tan, Y.; Zhang, Q.; Jia, W.; Hua, Q. Staurosporine induced apoptosis rapidly downregulates TDP- 43 in glioma cells. Asian Pac. J. Cancer Prev. 2014, 15, 3575–3579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.L.; Li, Y.D.; Guo, F.J. Expression of TDP43 in cervical cancer. Int. J. Clin. Exp. Pathol. 2016, 9, 1467–1473. [Google Scholar]
- Zeng, Q.; Cao, K.; Liu, R.; Huang, J.; Xia, K.; Tang, J.; Chen, X.; Zhou, M.; Xie, H.; Zhou, J. Identification of TDP-43 as an oncogene in melanoma and its function during melanoma pathogenesis. Cancer Biol. 2017, 18, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zheng, J.; Xue, Y.; Qu, C.; Chen, J.; Wang, Z.; Li, Z.; Zhang, L.; Liu, Y. Inhibition of TDP43-Mediated SNHG12-miR-195-SOX5 Feedback Loop Impeded Malignant Biological Behaviors of Glioma Cells. Mol. Nucleic Acids 2018, 10, 142–158. [Google Scholar] [CrossRef]
- Guo, F.; Jiao, F.; Song, Z.; Li, S.; Liu, B.; Yang, H.; Zhou, Q.; Li, Z. Regulation of MALAT1 expression by TDP43 controls the migration and invasion of non-small cell lung cancer cells in vitro. Biochem. Biophys. Res. Commun. 2015, 465, 293–298. [Google Scholar] [CrossRef]
- Ke, H.; Zhao, L.; Zhang, H.; Feng, X.; Xu, H.; Hao, J.; Wang, S.; Yang, Q.; Zou, L.; Su, X.; et al. Loss of TDP43 inhibits progression of triple-negative breast cancer in coordination with SRSF3. Proc. Natl. Acad. Sci. USA 2018, 115, E3426–E3435. [Google Scholar] [CrossRef]
- Boxer, A.L.; Gold, M.; Huey, E.; Gao, F.B.; Burton, E.A.; Chow, T.; Kao, A.; Leavitt, B.R.; Lamb, B.; Grether, M.; et al. Frontotemporal degeneration, the next therapeutic frontier: Molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. 2013, 9, 176–188. [Google Scholar] [CrossRef]
- Varani, G.; Nagai, K. RNA recognition by RNP proteins during RNA processing. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 407–445. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.Y.; Cui, M.; Heldsinger, A.; Lemrow, S.M.; Loo, J.A.; Sannes-Lowery, K.A.; Sharmeen, L.; Czarnik, A.W. Inhibitors of protein-RNA complexation that target the RNA: Specific recognition of human immunodeficiency virus type 1 TAR RNA by small organic molecules. Biochem.-Us 1998, 37, 14204–14212. [Google Scholar] [CrossRef] [PubMed]
- Gayle, A.Y.; Baranger, A.M. Inhibition of the U1A–RNA Complex by an Aminoacridine Derivative. Bioorg. Med. Chem. Lett. 2002, 12, 2839–2842. [Google Scholar] [CrossRef]
- Cassel, J.A.; Blass, B.E.; Reitz, A.B.; Pawlyk, A.C. Development of a novel nonradiometric assay for nucleic acid binding to TDP-43 suitable for high-throughput screening using AlphaScreen technology. J. Biomol. Screen. 2010, 15, 1099–1106. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cassel, J.A.; McDonnell, M.E.; Velvadapu, V.; Andrianov, V.; Reitz, A.B. Characterization of a series of 4-aminoquinolines that stimulate caspase-7 mediated cleavage of TDP-43 and inhibit its function. Biochimie 2012, 94, 1974–1981. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vidal, R.L.; Matus, S.; Bargsted, L.; Hetz, C. Targeting autophagy in neurodegenerative diseases. Trends Pharm. Sci 2014, 35, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Bures, M.; Praestgaard, J.; Fesik, S.W. Privileged molecules for protein binding identified from NMR-based screening. J. Med. Chem 2000, 43, 3443–3447. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. NMR-based screening in drug discovery. Q Rev. Biophys. 1999, 32, 211–240. [Google Scholar] [CrossRef]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef]
- Fesik, S.W. NMR structure-based drug design. J. Biomol. NMR 1993, 3, 261–269. [Google Scholar] [CrossRef]
- Liu, J.; Gao, J.; Li, F.; Ma, R.; Wei, Q.; Wang, A.; Wu, J.; Ruan, K. NMR characterization of weak interactions between RhoGDI2 and fragment screening hits. Biochim. Et Biophys. Acta (Bba)-Gen. Subj. 2017, 1861, 3061–3070. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, F.; Bao, H.; Jiang, Y.; Zhang, S.; Ma, R.; Gao, J.; Wu, J.; Ruan, K. The polar warhead of a TRIM24 bromodomain inhibitor rearranges a water-mediated interaction network. FEBS J. 2017, 284, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, S.; Liu, M.; Liu, Y.; Nshogoza, G.; Gao, J.; Ma, R.; Yang, Y.; Wu, J.; Zhang, J.; et al. Structural plasticity of the TDRD3 Tudor domain probed by a fragment screening hit. FEBS J. 2018, 285, 2091–2103. [Google Scholar] [CrossRef] [PubMed]
- Ruan, K.; Gao, J.; Ma, R. NMR in Fragment Based Lead Discovery. Chin. J. Magn. Reson. 2012, 29, 163–181. [Google Scholar]
- Tang, H.; Nshogoza, G.; Liu, M.; Liu, Y.; Ruan, K.; Ma, R.; Gao, J. Identification of Novel Hits of the NSD1 SET Domain by NMR Fragment-Based Screening. Chin. J. Magn. Reson. 2018, 36, 148–154. [Google Scholar] [CrossRef]
- Gao, J.; Ma, R.; Wang, W.; Wang, N.; Sasaki, R.; Snyderman, D.; Wu, J.; Ruan, K. Automated NMR fragment based screening identified a novel interface blocker to the LARG/RhoA complex. PLoS ONE 2014, 9, e88098. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef]
- Kuo, P.H.; Chiang, C.H.; Wang, Y.T.; Doudeva, L.G.; Yuan, H.S. The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res. 2014, 42, 4712–4722. [Google Scholar] [CrossRef]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef]
- Viegas, A.; Manso, J.o.; Nobrega, F.L.; Cabrita, E.J. Saturation-Transfer Difference (STD) NMR: A Simple and Fast Method for Ligand Screening and Characterization of Protein Binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Dalvit, C.; Fogliatto, G.; Stewart, A.; Veronesi, M.; Stockman, B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Olejniczak, E.T.; Fesik, S.W. One-dimensional relaxation- and diffusion-edited NMR methods for screening compounds that bind to macromolecules. J. Am. Chem. Soc. 1997, 119, 12257–12261. [Google Scholar] [CrossRef]
- Zega, A. NMR Methods for Identification of False Positives in Biochemical Screens. J. Med. Chem. 2017, 60, 9437–9447. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2015, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.; ten Brink, T.; Cala, O.; Guichou, J.F.; Krimm, I. Protein–ligand structure guided by backbone and side-chain proton chemical shift perturbations. J. Biomol. Nmr. 2014, 60, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.; ten Brink, T.; Guichou, J.F.; Cala, O.; Krimm, I. Comparing Binding Modes of Analogous Fragments Using NMR in Fragment-Based Drug Design: Application to PRDX5. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef]
- Shortridge, M.D.; Hage, S.H.; Harbison, G.S.; Powers, R. Estimating Protein-Ligand Binding Affinity using High-Throughput Screening by NMR. J. Comb. Chem. 2008, 10, 948–958. [Google Scholar] [CrossRef]
- Mashalidis, E.H.; Sledz, P.; Lang, S.; Abell, C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat. Protoc. 2013, 8, 2309–2324. [Google Scholar] [CrossRef]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef]
- Wang, N.; Li, F.; Bao, H.; Li, J.; Wu, J.; Ruan, K. NMR Fragment Screening Hit Induces Plasticity of BRD7/9 Bromodomains. ChemBioChem 2016, 17, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Van Dijk, M.; Loureiro-Ferreira, N.; Van Der Schot, G.; De Vries, S.J.; Schmitz, C.; Van Der Zwan, J.; Boelens, R.; Giachetti, A.; Ferella, L.; et al. WeNMR: Structural Biology on the Grid. J. Grid Comput. 2012, 10, 743–767. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Cryst. D Biol. Cryst. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nshogoza, G.; Liu, Y.; Gao, J.; Liu, M.; Moududee, S.A.; Ma, R.; Li, F.; Zhang, J.; Wu, J.; Shi, Y.; et al. NMR Fragment-Based Screening against Tandem RNA Recognition Motifs of TDP-43. Int. J. Mol. Sci. 2019, 20, 3230. https://doi.org/10.3390/ijms20133230
Nshogoza G, Liu Y, Gao J, Liu M, Moududee SA, Ma R, Li F, Zhang J, Wu J, Shi Y, et al. NMR Fragment-Based Screening against Tandem RNA Recognition Motifs of TDP-43. International Journal of Molecular Sciences. 2019; 20(13):3230. https://doi.org/10.3390/ijms20133230
Chicago/Turabian StyleNshogoza, Gilbert, Yaqian Liu, Jia Gao, Mingqing Liu, Sayed Ala Moududee, Rongsheng Ma, Fudong Li, Jiahai Zhang, Jihui Wu, Yunyu Shi, and et al. 2019. "NMR Fragment-Based Screening against Tandem RNA Recognition Motifs of TDP-43" International Journal of Molecular Sciences 20, no. 13: 3230. https://doi.org/10.3390/ijms20133230
APA StyleNshogoza, G., Liu, Y., Gao, J., Liu, M., Moududee, S. A., Ma, R., Li, F., Zhang, J., Wu, J., Shi, Y., & Ruan, K. (2019). NMR Fragment-Based Screening against Tandem RNA Recognition Motifs of TDP-43. International Journal of Molecular Sciences, 20(13), 3230. https://doi.org/10.3390/ijms20133230