IL-13 Impairs Tight Junctions in Airway Epithelia

 ,
,
Abstract
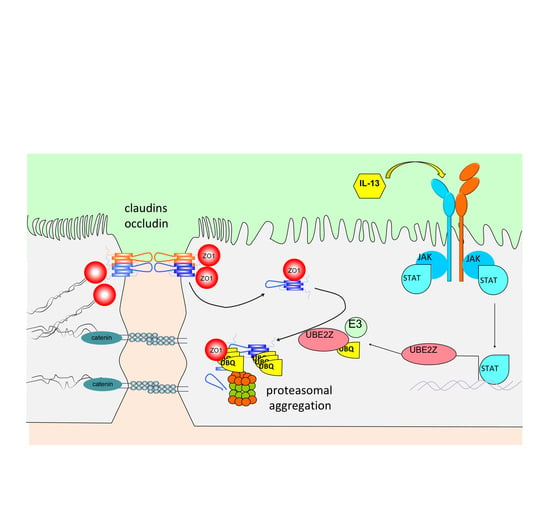
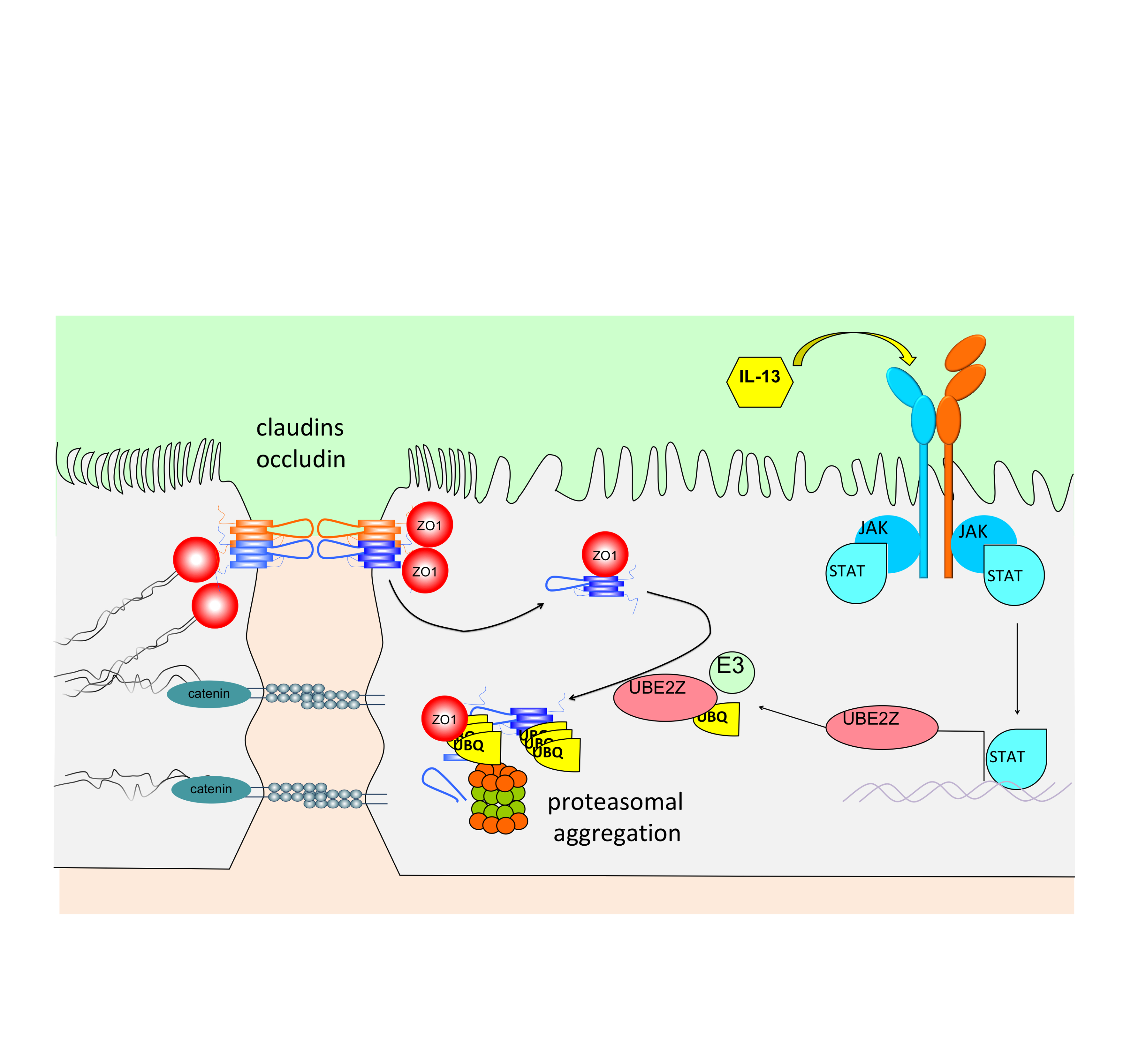{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
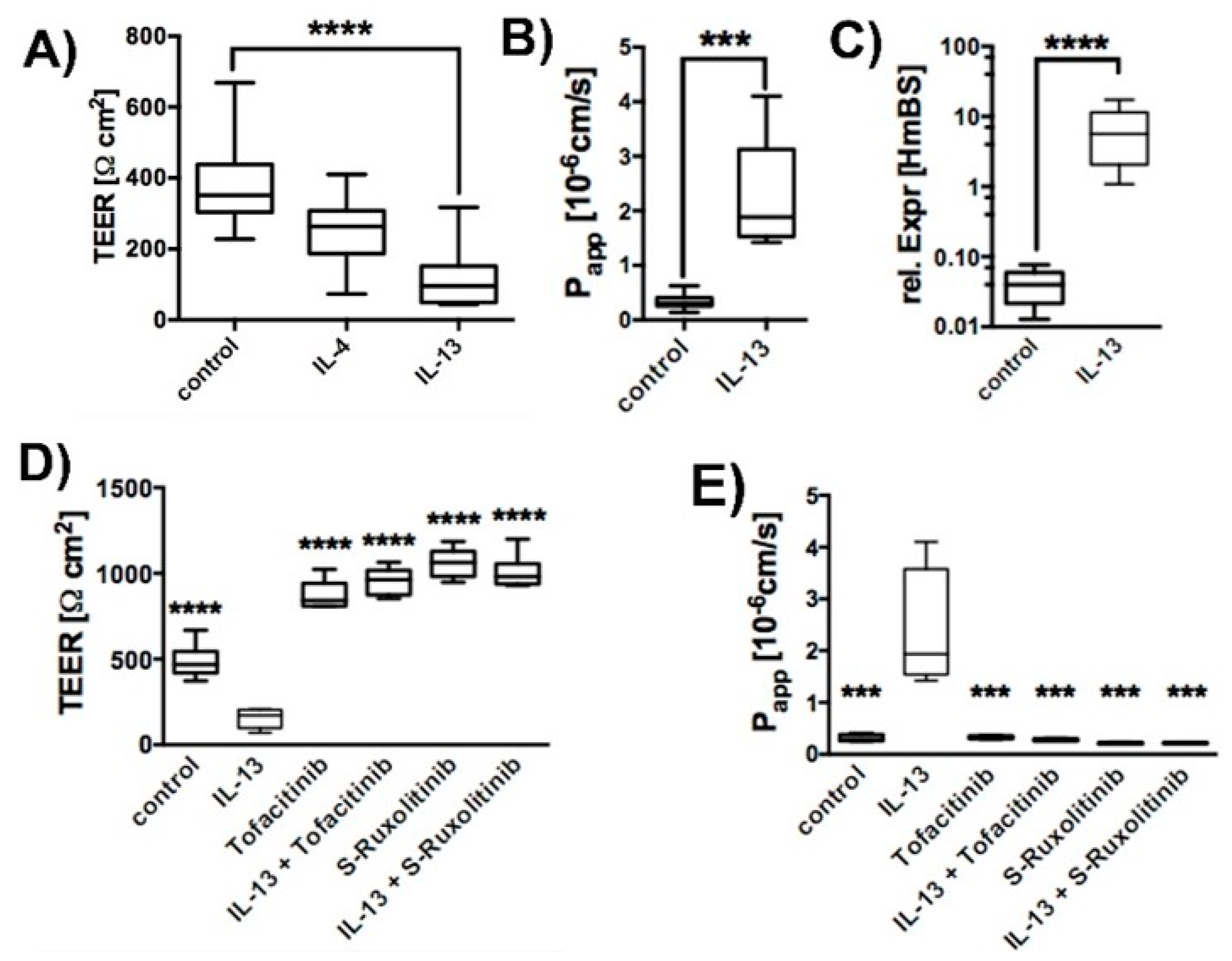
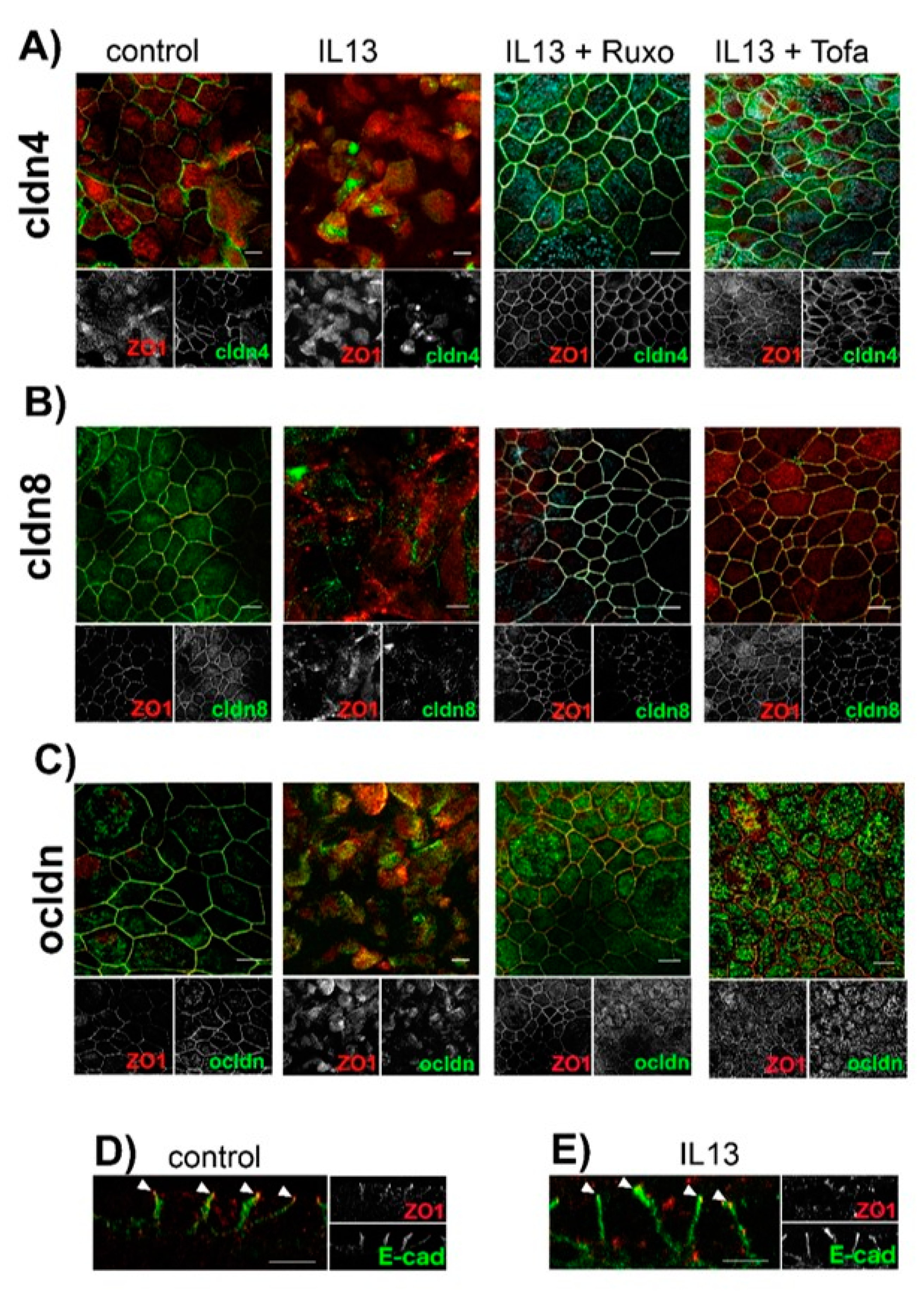
2.1. Effect of IL-13 on hTEpC Epithelia
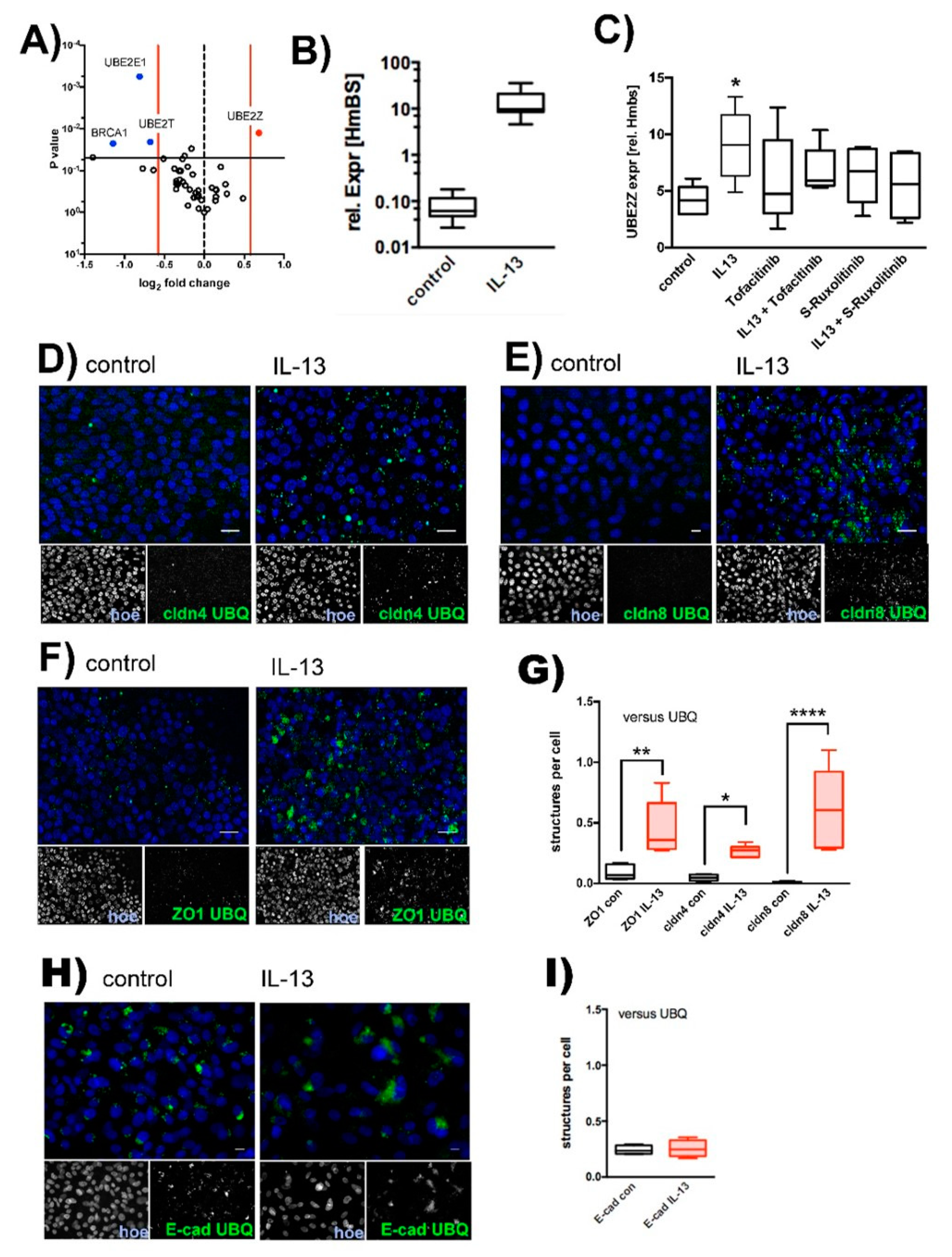
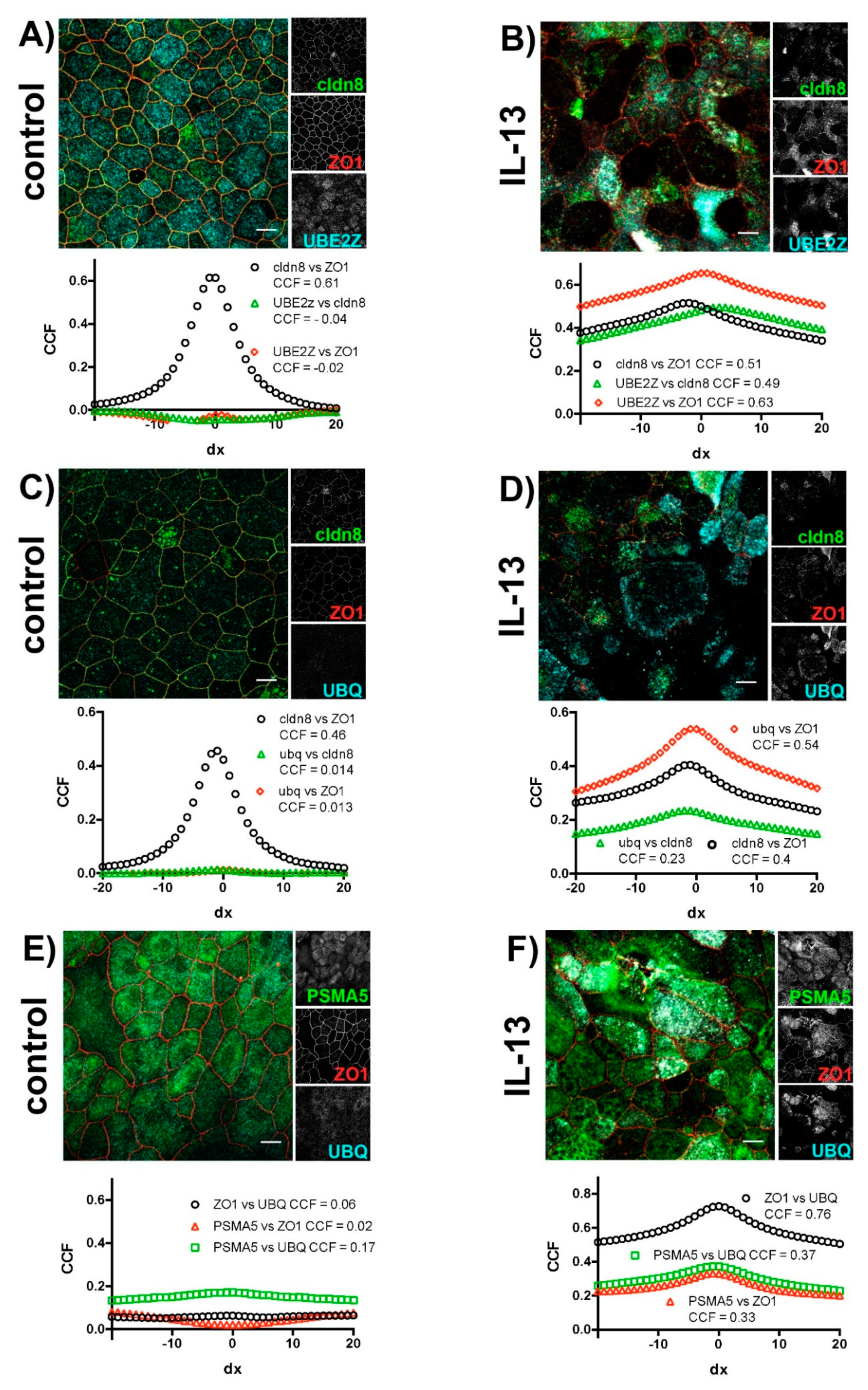
2.2. IL-13 Activates the Ubiquitin-Proteasome Pathway
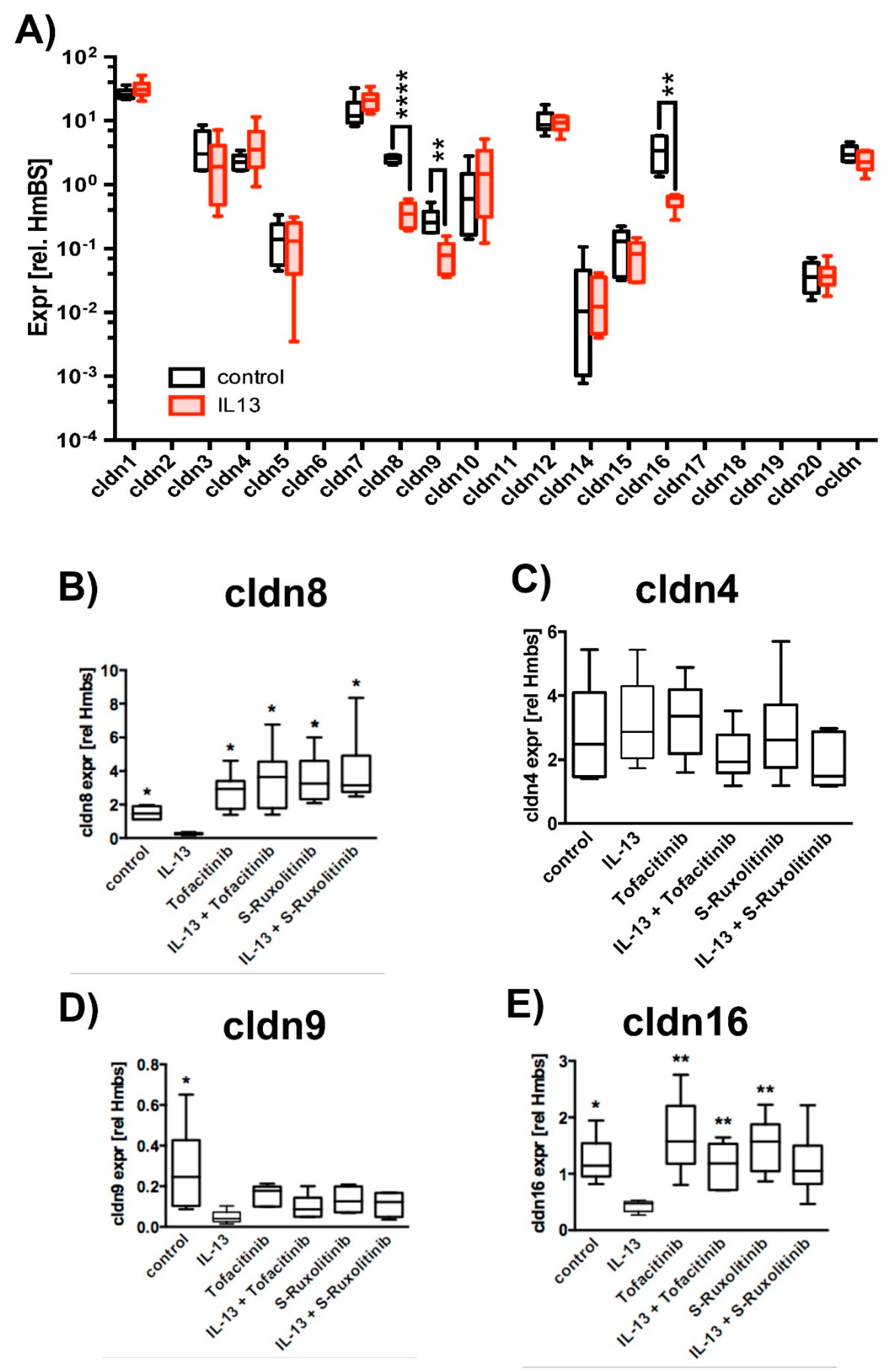
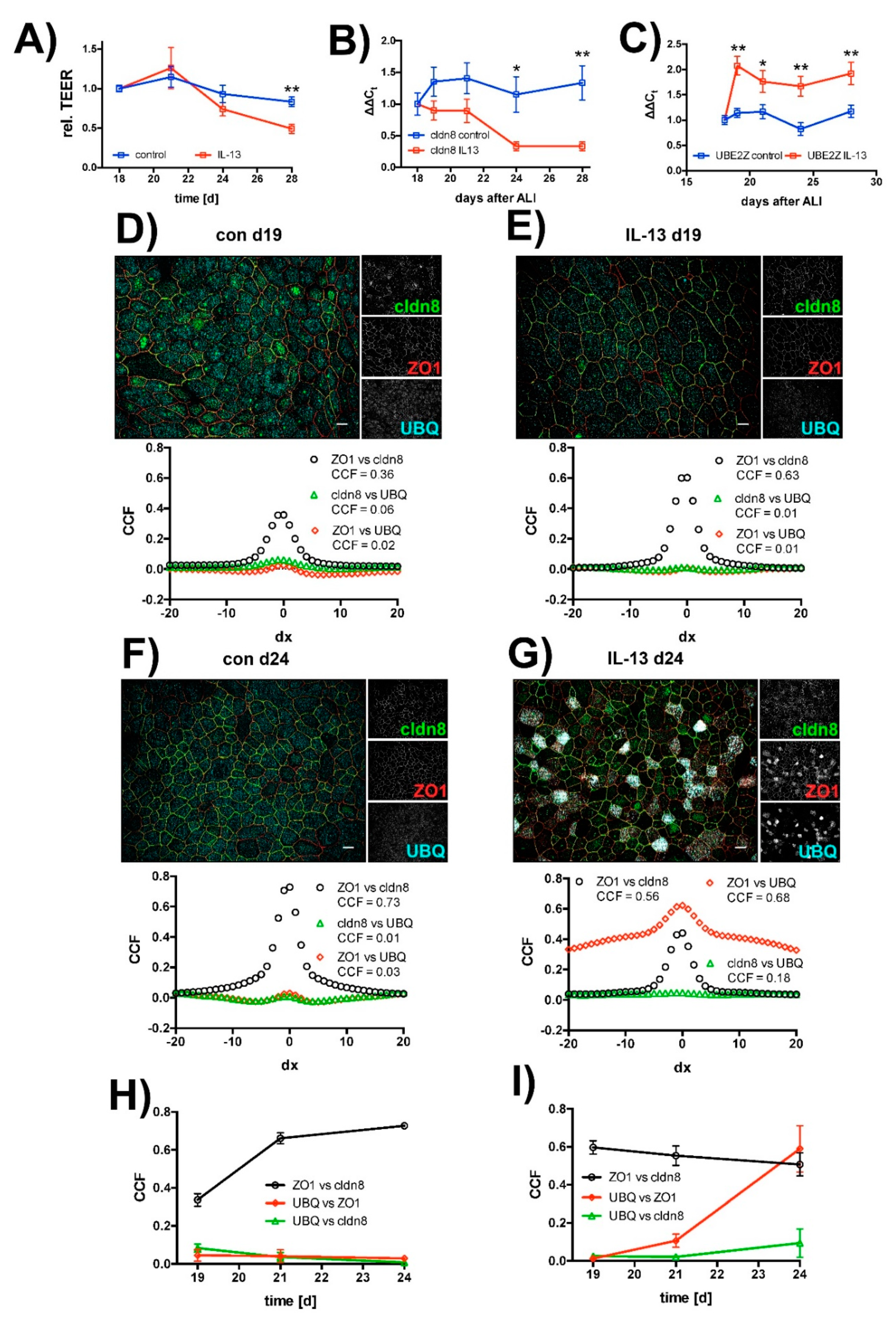
2.3. Effect of IL-13 on Mature hTEpC Epithelia
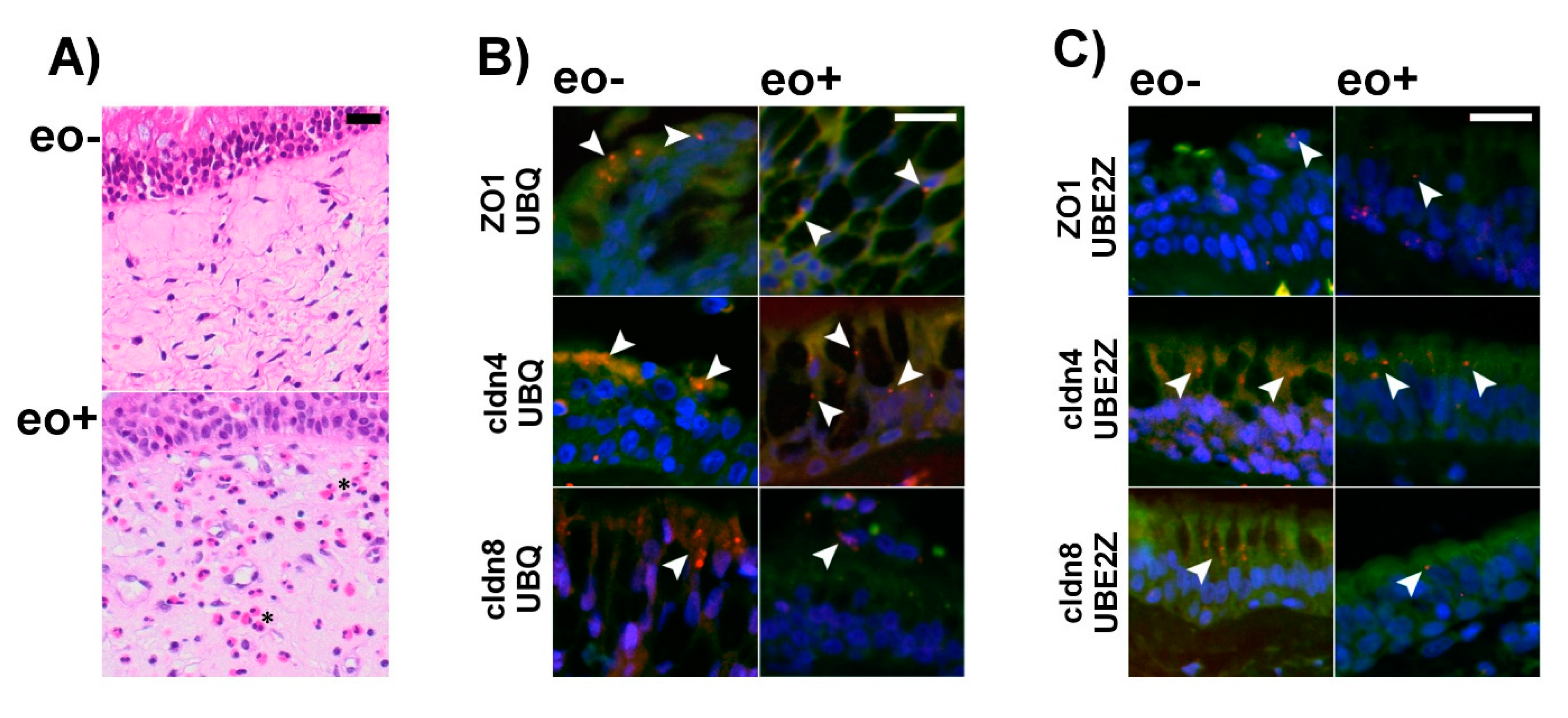
2.4. Ubiquitination of TJ Proteins in Sinunasal Epithelia
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Cell Culture
4.3. TEER Measurement
4.4. Paracellular Permeability
4.5. qRT-PCR
4.6. Immunocytochemistry
4.7. Proximity Ligation Assay
4.8. Immunohistochemistry
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALI | air-liquid interface |
| CCF | cross-correlation coefficient |
| cldn | claudin |
| E-cad | E-cadherin |
| hTEpC | human tracheal epithelial cells |
| IL | interleukin |
| JAK | janus kinase |
| JAM-A | junctional adhesion molecule A |
| MDCK | Madin-Darby kidney cells |
| muc5ac | mucin 5AC |
| ocldn | ocludin |
| Papp | apparent permeability coefficient |
| PLA | proximity ligation assay |
| PSMA5 | proteasomal subunit alpha type 5 |
| RT-PCR | reverse transcriptase PCR |
| STAT | signal transducer and activator of transcription |
| TEER | transepithelial electrical resistance |
| Th2 | T-helper cell type 2 |
| TJ | tight junction |
| UBE2Z | ubiquitin conjugating enzyme 2Z |
| UBQ | ubiquitin |
| ZO1 | zonula occludens 1 protein |
References
- Lötvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F., Jr.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 2011, 127, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Humbert, M.; Buhl, R.; Cruz, A.A.; Inoue, H.; Korom, S.; Hanania, N.A.; Nair, P. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: Current knowledge and therapeutic implications. Clin. Exp. Allergy 2017, 47, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G. The concept of type-1 and type-2 helper T cells and their cytokines in humans. Int. Rev. Immunol. 1998, 16, 427–455. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Homer, R.J.; Wang, Z.; Chen, Q.; Geba, G.P.; Wang, J.; Zhang, Y.; Elias, J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Investig. 1999, 103, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M.; Luyimbazi, J.; Xu, X.; Schofield, B.; Neben, T.Y.; Karp, C.L.; Donaldson, D.D. Interleukin-13: Central mediator of allergic asthma. Science 1998, 282, 2258–2261. [Google Scholar] [CrossRef] [PubMed]
- Hanania, N.A.; Noonan, M.; Corren, J.; Korenblat, P.; Zheng, Y.; Fischer, S.K.; Cheu, M.; Putnam, W.S.; Murray, E.; Scheerens, H.; et al. Lebrikizumab in moderate-to-severe asthma: Pooled data from two randomised placebo-controlled studies. Thorax 2015, 70, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Brightling, C.E.; Chanez, P.; Leigh, R.; O’Byrne, P.M.; Korn, S.; She, D.; May, R.D.; Streicher, K.; Ranade, K.; Piper, E. Efficacy and safety of tralokinumab in patients with severe uncontrolled asthma: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 692–701. [Google Scholar] [CrossRef]
- Baverel, P.G.; White, N.; Vicini, P.; Karlsson, M.O.; Agoram, B. Dose-exposure-response relationship of the investigational anti-interleukin-13 monoclonal antibody tralokinumab in patients with severe, uncontrolled asthma. Clin. Pharmacol. Ther. 2018, 103, 826–835. [Google Scholar] [CrossRef]
- Blease, K.; Jakubzick, C.; Westwick, J.; Lukacs, N.; Kunkel, S.L.; Hogaboam, C.M. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J. Immunol. 2001, 166, 5219–5224. [Google Scholar] [CrossRef]
- Tripp, C.S.; Cuff, C.; Campbell, A.L.; Hendrickson, B.A.; Voss, J.; Melim, T.; Wu, C.; Cherniack, A.D.; Kim, K. RPC4046, a novel anti-interleukin-13 antibody, blocks IL-13 binding to IL-13 α1 and α2 receptors: A randomized, double-blind, placebo-controlled, dose-escalation first-in-human study. Adv. Ther. 2017, 34, 1364–1381. [Google Scholar] [CrossRef]
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: Role of cigarette smoke exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, A.; Carico, E. Role of HIF-1 in cancer progression: Novel insights. A review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Tonini, G.; D’Onofrio, L.; Dell’Aquila, E.; Pezzuto, A. New molecular insights in tobacco-induced lung cancer. Future Oncol. 2013, 9, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Liao, H.; Zhang, B.; Pan, Y.; Kong, Y.; Liu, W.; Yang, P.; Huo, Z.; Cao, Z.; Zhou, Q. Cigarette smoke extracts induce overexpression of the proto-oncogenic gene interleukin-13 receptor α2 through activation of the PKA-CREB signaling pathway to trigger malignant transformation of lung vascular endothelial cells and angiogenesis. Cell. Signal. 2017, 31, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Venkayya, R.; Lam, M.; Willkom, M.; Grünig, G.; Corry, D.B.; Erle, D.J. The Th2 lymphocyte products IL-4 and IL-13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am. J. Respir. Cell Mol. Biol. 2002, 26, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Morse, B.; Sypek, J.P.; Donaldson, D.D.; Haley, K.J.; Lilly, C.M. Effects of IL-13 on airway responses in the guinea pig. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L44–L49. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, V.E.; Thompson, K.E.; Neuland, K.; Jaramillo, A.-M.; Fois, G.; Schmidt, H.; Wittekindt, O.H.; Han, W.; Tuvim, M.J.; Dickey, B.F.; et al. Inflammation-induced upregulation of P2X4 expression augments mucin secretion in airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L58–L70. [Google Scholar] [CrossRef]
- Kanoh, S.; Tanabe, T.; Rubin, B.K. IL-13-induced MUC5AC production and goblet cell differentiation is steroid resistant in human airway cells. Clin. Exp. Allergy 2011, 41, 1747–1756. [Google Scholar] [CrossRef]
- Kistemaker, L.E.M.; Hiemstra, P.S.; Bos, I.S.T.; Bouwman, S.; van den Berge, M.; Hylkema, M.N.; Meurs, H.; Kerstjens, H.A.M.; Gosens, R. Tiotropium attenuates IL-13-induced goblet cell metaplasia of human airway epithelial cells. Thorax 2015, 70, 668–676. [Google Scholar] [CrossRef]
- Zhang, F.-Q.; Han, X.-P.; Zhang, F.; Ma, X.; Xiang, D.; Yang, X.-M.; Ou-Yang, H.-F.; Li, Z. Therapeutic efficacy of a co-blockade of IL-13 and IL-25 on airway inflammation and remodeling in a mouse model of asthma. Int. Immunopharmacol. 2017, 46, 133–140. [Google Scholar] [CrossRef]
- Ohashi, Y.; Motojima, S.; Fukuda, T.; Makino, S. Airway hyperresponsiveness, increased intracellular spaces of bronchial epithelium, and increased infiltration of eosinophils and lymphocytes in bronchial mucosa in asthma. Am. Rev. Respir. Dis. 1992, 145, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Wise, S.K.; Laury, A.M.; Katz, E.H.; Den Beste, K.A.; Parkos, C.A.; Nusrat, A. Interleukin-4 and interleukin-13 compromise the sinonasal epithelial barrier and perturb intercellular junction protein expression. Int. Forum Allergy Rhinol. 2014, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, P.; Wawrzyniak, M.; Wanke, K.; Sokolowska, M.; Bendelja, K.; Rückert, B.; Globinska, A.; Jakiela, B.; Kast, J.I.; Idzko, M.; et al. Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients. J. Allergy Clin. Immunol. 2017, 139, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Sweerus, K.; Lachowicz-Scroggins, M.; Gordon, E.; LaFemina, M.; Huang, X.; Parikh, M.; Kanegai, C.; Fahy, J.V.; Frank, J.A. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J. Allergy Clin. Immunol. 2016, 139, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Saatian, B.; Rezaee, F.; Desando, S.; Emo, J.; Chapman, T.; Knowlden, S.; Georas, S.N. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers 2013, 1, e24333. [Google Scholar] [CrossRef] [PubMed]
- Ahdieh, M.; Vandenbos, T.; Youakim, A. Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-gamma. Am. J. Physiol. Cell Physiol. 2001, 281, C2029–C2038. [Google Scholar] [CrossRef] [PubMed]
- Günzel, D. Claudins: Vital partners in transcellular and paracellular transport coupling. Pflug. Arch. Eur. J. Physiol. 2017, 469, 35–44. [Google Scholar] [CrossRef]
- Schlingmann, B.; Molina, S.A.; Koval, M. Claudins: Gatekeepers of lung epithelial function. Semin. Cell Dev. Biol. 2015, 42, 47–57. [Google Scholar] [CrossRef]
- Den Beste, K.A.; Hoddeson, E.K.; Parkos, C.A.; Nusrat, A.; Wise, S.K. Epithelial permeability alterations in an in vitro air-liquid interface model of allergic fungal rhinosinusitis. Int. Forum Allergy Rhinol. 2013, 3, 19–25. [Google Scholar] [CrossRef]
- Holter, J.F.; Weiland, J.E.; Pacht, E.R.; Gadek, J.E.; Davis, W.B. Protein permeability in the adult respiratory distress syndrome. Loss of size selectivity of the alveolar epithelium. J. Clin. Invest. 1986, 78, 1513–1522. [Google Scholar] [CrossRef]
- Ritchie, A.I.; Jackson, D.J.; Edwards, M.R.; Johnston, S.L. Airway epithelial orchestration of innate immune function in response to virus infection. A focus on asthma. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 1), S55–S63. [Google Scholar]
- Meyer, D.M.; Jesson, M.I.; Li, X.; Elrick, M.M.; Funckes-Shippy, C.L.; Warner, J.D.; Gross, C.J.; Dowty, M.E.; Ramaiah, S.K.; Hirsch, J.L.; et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm. 2010, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Pfaffenrot, E.; Bauer, S.; Laufer, S.A. Design and synthesis of tricyclic jak3 inhibitors with picomolar affinities as novel molecular probes. ChemMedChem 2014, 9, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: Preclinical characterization of INCB028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Renigunta, A.; Yang, J.; Waldegger, S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc. Natl. Acad. Sci. USA 2010, 107, 18010–18015. [Google Scholar] [CrossRef]
- Kielgast, F.; Schmidt, H.; Braubach, P.; Winkelmann, V.E.; Thompson, K.E.; Frick, M.; Dietl, P.; Wittekindt, O.H. Glucocorticoids regulate tight junction permeability of lung epithelia by modulating claudin 8. Am. J. Respir. Cell Mol. Biol. 2016, 54, 707–717. [Google Scholar] [CrossRef]
- Nelson, W.J.; Shore, E.M.; Wang, A.Z.; Hammerton, R.W. Identification of a membrane-cytoskeleton complex containing the cell adhesion molecule uvomorulin (E-Cadherin), ankyrin, and fodrin in Madin-Darby canine kidney epithelial cells. J. Cell Biol. 1990, 110, 349–357. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Gong, Y.; Wang, J.; Yang, J.; Gonzales, E.; Perez, R.; Hou, J. KLHL3 regulates paracellular chloride transport in the kidney by ubiquitination of claudin-8. Proc. Natl. Acad. Sci. USA 2015, 112, 4340–4345. [Google Scholar] [CrossRef]
- Lui, W.Y.; Lee, W.M. Regulation of junction dynamics in the testis—Transcriptional and post-translational regulations of cell junction proteins. Mol. Cell. Endocrinol. 2006, 250, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Wing, Y.L.; Lee, W.M. cAMP perturbs inter-Sertoli tight junction permeability barrier in vitro via its effect on proteasome-sensitive ubiquitination of occludin. J. Cell. Physiol. 2005, 203, 564–572. [Google Scholar]
- Mandel, I.; Paperna, T.; Volkowich, A.; Merhav, M.; Glass-Marmor, L.; Miller, A. The ubiquitin-proteasome pathway regulates claudin 5 degradation. J. Cell. Biochem. 2012, 113, 2415–2423. [Google Scholar] [CrossRef]
- Van Campenhout, C.A.; Eitelhuber, A.; Gloeckner, C.J.; Giallonardo, P.; Gegg, M.; Oller, H.; Grant, S.G.N.; Krappmann, D.; Ueffing, M.; Lickert, H. Dlg3 Trafficking and apical tight junction formation is regulated by Nedd4 and Nedd4-2 E3 Ubiquitin ligases. Dev. Cell 2011, 21, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Traweger, A.; Fang, D.; Liu, Y.C.; Stelzhammer, W.; Krizbai, I.A.; Fresser, F.; Bauer, H.C.; Bauer, H. The tight junction-specific protein occludin is a functional target of the E3 ubiquitin-protein ligase itch. J. Biol. Chem. 2002, 277, 10201–10208. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; van Binnendijk, E.P.; Hornsby, C.D.; van der Voort, H.T.; Krozowski, Z.S.; de Kloet, E.R.; van Driel, R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 1996, 109, 787–792. [Google Scholar] [PubMed]
- Ghebre, M.A.; Bafadhel, M.; Desai, D.; Cohen, S.E.; Newbold, P.; Rapley, L.; Woods, J.; Rugman, P.; Pavord, I.D.; Newby, C.; et al. Biological clustering supports both “dutch” and “british” hypotheses of asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 135, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Sugita, K.; Steer, C.A.; Martinez-Gonzalez, I.; Altunbulakli, C.; Morita, H.; Castro-Giner, F.; Kubo, T.; Wawrzyniak, P.; Rückert, B.; Sudo, K.; et al. Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL-13 in asthmatic patients. J. Allergy Clin. Immunol. 2018, 141, 300–310. [Google Scholar] [CrossRef] [PubMed]
- May, R.D.; Fung, M. Strategies targeting the IL-4/IL-13 axes in disease. Cytokine 2015, 75, 89–116. [Google Scholar] [CrossRef]
- Krug, S.M.; Bojarski, C.; Fromm, A.; Lee, I.M.; Dames, P.; Richter, J.F.; Turner, J.R.; Fromm, M.; Schulzke, J.-D. Tricellulin is regulated via interleukin-13-receptor α2, affects macromolecule uptake, and is decreased in ulcerative colitis. Mucosal Immunol. 2018, 11, 345–356. [Google Scholar] [CrossRef]
- Skieterska, K.; Rondou, P.; Van Craenenbroeck, K. Regulation of G protein-coupled receptors by ubiquitination. Int. J. Mol. Sci. 2017, 18, 923. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L. Ubiquitin-dependent internalization regulation of plasma membrane proteins. FASEB J. 1997, 11, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Widagdo, J.; Guntupalli, S.; Jang, S.E.; Anggono, V. Regulation of AMPA receptor trafficking by protein ubiquitination. Front. Mol. Neurosci. 2017, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iwamoto, N.; Sasaki, H.; Ohashi, M.; Oda, Y.; Tsukita, S.; Furuse, M. The E3 ubiquitin ligase LNX1p80 promotes the removal of claudins from tight junctions in MDCK cells. J. Cell Sci. 2009, 122, 985–994. [Google Scholar] [CrossRef]
- Cong, X.; Zhang, Y.; Li, J.; Mei, M.; Ding, C.; Xiang, R.-L.; Zhang, L.-W.; Wang, Y.; Wu, L.-L.; Yu, G.-Y. Claudin-4 is required for modulation of paracellular permeability by muscarinic acetylcholine receptor in epithelial cells. J. Cell Sci. 2015, 128, 2271–2286. [Google Scholar] [CrossRef]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef]
- Wetzel, F.; Mittag, S.; Cano-Cortina, M.; Wagner, T.; Krämer, O.H.; Niedenthal, R.; Gonzalez-Mariscal, L.; Huber, O. SUMOylation regulates the intracellular fate of ZO-2. Cell. Mol. Life Sci. 2016, 74, 373–392. [Google Scholar] [CrossRef]
- Dang, H.; Klokk, T.I.; Schaheen, B.; McLaughlin, B.M.; Thomas, A.J.; Durns, T.A.; Bitler, B.G.; Sandvig, K.; Fares, H. Derlin-dependent retrograde transport from endosomes to the golgi apparatus. Traffic 2011, 12, 1417–1431. [Google Scholar] [CrossRef]
- Schaheen, B.; Dang, H.; Fares, H. Derlin-dependent accumulation of integral membrane proteins at cell surfaces. J. Cell Sci. 2009, 122, 2228–2239. [Google Scholar] [CrossRef]
- Gu, X.; Zhao, F.; Zheng, M.; Fei, X.; Chen, X.; Huang, S.; Xie, Y.; Mao, Y. Cloning and characterization of a gene encoding the human putative ubiquitin conjugating enzyme E2Z (UBE2Z). Mol. Biol. Rep. 2007, 34, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, B.; Sun, L.; Bhuripanyo, K.; Wang, Y.; Bi, Y.; Davuluri, R.V.; Duong, D.M.; Nanavati, D.; Yin, J.; et al. Orthogonal ubiquitin transfer identifies ubiquitination substrates under differential control by the two ubiquitin activating enzymes. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, X.; Gygi, S.P.; Harper, J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 2007, 447, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Nijhuis, A.; Kumagai, T.; Lindsay, J.; Silver, A. Defects in the adherens junction complex (E-cadherin/ β-catenin) in inflammatory bowel disease. Cell Tissue Res. 2015, 360, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, M.V.; Salazar, I.L.; Curcio, M.; Canzoniero, L.M.T.; Duarte, C.B. Role of the ubiquitin-proteasome system in brain ischemia: Friend or foe? Prog. Neurobiol. 2014, 112, 50–69. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.H.; De Poot, S.A.H.; Lee, J.H.; Kim, J.H.; Han, D.H.; Kim, Y.K.; Finley, D.; Lee, M.J. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, H.O.; Tamura, T.; Ugai, S.; Kagawa, S.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Demonstration that a human 26S proteolytic complex consists of a proteasome and multiple associated protein components and hydrolyzes ATP and ubiquitin-ligated proteins by closely linked mechanisms. Eur. J. Biochem. 1992, 206, 567–578. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I. Adapting proteostatis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Inoue, S.; Nakase, H.; Matsuura, M.; Mikami, S.; Ueno, S.; Uza, N.; Chiba, T. The effect of proteasome inhibitor MG132 on experimental inflammatory bowel disease. Clin. Exp. Immunol. 2009, 156, 172–182. [Google Scholar] [CrossRef]
- Vadász, I.; Weiss, C.H.; Sznajder, J.I. Ubiquitination and proteolysis in acute lung injury. Chest 2012, 141, 763–771. [Google Scholar] [CrossRef][Green Version]
- Stepaniants, S.; Wang, I.M.; Boie, Y.; Mortimer, J.; Kennedy, B.; Elliott, M.; Hayashi, S.; Luo, H.; Wong, J.; Loy, L.; et al. Genes related to emphysema are enriched for ubiquitination pathways. BMC Pulm. Med. 2014, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Min, T.; Bodas, M.; Mazur, S.; Vij, N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 2011, 89, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Tran, I.; Ji, C.; Ni, I.; Min, T.; Tang, D.; Vij, N. Role of cigarette smoke-induced aggresome formation in chronic obstructive pulmonary disease-emphysema pathogenesis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, H.J.; Park, M.K.; Huh, J.W.; Park, H.Y.; Ha, S.Y.; Shin, J.H.; Lee, Y.S. Mitochondrial E3 ubiquitin protein ligase 1 mediates cigarette smoke-induced endothelial cell death and dysfunction. Am. J. Respir. Cell Mol. Biol. 2016, 54, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Li, G.; Flodby, P.; Luo, J.; Kage, H.; Sipos, A.; Gao, D.; Ji, Y.; Beard, L.M.L.; Marconett, C.N.; DeMaio, L.; et al. Knockout mice reveal key roles for Claudin 18 in alveolar barrier properties and fluid homeostasis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Kage, H.; Flodby, P.; Gao, D.; Kim, Y.H.; Marconett, C.N.; DeMaio, L.; Kim, K.-J.; Crandall, E.D.; Borok, Z. Claudin 4 knockout mice: Normal physiological phenotype with increased susceptibility to lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L524–L536. [Google Scholar] [CrossRef]
- Fulkerson, P.C.; Fischetti, C.A.; Hassman, L.M.; Nikolaidis, N.M.; Rothenberg, M.E. Persistent effects induced by IL-13 in the lung. Am. J. Respir. Cell Mol. Biol. 2006, 35, 337–346. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, H.; Braubach, P.; Schilpp, C.; Lochbaum, R.; Neuland, K.; Thompson, K.; Jonigk, D.; Frick, M.; Dietl, P.; Wittekindt, O.H. IL-13 Impairs Tight Junctions in Airway Epithelia. Int. J. Mol. Sci. 2019, 20, 3222. https://doi.org/10.3390/ijms20133222
Schmidt H, Braubach P, Schilpp C, Lochbaum R, Neuland K, Thompson K, Jonigk D, Frick M, Dietl P, Wittekindt OH. IL-13 Impairs Tight Junctions in Airway Epithelia. International Journal of Molecular Sciences. 2019; 20(13):3222. https://doi.org/10.3390/ijms20133222
Chicago/Turabian StyleSchmidt, Hanna, Peter Braubach, Carolin Schilpp, Robin Lochbaum, Kathrin Neuland, Kristin Thompson, Danny Jonigk, Manfred Frick, Paul Dietl, and Oliver H. Wittekindt. 2019. "IL-13 Impairs Tight Junctions in Airway Epithelia" International Journal of Molecular Sciences 20, no. 13: 3222. https://doi.org/10.3390/ijms20133222
APA StyleSchmidt, H., Braubach, P., Schilpp, C., Lochbaum, R., Neuland, K., Thompson, K., Jonigk, D., Frick, M., Dietl, P., & Wittekindt, O. H. (2019). IL-13 Impairs Tight Junctions in Airway Epithelia. International Journal of Molecular Sciences, 20(13), 3222. https://doi.org/10.3390/ijms20133222