3D-PP: A Tool for Discovering Conserved Three-Dimensional Protein Patterns

 ,
, 

Abstract
1. Introduction
2. Results
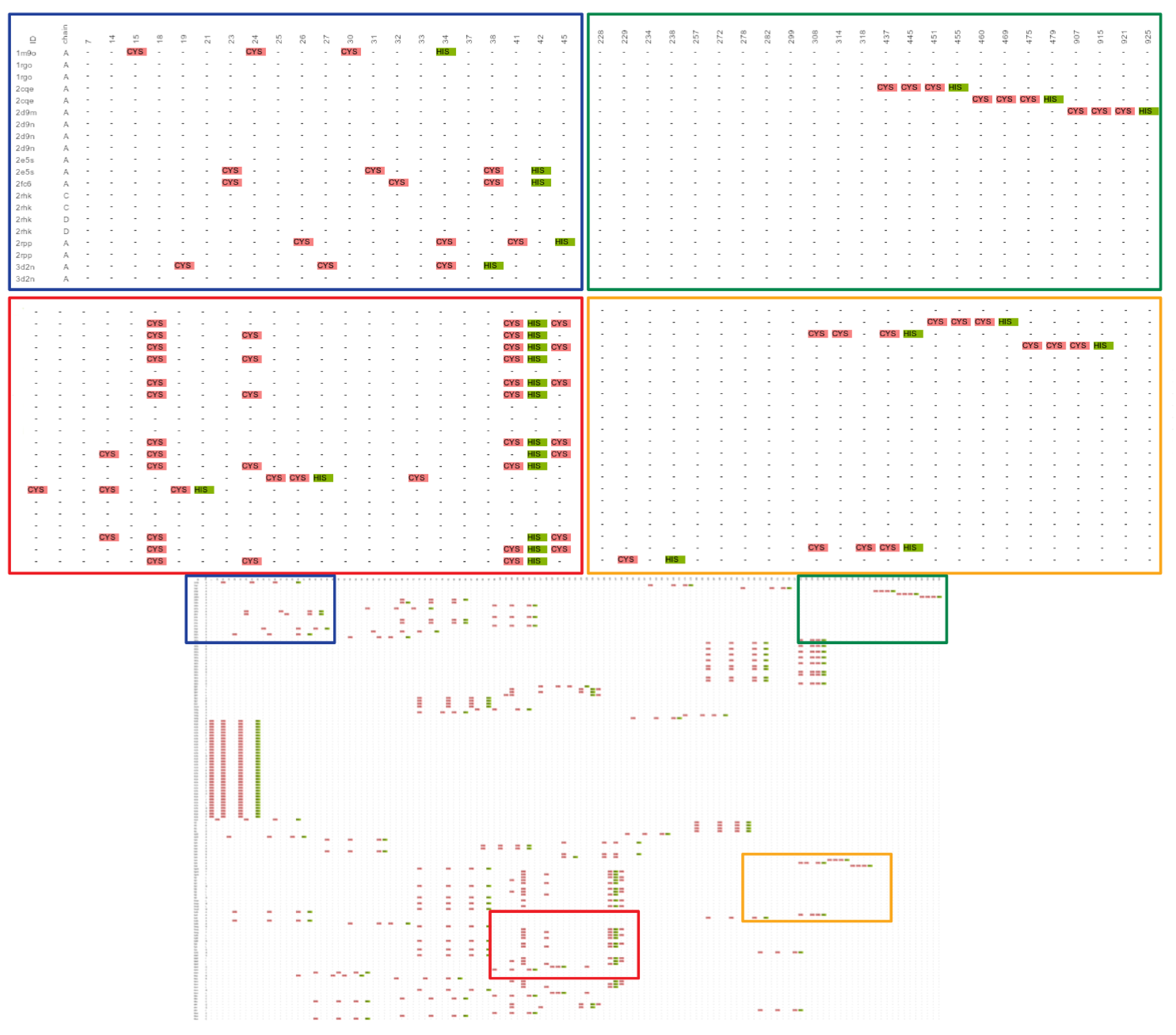
2.1. Known Small 3D-Patterns
2.1.1. Putative New Zinc Ion Coordination Sites
2.1.2. Promiscuous Binding Sites
2.1.3. Not Found Patterns
2.2. Serotonin Target Proteins
2.3. Finding/Discovering Unknown 3D-Patterns on Homology Model Structures
2.4. Comparison with other Methods for the Search and Description of Amino Acid Patterns
3. Materials and Methods
- Spacing Threshold (): This value is used to create the Virtual Grid of Coordinates and defines, how broad and rigorous will be the exploration of 3D-patterns. For instance, a = 0.5, means that every 0.5 Å in the 3D space of each protein structure, a new virtual coordinate of reference will be created. In all cases analysed in this work (Zinc finger C3H1-type containing proteins, serotonin target proteins and structures obtained from homology models), values were 0.8, 2 and 0.8 Å, respectively.
- Radius Threshold (): This term represents the limits of the size of the 3D-patterns searched. Low values are used to detect small binding sites ( e.g., 3 Å), whereas high values allow identification of bigger sites (e.g., 7 Å). In the two cases analyzed in this work (Zinc finger C3H1-type containing proteins, serotonin target proteins and structures obtained from homology models), values were 3, 5 and 2 Å, respectively.
- Displacement Threshold (): This value is used to expand the size and shape for the exploration of the 3D-patterns. By default, this value is set in 0, which means that only the spherical 3D-patterns are searched. If the user changes this value; for example, = 2, two new virtual centers will be considered for the searching of 3D-patterns. This option allows the obtaining of seven new elliptical/oval zones that will be explored to detect non spherical 3D-patterns (Supplementary Data, Figure S3).
- RMSD Threshold (): This value is used for clustering the 3D-patterns detected and represents a measure of structural variability for the sites composing each 3D-pattern. As mentioned in the Results, this parameter allows the comparison of a 3D-pattern with those, containing the same components (i.e., amino acid residues), previously found by 3D-PP. Thus, if the new site exceeds the threshold values defined by the user () when comparing it with the previously found site, a new cluster of the same 3D-pattern is created. Otherwise, the new 3D-pattern is included in the same cluster as that previously found. Therefore, this parameter is crucial for 3D-PP accuracy since it allows discrimination between 3D-patterns that contain similar components but exhibit a different topological conformations (i.e., amino acid residues which are not in the same spatial localization/order).
- Minimum Coverage (): This value allows the showing of only the 3D-patterns with a coverage value equal to or higher than .
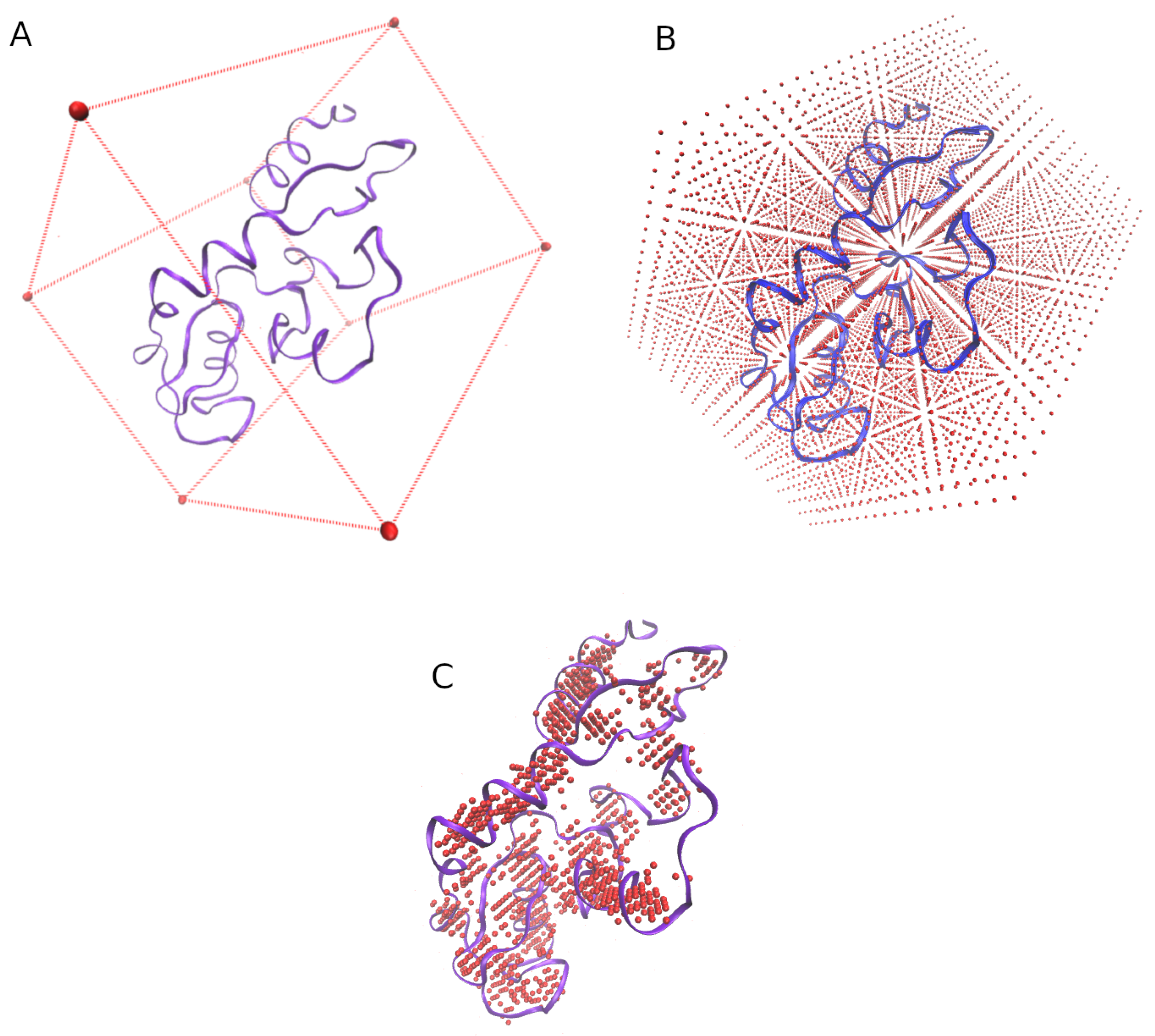
3.1. Grid of Virtual Coordinates
- the min and max values of the X, Y, and Z axis of each structure are obtained.
- a virtual box whose size is determined by the previous values is defined (Figure 7A).
- the virtual box is filled by reference coordinates (x, y, z) distanced between them by an user-defined value (e.g., = 2 Å, Figure 7B).
- only the reference coordinates that show at least four residues surrounding (at a user-defined distance, e.g., = 5 Å) will be considered for the final grid (Figure 7C.)
3.2. Protein Preprocessing
- residues of the proteins surrounding each coordinate of the until a user-defined distance () are grouped.
- groups of residues with at least four components are considered as a site.
- a vector with the list of residues is defined for each site. Then, the sites are transformed into a representation of components through a sorted alphabetical list which contains the one letter code of the amino acid and the amount of occurrences of the same amino acid (e.g., the site “H31:I32:K10:K90:L11:L12:L7:P92:S3:S8:T9” is transformed into “1H1I2K3L1P2S1T”).
- If two different sites match in their representation of components (e.g., “1H1I2K3L1P2S1T”), the RMSD between these two sites is measured. If the RMSD exceeds the threshold values defined by the user (), a new cluster of the same 3D-pattern is created. On the contrary, a new site is added to the current cluster. This step is implemented to avoid two sites having the same components but different 3D conformations, being grouped in the same cluster. It should be noted that if the user set too permissive values (high values), there are more possibilities for grouping sites with different structural topologies; thus, many false positives can occur.
3.3. Creation of the Graph Databases
3.4. Result Visualization
- : The number of proteins in which a specific 3D-pattern was detected.
- : The number of proteins in which a specific 3D-pattern was not detected.
- : Level of conservation of a 3D-pattern in the set of proteins evaluated. The is calculated as follow:A high PCv value (e.g., 80%) indicates that a pattern containing a certain type of residues is found in many proteins (e.g., 80% of the proteins analyzed). It is worth noting that the sites composing a 3D pattern found do not necessarily exhibit the same structural topology in all the proteins in which such a pattern occurs.
- : Amount of sites (arrangement of residues) which are part of a specific 3D-pattern.
- : This value represents the structural variability of a 3D-pattern. Thus, a low number of clusters denotes low variability and, on the contrary, a high number of clusters is indicative of several structural conformations (with different topologies) of sites forming a 3D-pattern.
- : Represents the cluster with the highest coverage on each 3D-pattern.
- : Amount of sites (arrangement of residues) which are part of a specific cluster.
- : The number of proteins that contain a particular 3D-pattern.
- : Level of conservation of a cluster in the set of proteins belonging to a particular 3D-pattern. The is calculated as follows:A high CCv value (e.g., 80%) indicates that a pattern with the same structural topology is present in most of the proteins (80%) of the corresponding cluster.
- : This button shows a multiple sequences-based alignment of the residues of each site of a specific cluster.
- : This button displays a jsmol viewer with multiple structural-based alignments of the residues of each site of a particular cluster.
- : Information of the name and number of the residues forming the site.
- : Name of the protein where the site was detected. This variable can be the PDBid or the name of the file, in the case of homology models.
- : The chain where the site was detected.
- : Root mean square deviation of atomic positions of the particular site against the reference site.
- : Referential coordinate of the from where a specific site was detected.
- : This button shows a jsmol viewer loading the protein and highlighting the residues corresponding to a particular site.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jasial, S.; Hu, Y.; Bajorath, J. Determining the degree of promiscuity of extensively assayed compounds. PLoS ONE 2016, 11, e153873. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Yang, B. Rational application of drug promiscuity in medicinal chemistry. Future Med. Chem. 2018, 10, 1835–1851. [Google Scholar] [CrossRef] [PubMed]
- Mencher, S.K.; Wang, L.G. Promiscuous drugs compared to selective drugs (promiscuity can be a virtue). BMC Clin. Pharmacol. 2005, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.U. Polypharmacology—Foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mishra, C.B.; Tiwari, M. Polypharmacological Drugs in the Treatment of Epilepsy: The Comprehensive Review of Marketed and New Emerging Molecules. Curr. Pharm. Des. 2016, 22, 3212–3225. [Google Scholar] [CrossRef] [PubMed]
- Antolin, A.A.; Workman, P.; Mestres, J.; Al-Lazikani, B. Polypharmacology in Precision Oncology: Current Applications and Future Prospects. Curr. Pharm. Des. 2016, 1, 6935–6945. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.R.; Ciesla, A.; Strutzenberg, T.S.; Novick, S.J.; He, Y.; Garcia-Ordonez, R.; Frkic, R.L.; Bruning, J.B.; Kamenecka, T.M.; Griffin, P.R. A unique polypharmacology nuclear receptor modulator blocks inflammatory signaling pathways. ACS Chem. Biol. 2019, 14, 1051–1062. [Google Scholar] [CrossRef]
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef]
- Chaudhari, R.; Tan, Z.; Huang, B.; Zhang, S. Computational polypharmacology: A new paradigm for drug discovery. Expert Opin. Drug Discov. 2017, 12, 279–291. [Google Scholar] [CrossRef]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence—A study of structural response in protein cores. Proteins Struct. Funct. Bioinform. 2009, 77, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Ingles-Prieto, A.; Ibarra-Molero, B.; Delgado-Delgado, A.; Perez-Jimenez, R.; Fernandez, J.M.; Gaucher, E.A.; Sanchez-Ruiz, J.M.; Gavira, J.A. Conservation of protein structure over four billion years. Structure 2013, 21, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Govindaraj, R.G.; Lemoine, J.M.; Wu, H.C.; Brylinski, M. DeepDrug3D: Classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput. Biol. 2019, 15, e1006718. [Google Scholar] [CrossRef] [PubMed]
- Skolnick, J.; Gao, M.; Roy, A.; Srinivasan, B.; Zhou, H. Implications of the small number of distinct ligand binding pockets in proteins for drug discovery, evolution and biochemical function. Bioorg. Med. Chem. Lett. 2015, 25, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Ehrt, C.; Brinkjost, T.; Koch, O. Impact of Binding Site Comparisons on Medicinal Chemistry and Rational Molecular Design. J. Med. Chem. 2016, 59, 4121–4151. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Kuhn, D.; Klebe, G. A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol. 2002, 323, 387–406. [Google Scholar] [CrossRef]
- Jambon, M.; Andrieu, O.; Combet, C.; Del??age, G.; Delfaud, F.; Geourjon, C. The SuMo server: 3D search for protein functional sites. Bioinformatics 2005, 21, 3929–3930. [Google Scholar] [CrossRef] [PubMed]
- Nebel, J.C.; Herzyk, P.; Gilbert, D.R. Automatic generation of 3D motifs for classification of protein binding sites. BMC Bioinform. 2007, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Nadzirin, N.; Willett, P.; Artymiuk, P.J.; Firdaus-Raih, M. IMAAAGINE: A webserver for searching hypothetical 3D amino acid side chain arrangements in the Protein Data Bank. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef]
- Kirshner, D.A.; Nilmeier, J.P.; Lightstone, F.C. Catalytic site identification—A web server to identify catalytic site structural matches throughout PDB. Nucleic Acids Res. 2013, 41, 256–265. [Google Scholar] [CrossRef]
- Ghoorah, A.W.; Devignes, M.D.; Smaïl-Tabbone, M.; Ritchie, D.W. KBDOCK 2013: A spatial classification of 3D protein domain family interactions. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M. eMatchSite: Sequence Order-Independent Structure Alignments of Ligand Binding Pockets in Protein Models. PLoS Comput. Biol. 2014, 10, e1003829. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Pravda, L.; Svobodová Vařeková, R.; Ionescu, C.M.; Koča, J. PatternQuery: Web application for fast detection of biomacromolecular structural patterns in the entire Protein Data Bank. Nucleic Acids Res. 2015, 43, 383–388. [Google Scholar] [CrossRef]
- Konc, J.; Janežič, D. ProBiS tools (algorithm, database, and web servers) for predicting and modeling of biologically interesting proteins. Prog. Biophys. Mol. Biol. 2017, 128, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Awale, M.; Reymond, J.L. Web-based tools for polypharmacology prediction. Methods Mol. Biol. 2019, 1888, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.B. Detection of protein three-dimensional side-chain patterns: New examples of convergent evolution. J. Mol. Biol. 1998, 279, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.; Russell, R.B. Annotation in three dimensions. PINTS: Patterns in Non-homologous Tertiary Structures. Nucleic Acids Res. 2003, 31, 3341–3344. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, R.V.; Artymiuk, P.J.; Willett, P. Searching for patterns of amino acids in 3D protein structures. J. Chem. Inf. Comput. Sci. 2003, 43, 412–421. [Google Scholar] [CrossRef]
- Polacco, B.J.; Babbitt, P.C. Automated discovery of 3D motifs for protein function annotation. Bioinformatics 2006, 22, 723–730. [Google Scholar] [CrossRef]
- Shulman-Peleg, A.; Shatsky, M.; Nussinov, R.; Wolfson, H.J. MultiBind and MAPPIS: Webservers for multiple alignment of protein 3D-binding sites and their interactions. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef]
- Nadzirin, N.; Gardiner, E.J.; Willett, P.; Artymiuk, P.J.; Firdaus-Raih, M. SPRITE and ASSAM: Web servers for side chain 3D-motif searching in protein structures. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Vivanco, G.; Valdés-Jiménez, A.; Besoaín, F.; Reyes-Parada, M. Geomfinder: A multi-feature identifier of similar three-dimensional protein patterns: A ligand-independent approach. J. Cheminform. 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Vivanco, G.; Fierro, A.; Moya, P.; Iturriaga-Vásquez, P.; Reyes-Parada, M. 3D similarities between the binding sites of monoaminergic target proteins. PLoS ONE 2018, 13, e200637. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Bazan, N.; Gómez-Villamor, S.; Escalé-Claveras, F. DEX: A high-performance graph database management system. In Proceedings of the 2011 IEEE 27th International Conference on Data Engineering Workshops, Hannover, Germany, 11–16 April 2011. [Google Scholar] [CrossRef]
- Have, C.T.; Jensen, L.J.; Wren, J. Are graph databases ready for bioinformatics? Bioinformatics 2013, 29, 3107–3108. [Google Scholar] [CrossRef] [PubMed]
- Lysenko, A.; Roznovăţ, I.A.; Saqi, M.; Mazein, A.; Rawlings, C.J.; Auffray, C.; Auffray, C.; Charron, D.; Hood, L.; Hood, L.; et al. Representing and querying disease networks using graph databases. BioData Min. 2016, 9, 23. [Google Scholar] [CrossRef]
- Ghrab, A.; Romero, O.; Skhiri, S.; Vaisman, A.A.; Zimányi, E. GRAD: On Graph Database Modeling. arXiv 2016, arXiv:1602.00503. [Google Scholar]
- Messina, A.; Fiannaca, A.; La Paglia, L.; La Rosa, M.; Urso, A. BioGraph: A web application and a graph database for querying and analyzing bioinformatics resources. Bmc Syst. Biol. 2018, 12 (Suppl. 5), 98. [Google Scholar] [CrossRef]
- Fabregat, A.; Korninger, F.; Viteri, G.; Sidiropoulos, K.; Marin-Garcia, P.; Ping, P.; Wu, G.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome graph database: Efficient access to complex pathway data. PLoS Comput. Biol. 2018, 14, e1005968. [Google Scholar] [CrossRef]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Qiu, R.; Tang, Y.; Wang, S. Cadmium-zinc exchange and their binary relationship in the structure of Zn-related proteins: A mini review. Metallomics 2014, 6, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Béchade, C.; Roumier, A. Dimers of serotonin receptors: Impact on ligand affinity and signaling. Biochimie 2019, 161, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Jonnakuty, C.; Gragnoli, C. What do we know about serotonin? J. Cell. Physiol. 2008, 217, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Felts, B.; Pramod, A.B.; Sandtner, W.; Burbach, N.; Bulling, S.; Sitte, H.H.; Henry, L.K. The two Na+ sites in the human serotonin transporter play distinct roles in the ion coupling and electrogenicity of transport. J. Biol. Chem. 2014, 289, 1825–1840. [Google Scholar] [CrossRef] [PubMed]
- Rannversson, H.; Andersen, J.; Bang-Andersen, B.; Strømgaard, K. Mapping the Binding Site for Escitalopram and Paroxetine in the Human Serotonin Transporter Using Genetically Encoded Photo-Cross-Linkers. ACS Chem. Biol. 2017, 12, 2558–2562. [Google Scholar] [CrossRef] [PubMed]
- Westkaemper, R.B.; Roth, B.L. Structure and Function Reveal Insights in the Pharmacology of 5-HT Receptor Subtypes. In The Serotonin Receptors; Humana Press: Totowa, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Zapata-Torres, G.; Fierro, A.; Miranda-Rojas, S.; Guajardo, C.; Saez-Briones, P.; Salgado, J.C.; Celis-Barros, C. Influence of protonation on substrate and inhibitor interactions at the active site of human monoamine oxidase-A. J. Chem. Inf. Model. 2012, 52, 1213–1221. [Google Scholar] [CrossRef]
- Christopoulos, A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.C.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Wilkins, A.D.; Bachman, B.J.; Erdin, S.; Lichtarge, O. The use of evolutionary patterns in protein annotation. Curr. Opin. Struct. Biol. 2012, 22, 316–325. [Google Scholar] [CrossRef]
- Bennionn, B.J.; Daggett, V. Protein conformation and diagnostic tests: The prion protein. Clin. Chem. 2002, 48, 2105–2114. [Google Scholar]
- Madej, T.; Lanczycki, C.J.; Zhang, D.; Thiessen, P.A.; Geer, R.C.; Marchler-Bauer, A.; Bryant, S.H. MMDB and VAST+: Tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014, 42, D297–D303. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | PROSITE (A) | 3D-PP (B) | PDBsum(C) | A & B | A & C | B & C | A & B & C |
|---|---|---|---|---|---|---|---|
| amount of sites | 125 | 152 | 124 | 123 | 124 | 122 | 122 |
| % | 100% | – | – | 94.8% | 99.2% | 97.6% | 97.6% |
| Tool | X-ray/Homology Model | Require Known Pattern/Ligand | Comments |
|---|---|---|---|
| Catalytic site identification—a web server to identify catalytic site structural matches throughout PDB [20] | Yes/No | Yes | The most similar section to replicate our experiments with this tool is “Find CSA catalytic sites in your proteins.” However, it was not possible to obtain the results because only one PDB structure can be processed at the same time. When we tried to upload a homology model the server returned a “Server Error (500)”. |
| MMDB and VAST+ [55] | Yes/No | Yes | This tool allows the finding of 3D-patterns of amino acids in only one PDB structure at the same time. It is not possible to identify conserved 3D-patterns on a set of protein structures. |
| IMAAAGINE [19] | Yes/No | Yes | This tool allows searching 3D-patterns of amino acids in the entire PDB database. The user must define a structural description of query. It is not possible to identify conserved 3D-patterns on a set of protein structures. |
| PatternQuery [23] | Yes/No | Yes | This tool allows for the searching of 3D-patterns of amino acids in the PDB database or in a particular data set of protein structures. The user must define a detailed structural description of the query (known 3D-pattern). It is not possible to identify conserved 3D-patterns on a set of protein structures. |
| ProBiS [24] | Yes/Yes | No | This tool search all the 3D-patterns of amino acids associated to a ligand or functional annotations, present in a queried protein structure. Then these 3D-patterns are searched on the entire PDB database. It is not possible to identify conserved 3D-patterns on a set of protein structures. |
| Geomfinder [32] | Yes/Yes | No | This tool compares all the possible 3D-patterns of amino acids of one protein structure with all the possible 3D-patterns of a second protein structure. The identification of the 3D-patterns works separately on each pair of protein structures, and the results are not matched. Therefore, it is not possible to identify conserved 3D-patterns in a set of protein structures. |
| MultiBind [30] | Yes/Yes | No | This tool identifies similar 3D-patterns among a list of PDBids. With this tool, we could find conserved 3D-patterns, but the server did not work with our data sets. In the case of the homology models, the measures can not be assessed because our structures do not contains ligands. The server returns the following comment “No valid ligand. Can not define the query binding site”. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdés-Jiménez, A.; Larriba-Pey, J.-L.; Núñez-Vivanco, G.; Reyes-Parada, M. 3D-PP: A Tool for Discovering Conserved Three-Dimensional Protein Patterns. Int. J. Mol. Sci. 2019, 20, 3174. https://doi.org/10.3390/ijms20133174
Valdés-Jiménez A, Larriba-Pey J-L, Núñez-Vivanco G, Reyes-Parada M. 3D-PP: A Tool for Discovering Conserved Three-Dimensional Protein Patterns. International Journal of Molecular Sciences. 2019; 20(13):3174. https://doi.org/10.3390/ijms20133174
Chicago/Turabian StyleValdés-Jiménez, Alejandro, Josep-L. Larriba-Pey, Gabriel Núñez-Vivanco, and Miguel Reyes-Parada. 2019. "3D-PP: A Tool for Discovering Conserved Three-Dimensional Protein Patterns" International Journal of Molecular Sciences 20, no. 13: 3174. https://doi.org/10.3390/ijms20133174
APA StyleValdés-Jiménez, A., Larriba-Pey, J.-L., Núñez-Vivanco, G., & Reyes-Parada, M. (2019). 3D-PP: A Tool for Discovering Conserved Three-Dimensional Protein Patterns. International Journal of Molecular Sciences, 20(13), 3174. https://doi.org/10.3390/ijms20133174