Implications of Diet and The Gut Microbiome in Neuroinflammatory and Neurodegenerative Diseases

{kind=link}
Abstract
1. Introduction
2. The Role of Diet in Dysbiosis Onset
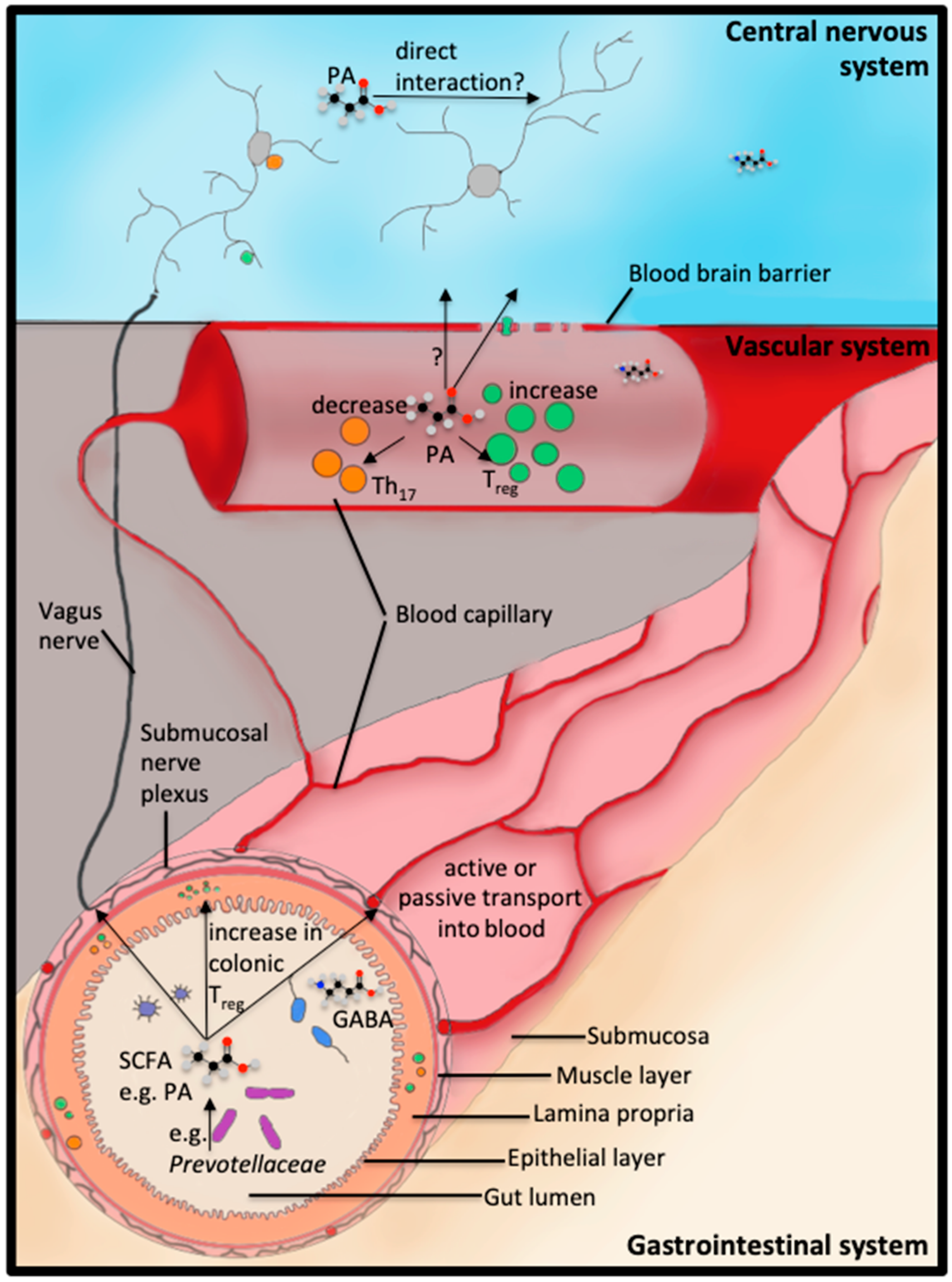
3. Dysbiosis in Neurodegenerative Disorders
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IBD | Inflammatory bowel disease |
| Treg | Regulatory T cells |
| MS | Multiple sclerosis |
| ASD | Autism spectrum disorder |
| HPA | Hypothalamic–pituitary–adrenal axis |
| SCFAs | Short chain fatty acids |
| HDAC | Histone deacetylase |
| PA | Propionic acid |
| CNS | Central nervous system |
| LCFA | Long chain fatty acids |
| Th | T-helper |
| TCR | T cell-Receptor |
| EAE | Experimental autoimmune encephalomyelits |
| PD | Parkinson’s disease |
| AD | Alzheimer’s disease |
| IL | Interleukin |
| GABA | γ-Aminobutyric acid |
| GPR | G-protein coupled receptor |
| HAT | Histone acetyltransferase |
| HOMA- | Homoeostasis model assessment-estimated |
| Nrf2 | Nuclear factor, erythroid 2 like 2 |
| IR | Insulin resistance |
| ENS | Enteric nervous system |
| NAD+ | Nicotiamide adenine dinucleotide |
References
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut. Microbes. 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef]
- Bonder, M.J.; Kurilshikov, A.; Tigchelaar, E.F.; Mujagic, Z.; Imhann, F.; Vila, A.V.; Deelen, P.; Vatanen, T.; Schirmer, M.; Smeekens, S.P.; et al. The effect of host genetics on the gut microbiome. Nat. Genet. 2016, 48, 1407–1412. [Google Scholar] [CrossRef]
- De Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Butto, L.F.; Schaubeck, M.; Haller, D. Mechanisms of Microbe-Host Interaction in Crohn’s Disease: Dysbiosis vs. Pathobiont Selection. Front. Immunol. 2015, 6, 555. [Google Scholar] [CrossRef] [PubMed]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.P.; Ugarte, E.; Munoz-Tamayo, R.; Paslier, D.L.; Nalin, R.; et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef]
- Verdu, E.F.; Galipeau, H.J.; Jabri, B. Novel players in coeliac disease pathogenesis: Role of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Karell, K.; Louka, A.S.; Moodie, S.J.; Ascher, H.; Clot, F.; Greco, L.; Ciclitira, P.J.; Sollid, L.M.; Partanen, J.; European Genetics Cluster on Celiac, D. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: Results from the European Genetics Cluster on Celiac Disease. Hum. Immunol. 2003, 64, 469–477. [Google Scholar] [CrossRef]
- Sanchez, E.; Donat, E.; Ribes-Koninckx, C.; Fernandez-Murga, M.L.; Sanz, Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl. Environ. Microbiol. 2013, 79, 5472–5479. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalances in faecal and duodenal Bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol. 2008, 8, 232. [Google Scholar] [CrossRef]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J. Clin. Pathol. 2009, 62, 264–269. [Google Scholar] [CrossRef]
- Di Cagno, R.; De Angelis, M.; De Pasquale, I.; Ndagijimana, M.; Vernocchi, P.; Ricciuti, P.; Gagliardi, F.; Laghi, L.; Crecchio, C.; Guerzoni, M.E.; et al. Duodenal and faecal microbiota of celiac children: Molecular, phenotype and metabolome characterization. BMC Microbiol. 2011, 11, 219. [Google Scholar] [CrossRef]
- De Palma, G.; Nadal, I.; Medina, M.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2010, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Kaukinen, K.; Tuovinen, E.; Collin, P.; Lindfors, K.; Partanen, J.; Maki, M.; Matto, J. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm. Bowel. Dis. 2013, 19, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Nadal, I.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 2007, 56, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Bramanti, A.; Cantone, M.; Pennisi, G.; Bella, R.; Lanza, G. Neurophysiology of the “Celiac Brain”: Disentangling Gut-Brain Connections. Front. Neurosci. 2017, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; Labaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism--comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Khoruts, A.; Sadowsky, M.J. Understanding the mechanisms of faecal microbiota transplantation. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Stilling, R.M.; Bordenstein, S.R.; Dinan, T.G.; Cryan, J.F. Friends with social benefits: Host-microbe interactions as a driver of brain evolution and development? Front. Cell Infect Microbiol. 2014, 4, 147. [Google Scholar] [CrossRef]
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [PubMed]
- Raina, J.B.; Eme, L.; Pollock, F.J.; Spang, A.; Archibald, J.M.; Williams, T.A. Symbiosis in the microbial world: From ecology to genome evolution. Biol. Open 2018, 7. [Google Scholar] [CrossRef]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef]
- Willyard, C. Could baby’s first bacteria take root before birth? Nature 2018, 553, 264–266. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef]
- Perez-Munoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef]
- Huurre, A.; Kalliomaki, M.; Rautava, S.; Rinne, M.; Salminen, S.; Isolauri, E. Mode of delivery—Effects on gut microbiota and humoral immunity. Neonatology 2008, 93, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.; De Grandi, R.; Peroni, D.G.; Grossi, E.; Facchin, V.; Comberiati, P.; Drago, L. Impact of delivery mode on the colostrum microbiota composition. BMC Microbiol. 2017, 17, 205. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Harmsen, H.J.; Wildeboer-Veloo, A.C.; Raangs, G.C.; Wagendorp, A.A.; Klijn, N.; Bindels, J.G.; Welling, G.W. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef]
- Pusceddu, M.M.; El Aidy, S.; Crispie, F.; O’Sullivan, O.; Cotter, P.; Stanton, C.; Kelly, P.; Cryan, J.F.; Dinan, T.G. N-3 Polyunsaturated Fatty Acids (PUFAs) Reverse the Impact of Early-Life Stress on the Gut Microbiota. PLoS ONE 2015, 10, e0139721. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Morris, G.; Berk, M.; Carvalho, A.; Caso, J.R.; Sanz, Y.; Walder, K.; Maes, M. The Role of the Microbial Metabolites Including Tryptophan Catabolites and Short Chain Fatty Acids in the Pathophysiology of Immune-Inflammatory and Neuroimmune Disease. Mol. Neurobiol. 2017, 54, 4432–4451. [Google Scholar] [CrossRef]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Salminen, S.; Bouley, C.; Boutron-Ruault, M.C.; Cummings, J.H.; Franck, A.; Gibson, G.R.; Isolauri, E.; Moreau, M.C.; Roberfroid, M.; Rowland, I. Functional food science and gastrointestinal physiology and function. Br. J. Nutr. 1998, 80 Suppl 1, S147–S171. [Google Scholar] [CrossRef]
- Fresia Fernandez, M.D.C. Vitamin K composition of anaerobic gut bacteria. FEMS Microbiol. Lett. 1987, 41, 175–180. [Google Scholar] [CrossRef]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Stampfer, M.J.; Hu, F.B.; Giovannucci, E.; Rimm, E.; Manson, J.E.; Hennekens, C.H.; Willett, W.C. Whole-grain consumption and risk of coronary heart disease: Results from the Nurses’ Health Study. Am. J. Clin. Nutr. 1999, 70, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Montonen, J.; Knekt, P.; Jarvinen, R.; Aromaa, A.; Reunanen, A. Whole-grain and fiber intake and the incidence of type 2 diabetes. Am. J. Clin. Nutr. 2003, 77, 622–629. [Google Scholar] [CrossRef]
- Harris, K.A.; Kris-Etherton, P.M. Effects of whole grains on coronary heart disease risk. Curr. Atheroscler. Rep. 2010, 12, 368–376. [Google Scholar] [CrossRef]
- Post, R.E.; Mainous, A.G., 3rd; King, D.E.; Simpson, K.N. Dietary fiber for the treatment of type 2 diabetes mellitus: A meta-analysis. J. Am. Board Fam. Med. 2012, 25, 16–23. [Google Scholar] [CrossRef]
- Aoyama, M.; Kotani, J.; Usami, M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition 2010, 26, 653–661. [Google Scholar] [CrossRef]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; de Roos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Backhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Zinocker, M.K.; Lindseth, I.A. The Western Diet-Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients 2018, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Neves, F.A.; Cortez, E.; Bernardo, A.F.; Mattos, A.B.; Vieira, A.K.; Malafaia Tde, O.; Thole, A.A.; Rodrigues-Cunha, A.C.; Garcia-Souza, E.P.; Sichieri, R.; et al. Heart energy metabolism impairment in Western-diet induced obese mice. J. Nutr. Biochem. 2014, 25, 50–57. [Google Scholar] [CrossRef]
- Harkiolaki, M.; Holmes, S.L.; Svendsen, P.; Gregersen, J.W.; Jensen, L.T.; McMahon, R.; Friese, M.A.; van Boxel, G.; Etzensperger, R.; Tzartos, J.S.; et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 2009, 30, 348–357. [Google Scholar] [CrossRef]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mahler, A.; Balogh, A.; Marko, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Panigrahi, P.; Parida, S.; Nanda, N.C.; Satpathy, R.; Pradhan, L.; Chandel, D.S.; Baccaglini, L.; Mohapatra, A.; Mohapatra, S.S.; Misra, P.R.; et al. A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature 2017, 548, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Buys, N.J. Glucose- and glycaemic factor-lowering effects of probiotics on diabetes: A meta-analysis of randomised placebo-controlled trials. Br. J. Nutr. 2016, 115, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Guyonnet, D.; Schlumberger, A.; Mhamdi, L.; Jakob, S.; Chassany, O. Fermented milk containing Bifidobacterium lactis DN-173 010 improves gastrointestinal well-being and digestive symptoms in women reporting minor digestive symptoms: A randomised, double-blind, parallel, controlled study. Br. J. Nutr. 2009, 102, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Juul, F.E.; Garborg, K.; Bretthauer, M.; Skudal, H.; Oines, M.N.; Wiig, H.; Rose, O.; Seip, B.; Lamont, J.T.; Midtvedt, T.; et al. Fecal Microbiota Transplantation for Primary Clostridium difficile Infection. N. Engl. J. Med. 2018, 378, 2535–2536. [Google Scholar] [CrossRef] [PubMed]
- Fujiya, M.; Ueno, N.; Kohgo, Y. Probiotic treatments for induction and maintenance of remission in inflammatory bowel diseases: A meta-analysis of randomized controlled trials. Clin. J. Gastroenterol. 2014, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Zilberman-Schapira, G.; Suez, J.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Kotler, E.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.; et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell 2018, 174, 1388–1405.e21. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Zeevi, D.; Korem, T.; Segal, E.; Elinav, E. Taking it Personally: Personalized Utilization of the Human Microbiome in Health and Disease. Cell Host Microbe 2016, 19, 12–20. [Google Scholar] [CrossRef]
- Minato, T.; Maeda, T.; Fujisawa, Y.; Tsuji, H.; Nomoto, K.; Ohno, K.; Hirayama, M. Progression of Parkinson’s disease is associated with gut dysbiosis: Two-year follow-up study. PLoS ONE 2017, 12, e0187307. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Burmann, J.; Fassbender, K.; Schwiertz, A.; Schafer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Lubomski, M.; Tan, A.H.; Lim, S.Y.; Holmes, A.J.; Davis, R.L.; Sue, C.M. Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Roubaud-Baudron, C.; Krolak-Salmon, P.; Quadrio, I.; Megraud, F.; Salles, N. Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: Preliminary results. Neurobiol. Aging 2012, 33, 1009.e11-9. [Google Scholar] [CrossRef] [PubMed]
- Park, A.M.; Omura, S.; Fujita, M.; Sato, F.; Tsunoda, I. Helicobacter pylori and gut microbiota in multiple sclerosis versus Alzheimer’s disease: 10 pitfalls of microbiome studies. Clin. Exp. Neuroimmunol. 2017, 8, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, W.; Zhang, Y.; Lin, L.; Chen, J.; Zeng, Y.; Zheng, M.; Zhuang, Z.; Du, H.; Chen, R.; et al. IL-10 Promotes Neurite Outgrowth and Synapse Formation in Cultured Cortical Neurons after the Oxygen-Glucose Deprivation via JAK1/STAT3 Pathway. Sci. Rep. 2016, 6, 30459. [Google Scholar] [CrossRef]
- Siffrin, V.; Radbruch, H.; Glumm, R.; Niesner, R.; Paterka, M.; Herz, J.; Leuenberger, T.; Lehmann, S.M.; Luenstedt, S.; Rinnenthal, J.L.; et al. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity 2010, 33, 424–436. [Google Scholar] [CrossRef]
- Barrett, E.; Ross, R.P.; O’Toole, P.W.; Fitzgerald, G.F.; Stanton, C. gamma-Aminobutyric acid production by culturable bacteria from the human intestine. J. Appl. Microbiol. 2012, 113, 411–417. [Google Scholar] [CrossRef]
- Özogul. Production of biogenic amines by Morganella morganii, Klebsiella pneumoniae and Hafnia alvei using a rapid HPLC method. Eur. Food Res. Technol. 2004, 219, 465–469. [Google Scholar] [CrossRef]
- Shishov, V.A.; Kirovskaia, T.A.; Kudrin, V.S.; Oleskin, A.V. Amine neuromediators, their precursors, and oxidation products in the culture of Escherichia coli K-12. Prikl. Biokhim. Mikrobiol. 2009, 45, 550–554. [Google Scholar] [CrossRef]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli, R.; Pessione, E. The Neuro-endocrinological Role of Microbial Glutamate and GABA Signaling. Front. Microbiol. 2016, 7, 1934. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fei, Y.J.; Anderson, C.M.; Wake, K.A.; Miyauchi, S.; Huang, W.; Thwaites, D.T.; Ganapathy, V. Structure, function and immunolocalization of a proton-coupled amino acid transporter (hPAT1) in the human intestinal cell line Caco-2. J. Physiol. 2003, 546, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Howland, R.H. Vagus Nerve Stimulation. Curr. Behav. Neurosci. Rep. 2014, 1, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Mashimo, H.; Goyal, R.K. Musings on the wanderer: what’s new in our understanding of vago-vagal reflex? IV. Current concepts of vagal efferent projections to the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G357–G366. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef]
- Wu, T.; Rayner, C.K.; Young, R.L.; Horowitz, M. Gut motility and enteroendocrine secretion. Curr. Opin. Pharmacol. 2013, 13, 928–934. [Google Scholar] [CrossRef]
- Powell, N.; Walker, M.M.; Talley, N.J. The mucosal immune system: Master regulator of bidirectional gut-brain communications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Blackshaw, L.A.; Brookes, S.J.; Grundy, D.; Schemann, M. Sensory transmission in the gastrointestinal tract. Neurogastroenterol. Motil. 2007, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Knight, R.; Mazmanian, S.K.; Cryan, J.F.; Tillisch, K. Gut microbes and the brain: Paradigm shift in neuroscience. J. Neurosci. 2014, 34, 15490–15496. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed]
- Augenlicht, L.H.; Mariadason, J.M.; Wilson, A.; Arango, D.; Yang, W.; Heerdt, B.G.; Velcich, A. Short chain fatty acids and colon cancer. J. Nutr. 2002, 132, 3804S–3808S. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Howarth, G.S.; Butler, R.N. Short-chain fatty acids induce apoptosis in colon cancer cells associated with changes to intracellular redox state and glucose metabolism. Chemotherapy 2012, 58, 102–109. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Candido, E.P.; Reeves, R.; Davie, J.R. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 1978, 14, 105–113. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef]
- Harrison, I.F.; Dexter, D.T. Epigenetic targeting of histone deacetylase: Therapeutic potential in Parkinson’s disease? Pharmacol. Ther. 2013, 140, 34–52. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Jenq, R.; Mathew, A.V.; Koenigsknecht, M.; Hanash, A.; Toubai, T.; Oravecz-Wilson, K.; Wu, S.R.; Sun, Y.; Rossi, C.; et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 2016, 17, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug. Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Shen, S.; Dietz, K.; He, Y.; Howell, O.; Reynolds, R.; Casaccia, P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci. 2010, 13, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001, 413, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Young, D.; Gelovani, J.G.; Robinson, A.; Davidson, Y.; Herholz, K.; Mann, D.M. Histone deacetylase class II and acetylated core histone immunohistochemistry in human brains with Huntington’s disease. Brain Res. 2013, 1504, 16–24. [Google Scholar] [CrossRef]
- Narayan, P.J.; Lill, C.; Faull, R.; Curtis, M.A.; Dragunow, M. Increased acetyl and total histone levels in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2015, 74, 281–294. [Google Scholar] [CrossRef]
- Gebremedhin, K.G.; Rademacher, D.J. Histone H3 acetylation in the postmortem Parkinson’s disease primary motor cortex. Neurosci. Lett. 2016, 627, 121–125. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Perez-Mediavilla, A.; Frechilla, D.; Del Rio, J.; Garcia-Osta, A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 2009, 34, 1721–1732. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zheng, P.; Xie, Y.; Boz, Z.; Yu, Y.; Tang, R.; Jones, A.; Zheng, K.; Huang, X.F. Propionate Protects Haloperidol-Induced Neurite Lesions Mediated by Neuropeptide Y. Front. Neurosci. 2018, 12, 743. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Snelling, T.; Umlai, U.K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirschberg, S.; Gisevius, B.; Duscha, A.; Haghikia, A. Implications of Diet and The Gut Microbiome in Neuroinflammatory and Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3109. https://doi.org/10.3390/ijms20123109
Hirschberg S, Gisevius B, Duscha A, Haghikia A. Implications of Diet and The Gut Microbiome in Neuroinflammatory and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2019; 20(12):3109. https://doi.org/10.3390/ijms20123109
Chicago/Turabian StyleHirschberg, Sarah, Barbara Gisevius, Alexander Duscha, and Aiden Haghikia. 2019. "Implications of Diet and The Gut Microbiome in Neuroinflammatory and Neurodegenerative Diseases" International Journal of Molecular Sciences 20, no. 12: 3109. https://doi.org/10.3390/ijms20123109
APA StyleHirschberg, S., Gisevius, B., Duscha, A., & Haghikia, A. (2019). Implications of Diet and The Gut Microbiome in Neuroinflammatory and Neurodegenerative Diseases. International Journal of Molecular Sciences, 20(12), 3109. https://doi.org/10.3390/ijms20123109