Lithium and Not Acetoacetate Influences the Growth of Cells Treated with Lithium Acetoacetate


 and
and
Abstract
1. Introduction
2. Results
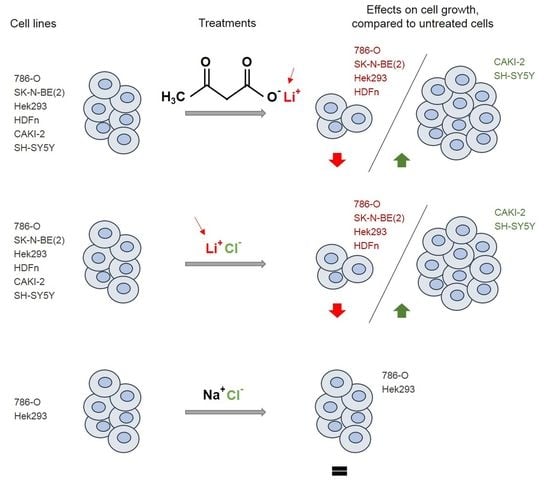
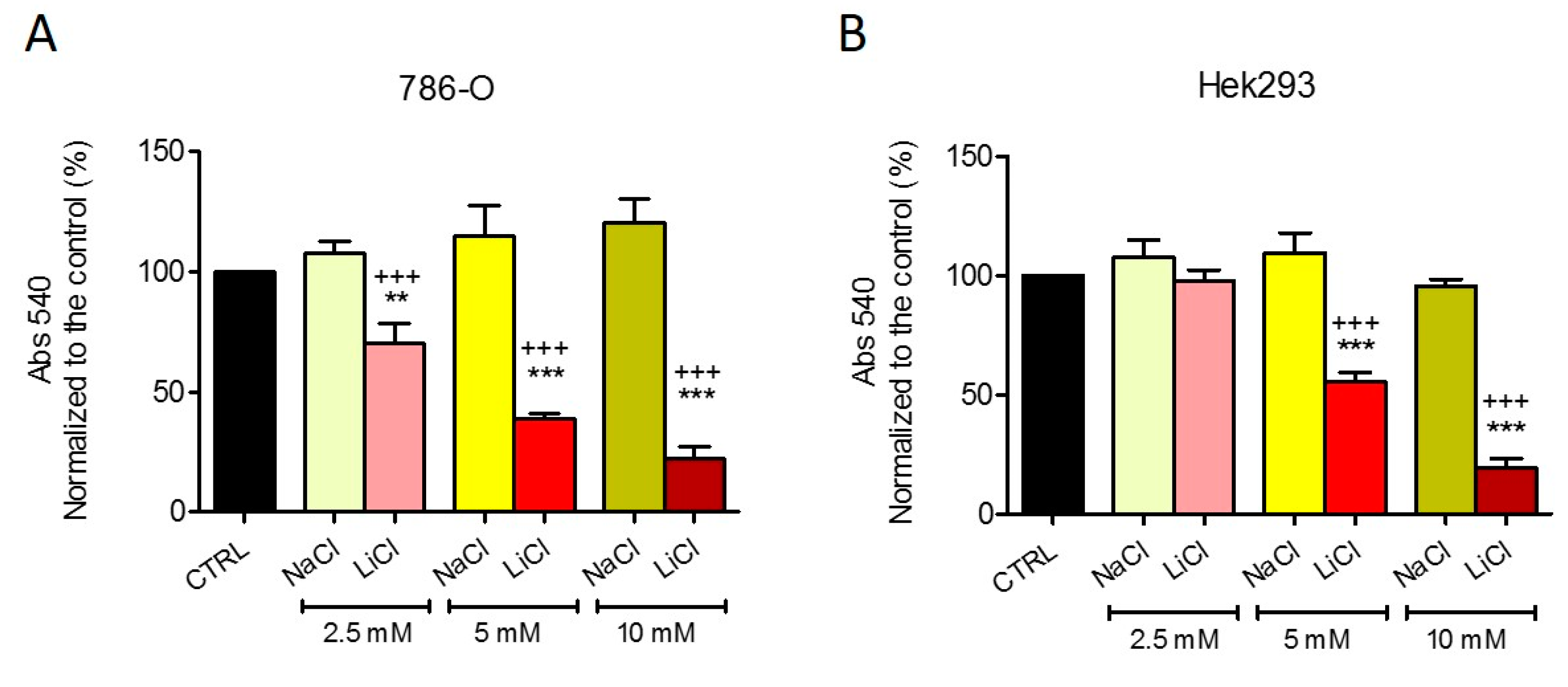
2.1. Lithium Alone is Responsible for the Effects of LiAcAc on Cell Growth
2.2. Chloride Does Not Affect Cell Growth
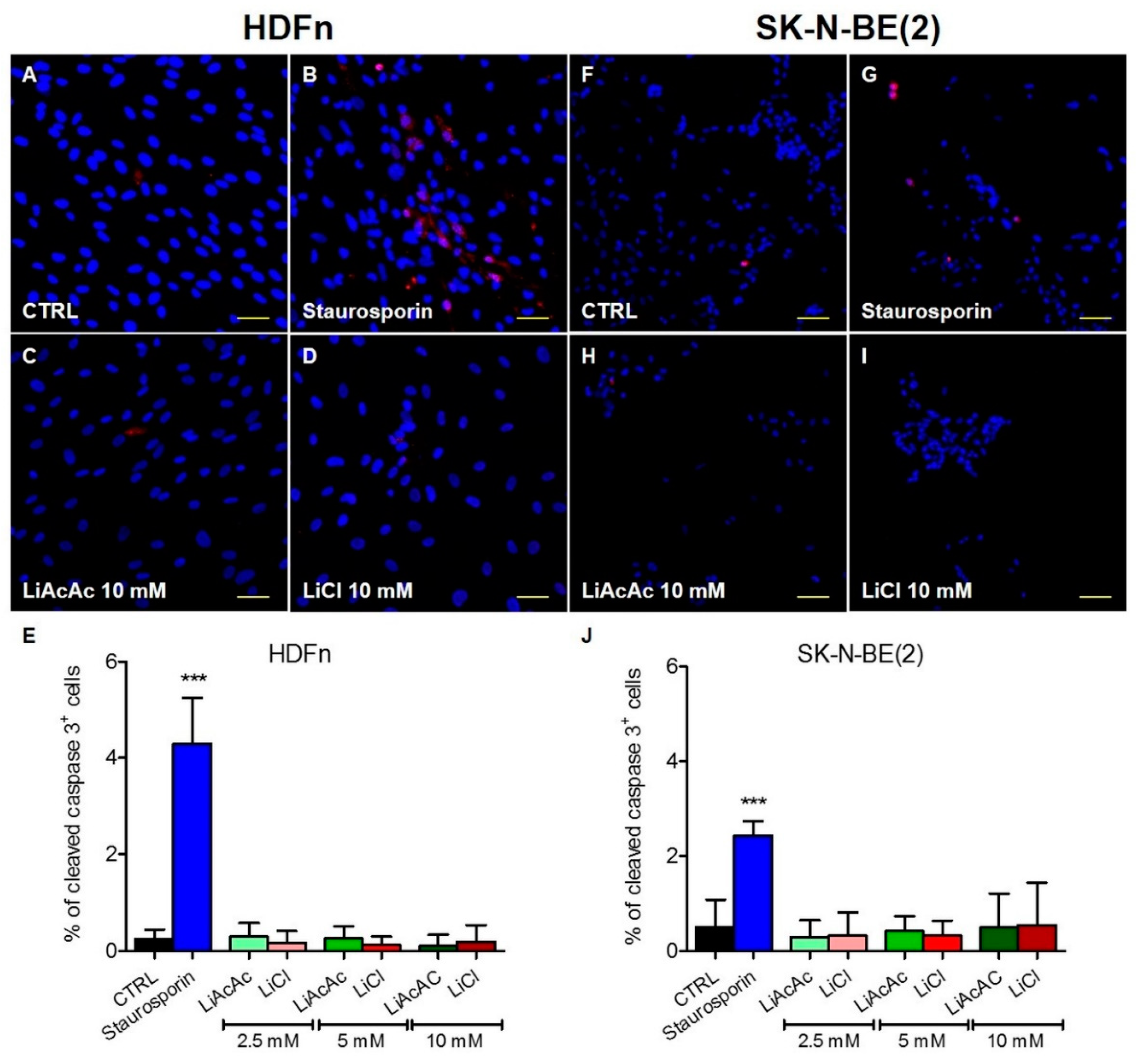
2.3. LiCl and LiAcAc Do Not Increase the Expression of Cleaved Caspase 3
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Crystal Violet Assay
4.3. Immunohistochemical Staining of Cleaved-Caspase 3
4.4. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-HB | 3-hydroxy-butyrate |
| AcAc | Acetoacetate |
| B3GALANT2 | Beta-1,3-N-acetylgalactosaminyltransferase 2 |
| HDFn | Normal human dermal fibroblasts |
| HEK | Human embryonic kidney |
| KB | ketone bodies |
| KD | ketogenic diet |
| NB | Neuroblastoma |
| RCC | Renal cell carcinoma |
References
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging (Albany NY) 2018, 10, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Vidali, S.; Aminzadeh, S.; Lambert, B.; Rutherford, T.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Mitochondria: The ketogenic diet—A metabolism-based therapy. Int. J. Biochem. Cell Biol. 2015, 63, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Poff, A.; Koutnik, A.P.; Egan, K.M.; Sahebjam, S.; D’Agostino, D.; Kumar, N.B. Targeting the Warburg effect for cancer treatment: Ketogenic diets for management of glioma. Semin. Cancer Biol. 2017, 56, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. The influence of ketogenic therapy on the 5 R’s of radiobiology. Int. J. Radiat. Biol. 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bartmann, C.; Janaki Raman, S.R.; Floter, J.; Schulze, A.; Bahlke, K.; Willingstorfer, J.; Strunz, M.; Wockel, A.; Klement, R.J.; Kapp, M.; et al. Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer Metab. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Lin, R.; Jin, L.; Zhao, L.; Kang, H.B.; Pan, Y.; Liu, S.; Qian, G.; Qian, Z.; Konstantakou, E.; et al. Prevention of Dietary-Fat-Fueled Ketogenesis Attenuates BRAF V600E Tumor Growth. Cell Metab. 2017, 25, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Franklin, S.T.; Young, J.W.; Nonnecke, B.J. Effects of ketones, acetate, butyrate, and glucose on bovine lymphocyte proliferation. J. Dairy Sci. 1991, 74, 2507–2514. [Google Scholar] [CrossRef]
- Gomez, E. Acetoacetate and beta-D-hydroxybutyrate as energy substrates during early bovine embryo development in vitro. Theriogenology 1997, 48, 63–74. [Google Scholar] [CrossRef]
- Maurer, G.D.; Brucker, D.P.; Bahr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef]
- Fine, E.J.; Miller, A.; Quadros, E.V.; Sequeira, J.M.; Feinman, R.D. Acetoacetate reduces growth and ATP concentration in cancer cell lines which over-express uncoupling protein 2. Cancer Cell Int. 2009, 9, 14. [Google Scholar] [CrossRef]
- Meroni, E.; Papini, N.; Criscuoli, F.; Casiraghi, M.C.; Massaccesi, L.; Basilico, N.; Erba, D. Metabolic Responses in Endothelial Cells Following Exposure to Ketone Bodies. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.S.; Hah, Y.S.; Nilufar, R.; Han, J.; Bong, J.H.; Kang, S.S.; Cho, G.J.; Choi, W.S. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J. Neurosci. Res. 2006, 83, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, II.; Powers, R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Skinner, R.; Trujillo, A.; Ma, X.; Beierle, E.A. Ketone bodies inhibit the viability of human neuroblastoma cells. J. Pediatr. Surg. 2009, 44, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The metabolism of tumors. New York: Richard R. Smith 1931, 129–169. [Google Scholar]
- Nowicki, M.O.; Dmitrieva, N.; Stein, A.M.; Cutter, J.L.; Godlewski, J.; Saeki, Y.; Nita, M.; Berens, M.E.; Sander, L.M.; Newton, H.B.; et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro. Oncol. 2008, 10, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, K.; Liu, X.; Liu, J.; Lu, X.; Tao, K.; Wang, G.; Wang, J. Lithium chloride suppresses colorectal cancer cell survival and proliferation through ROS/GSK-3beta/NF-kappaB signaling pathway. Oxid. Med. Cell. Longev. 2014, 2014, 241864. [Google Scholar] [CrossRef] [PubMed]
- Maeng, Y.S.; Lee, R.; Lee, B.; Choi, S.I.; Kim, E.K. Lithium inhibits tumor lymphangiogenesis and metastasis through the inhibition of TGFBIp expression in cancer cells. Sci. Rep. 2016, 6, 20739. [Google Scholar] [CrossRef]
- Han, S.; Meng, L.; Jiang, Y.; Cheng, W.; Tie, X.; Xia, J.; Wu, A. Lithium enhances the antitumour effect of temozolomide against TP53 wild-type glioblastoma cells via NFAT1/FasL signalling. Br. J. Cancer 2017, 116, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, M.; Goliaei, B.; Khodagholi, F.; Nikoofar, A. Lithium increases radiosensitivity by abrogating DNA repair in breast cancer spheroid culture. Arch. Iran. Med. 2014, 17, 352–360. [Google Scholar] [PubMed]
- Ozerdem, A.; Ceylan, D.; Targitay, B. The Relationship Between Lithium and Cancer Proliferation: A Case-Based Review of the Literature. Curr. Drug Metab. 2018, 19, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Kazachenko, A.V.; Vyssokikh, M.Y.; Vasileva, A.K.; Tcvirkun, D.V.; Isaev, N.K.; Kirpatovsky, V.I.; Zorov, D.B. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007, 72, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Jankauskas, S.S.; Zorov, S.D.; Babenko, V.A.; Skulachev, M.V.; Zorov, D.B. Lithium salts—Simple but magic. Biochemistry (Mosc) 2014, 79, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, E.; Arguello-Miranda, O.; Chiu, S.W.; Fazal, Z.; Kruczek, J.; Nunez-Corrales, S.; Pandit, S.; Pritchet, L. Towards a Unified Understanding of Lithium Action in Basic Biology and its Significance for Applied Biology. J. Membr. Biol. 2017, 250, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Undi, R.B.; Gutti, U.; Gutti, R.K. LiCl regulates mitochondrial biogenesis during megakaryocyte development. J. Trace Elem. Med. Biol. 2017, 39, 193–201. [Google Scholar] [CrossRef]
- Struewing, I.T.; Barnett, C.D.; Tang, T.; Mao, C.D. Lithium increases PGC-1alpha expression and mitochondrial biogenesis in primary bovine aortic endothelial cells. FEBS J. 2007, 274, 2749–2765. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, R.F.; Wang, Y.; Yuan, P.; Zhou, R.; Li, X.; Alesci, S.; Du, J.; Manji, H.K. Common effects of lithium and valproate on mitochondrial functions: Protection against methamphetamine-induced mitochondrial damage. Int. J. Neuropsychopharmacol. 2009, 12, 805–822. [Google Scholar] [CrossRef]
- Alural, B.; Ozerdem, A.; Allmer, J.; Genc, K.; Genc, S. Lithium protects against paraquat neurotoxicity by NRF2 activation and miR-34a inhibition in SH-SY5Y cells. Front. Cell Neurosci. 2015, 9, 209. [Google Scholar] [CrossRef]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cocheme, H.M.; et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Sun, X.; Xie, Y.; Liu, L.; Han, D.; Yao, Y.; Li, H.; Li, Z.; Xu, K. Lithium chloride inhibits cell survival, overcomes drug resistance, and triggers apoptosis in multiple myeloma via activation of the Wnt/beta-catenin pathway. Am. J. Transl. Res. 2018, 10, 2610–2618. [Google Scholar]
- Almond, M.K.; Smith, A.; Cohen, R.D. Ketone bodies promote a rapid rise in glutamate efflux from the isolated perfused rat liver without altering the rate of glutamine production. Amino Acids 1995, 9, 141–146. [Google Scholar] [PubMed]
- Sartorelli, P.; Paltrinieri, S.; Comazzi, S. Non-specific immunity and ketone bodies. II: In vitro studies on adherence and superoxide anion production in ovine neutrophils. J. Vet. Med. A. Physiol. Pathol. Clin. Med. 2000, 47, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bohlooli, M.; Ghaffari-Moghaddam, M.; Khajeh, M.; Shahraki-Fallah, G.; Haghighi-Kekhaiye, B.; Sheibani, N. The role of acetoacetate in Amadori product formation of human serum albumin. J. Photochem. Photobiol. B 2016, 163, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Thaler, S.; Choragiewicz, T.J.; Rejdak, R.; Fiedorowicz, M.; Turski, W.A.; Tulidowicz-Bielak, M.; Zrenner, E.; Schuettauf, F.; Zarnowski, T. Neuroprotection by acetoacetate and β-hydroxybutyrate against NMDA-induced RGC damage in rat—possible involvement of kynurenic acid. Graefes Arch. Clin. Exp. Ophthalmol. 2010, 248, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Kanikarla-Marie, P.; Jain, S.K. Hyperketonemia (acetoacetate) upregulates NADPH oxidase 4 and elevates oxidative stress, ICAM-1, and monocyte adhesivity in endothelial cells. Cell. Physiol. Biochem. 2015, 35, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Kannan, K.; Lim, G. Ketosis (acetoacetate) can generate oxygen radicals and cause increased lipid peroxidation and growth inhibition in human endothelial cells. Free Radic. Biol. Med. 1998, 25, 1083–1088. [Google Scholar] [CrossRef]
- Du, X.; Shi, Z.; Peng, Z.; Zhao, C.; Zhang, Y.; Wang, Z.; Li, X.; Liu, G.; Li, X. Acetoacetate induces hepatocytes apoptosis by the ROS-mediated MAPKs pathway in ketotic cows. J. Cell Physiol. 2017, 232, 3296–3308. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Meng, J.; Li, L.; Han, W.; Li, C.; Zhong, R.; Miao, X.; Cai, J.; Zhang, Y.; Zhu, D. Acetoacetate Accelerates Muscle Regeneration and Ameliorates Muscular Dystrophy in Mice. J. Biol. Chem. 2016, 291, 2181–2195. [Google Scholar] [CrossRef]
- Henry, R.R.; Brechtel, G.; Lim, K.H. Effects of ketone bodies on carbohydrate metabolism in non-insulin-dependent (type II) diabetes mellitus. Metabolism 1990, 39, 853–858. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Neureiter, D.; Skaria, T.; Wessler, S.; Cover, T.L.; Mayr, J.A.; Zimmermann, F.A.; Posselt, G.; Sperl, W.; Kofler, B. Oxidative Phosphorylation System in Gastric Carcinomas and Gastritis. Oxid. Med. Cell Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect of LiAcAc | Equimolar Controls | Ref. |
|---|---|---|
| LiAcAc or LiCl (1.5 mM) did not affect proliferation in breast cancer cells. | LiCl | [6] |
| LiAcAc but not LiCl increased BRAF positive melanoma tumor growth and increased MEK1 and ERK1/2 phosphorylation in the same cells. No effects is shown in BRAF negative melanomas. | LiCl | [7] |
| Both LiAcAc and LiCl (10 mM) inhibited bovine lymphocytes proliferation; the 3.125 mM concentration showed a trend to enhance the proliferation. | LiCl, NaCl | [8] |
| LiAcAc but not LiCl (3.6 mM) enhanced development of bovine embryos. Higher concentrations of both compounds were inhibitory. | LiCl | [9] |
| LiAcAc and LiCl (5 mM) neither affected glioma cells growth in normoxia nor in hypoxia conditions. | LiCl | [10] |
| LiAcAc (10 mM) significantly reduced cell growth and ATP and UCP2 production in several colon and breast cancer cell lines. | n.r. | [11] |
| LiAcAc (0.5–5 mM) reduced viability in dermal endothelium cells. A solution of 4 mM 3-HB and 1 mM LiAcAc induced moderate oxidative stress and reduced Nrf2 expression. | n.r. | [12] |
| LiAcAc (10–50 mM) reduced mouse hippocampal neuronal cell growth. LiAcAc (1–5 mM) protected the cells from glutamate induced toxicity. | n.r. | [13] |
| LiAcAc (5–20 mM) reduced pancreatic cancer cell growth. LiAcAc (10 and/or 20 mM) increased caspase 3/7 activity and reduced glucose and glutamine uptake, lactate release, ATP and ROS levels, and the expression of glycolytic enzymes. LiAcAc (20 mM) prevented cachexia. | n.r. | [14] |
| LiAcAc (13.9 mM) did not prevent the decrease in cell viability in neuroblastoma cells deprived of glucose but increased apoptosis. Normal fibroblasts were not affected. | n.r. | [15] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidali, S.; Aminzadeh-Gohari, S.; Vatrinet, R.; Iommarini, L.; Porcelli, A.M.; Kofler, B.; Feichtinger, R.G. Lithium and Not Acetoacetate Influences the Growth of Cells Treated with Lithium Acetoacetate. Int. J. Mol. Sci. 2019, 20, 3104. https://doi.org/10.3390/ijms20123104
Vidali S, Aminzadeh-Gohari S, Vatrinet R, Iommarini L, Porcelli AM, Kofler B, Feichtinger RG. Lithium and Not Acetoacetate Influences the Growth of Cells Treated with Lithium Acetoacetate. International Journal of Molecular Sciences. 2019; 20(12):3104. https://doi.org/10.3390/ijms20123104
Chicago/Turabian StyleVidali, Silvia, Sepideh Aminzadeh-Gohari, Renaud Vatrinet, Luisa Iommarini, Anna Maria Porcelli, Barbara Kofler, and René Günther Feichtinger. 2019. "Lithium and Not Acetoacetate Influences the Growth of Cells Treated with Lithium Acetoacetate" International Journal of Molecular Sciences 20, no. 12: 3104. https://doi.org/10.3390/ijms20123104
APA StyleVidali, S., Aminzadeh-Gohari, S., Vatrinet, R., Iommarini, L., Porcelli, A. M., Kofler, B., & Feichtinger, R. G. (2019). Lithium and Not Acetoacetate Influences the Growth of Cells Treated with Lithium Acetoacetate. International Journal of Molecular Sciences, 20(12), 3104. https://doi.org/10.3390/ijms20123104