Genome-Wide Analysis of ROS Antioxidant Genes in Resurrection Species Suggest an Involvement of Distinct ROS Detoxification Systems during Desiccation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
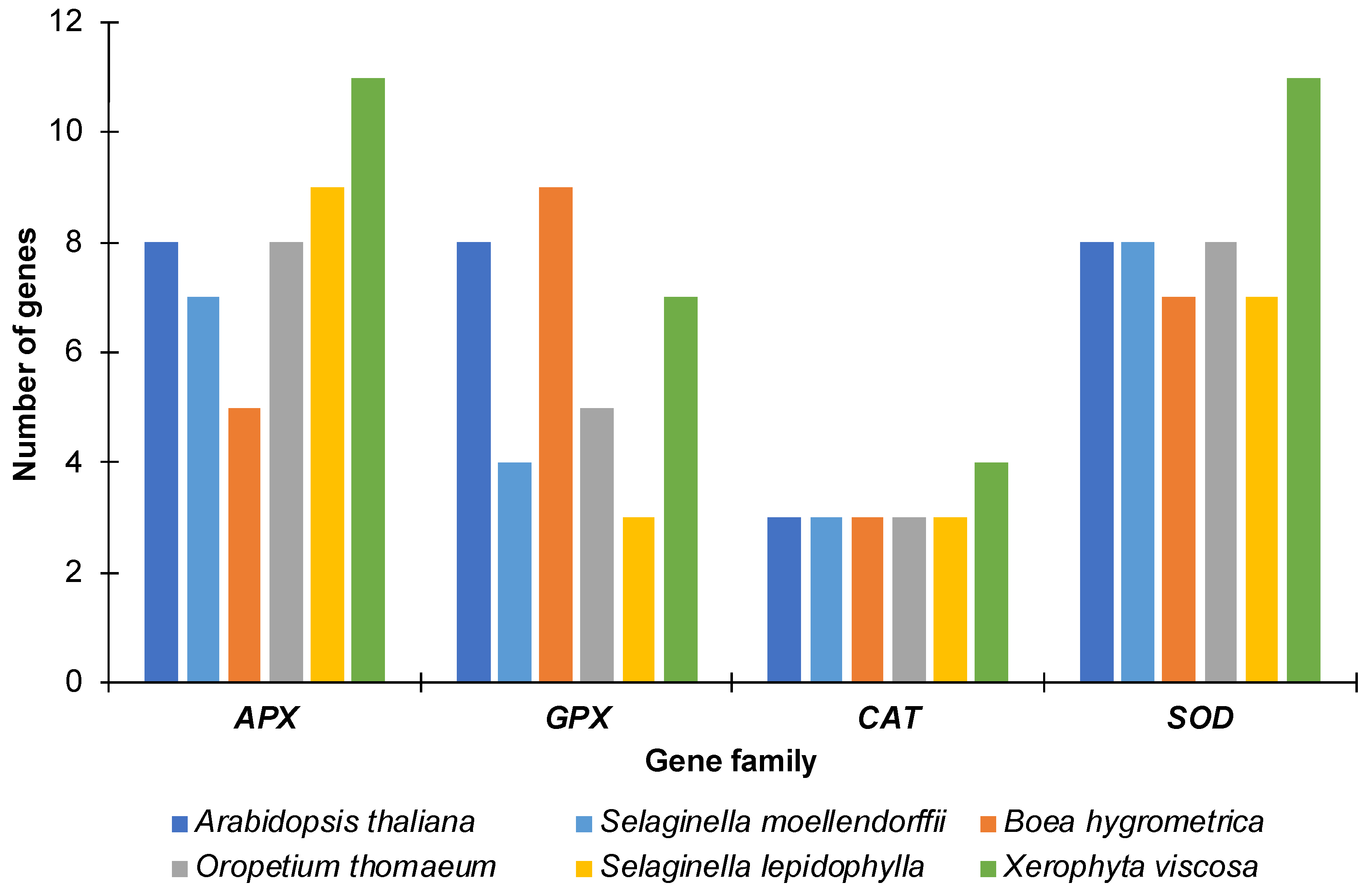
2.1. Genome-Wide Identification of APX, GPX, CAT and SOD Genes
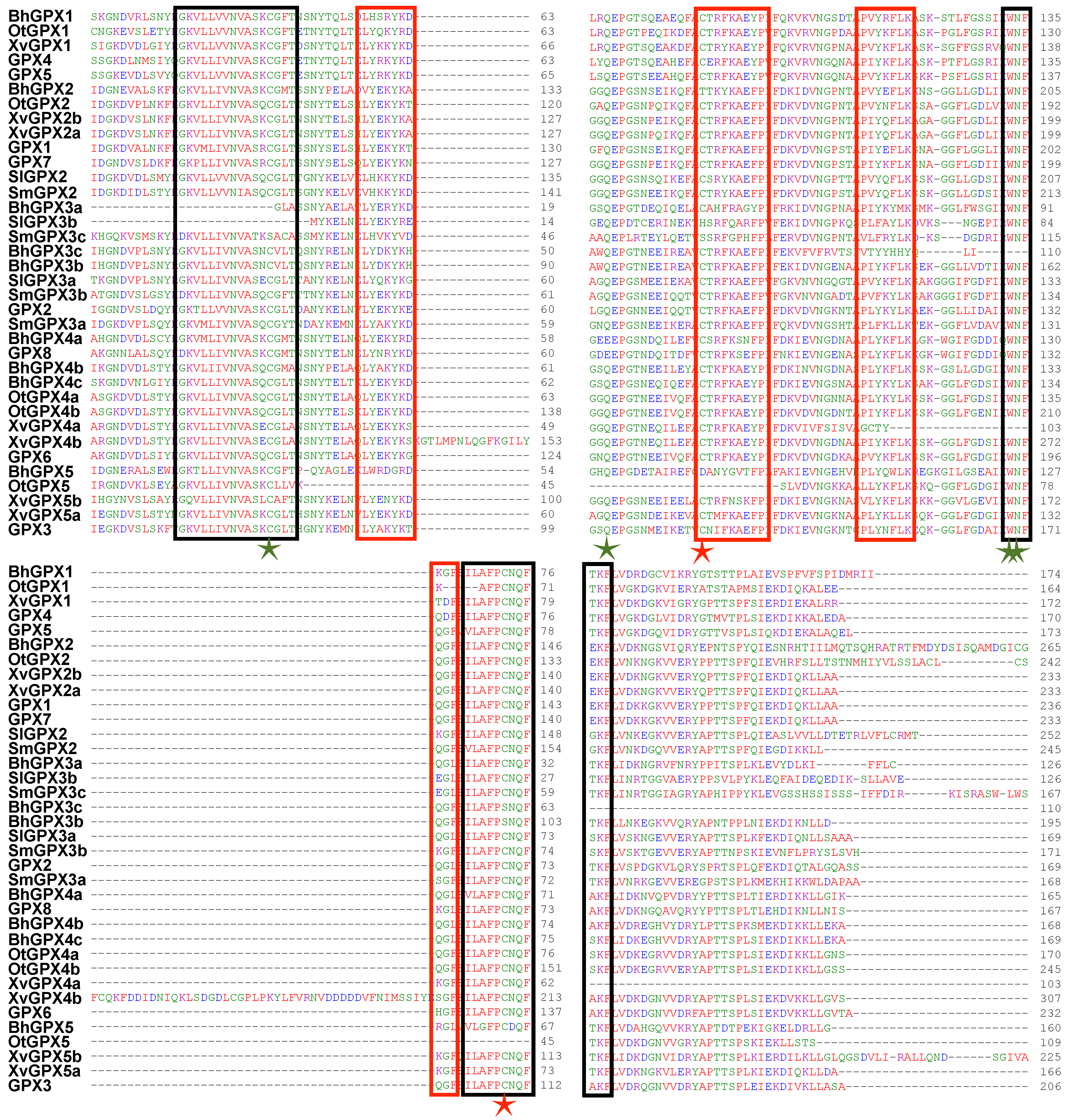
2.2. Phylogenetic Analysis
2.3. Gene Duplication Analysis
2.4. Identification of Exon-Intron Structure
2.5. Structural Organization and Conservation Patterns
2.6. Prediction of Cis-Acting Elements
2.7. miRNA Targeting of APX, GPX, CAT and SOD Genes
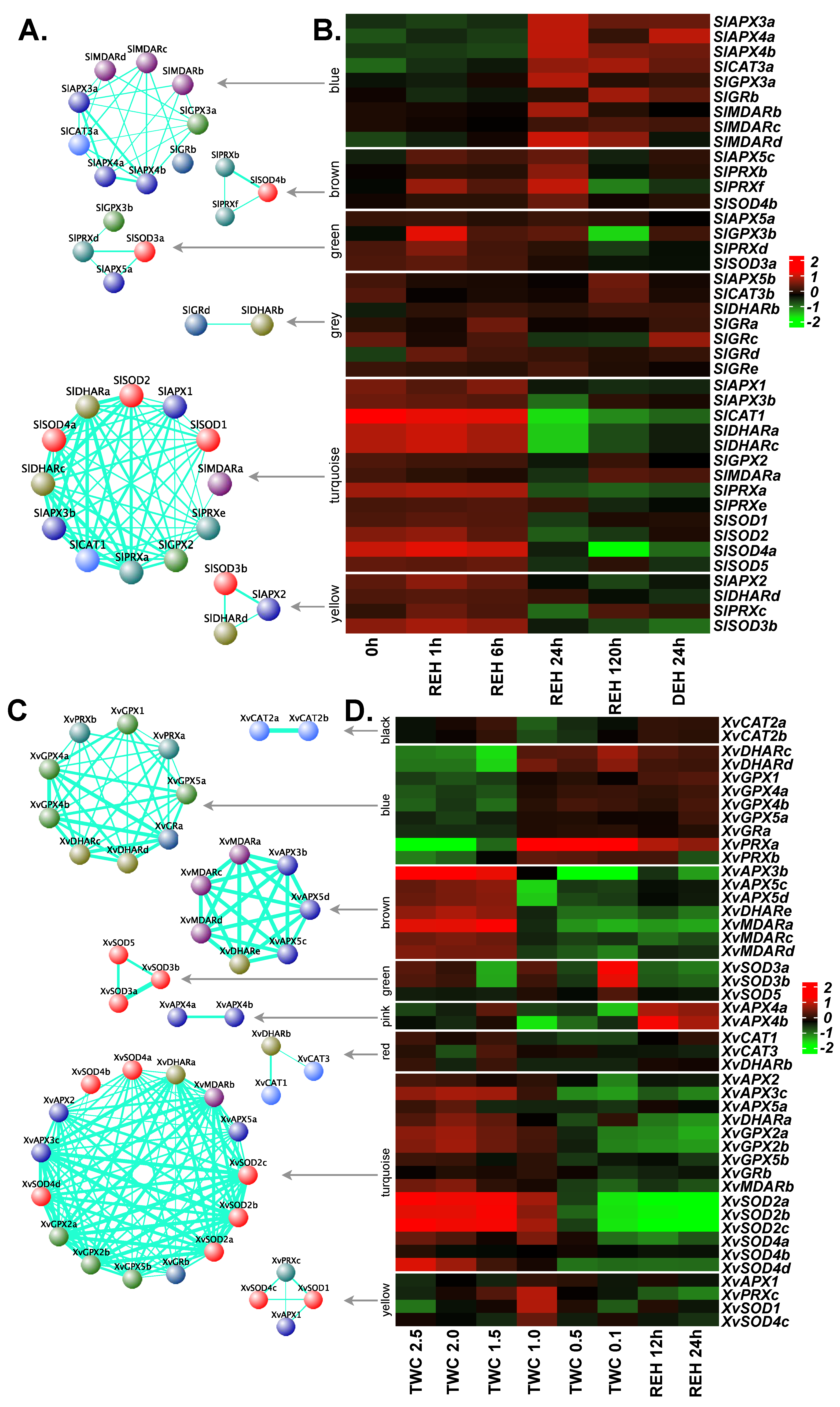
2.8. Expression Profiles of APX, GPX, CAT and SOD Genes in Response to Desiccation
2.9. Co-Expression Network Analysis for ROS-Scavenging Genes
3. Materials and Methods
3.1. Data Acquisition
3.2. Identification and Characterization of APX, GPX, CAT and SOD Genes
3.3. Phylogenetic and Gene Duplication Analysis
3.4. Chromosomal Distribution, Gene Structure, and Motif Analysis
3.5. Promoter Analysis and miRNA Prediction Analysis
3.6. Expression Profiling of APX, GPX, CAT and SOD Genes under Stress
3.7. Co-Expression Network Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gechev, T.S.; Van Breusegem, F.; Stone, J.M.; Denev, I.; Laloi, C. Reactive oxygen species as signals that modulate plant stress responses and programmed cell death. Bioessays 2006, 28, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Mhamdi, A.; Van Breusegem, F. Reactive oxygen species in plant development. Development 2018, 145, dev164376. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Alscher, R.G.; Erturk, N.; Heath, L.S. Role of superoxide dismutases (SODs) in controlling oxidative stress in plants. J. Exp. Bot. 2002, 53, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xia, M.; Chen, J.; Deng, F.; Yuan, R.; Zhang, X.; Shen, F. Genome-wide analysis of superoxide dismutase gene family in Gossypium raimondii and G. arboreum. Plant Gene 2016, 6, 18–29. [Google Scholar] [CrossRef]
- Nath, K.; Kumar, S.; Poudyal, R.S.; Yang, Y.N.; Timilsina, R.; Park, Y.S.; Nath, J.; Chauhan, P.S.; Pant, B.; Lee, C. Developmental stage-dependent differential gene expression of superoxide dismutase isoenzymes and their localization and physical interaction network in rice (Oryza sativa L.). Genes Genom. 2014, 36, 45–55. [Google Scholar] [CrossRef]
- Filiz, E.; Tombuloğlu, H. Genome-wide distribution of superoxide dismutase (SOD) gene families in Sorghum bicolor. Turk. J. Biol. 2015, 39, 49–59. [Google Scholar] [CrossRef]
- Teixeira, F.K.; Menezes-Benavente, L.; Galvão, V.C.; Margis, R.; Margis-Pinheiro, M. Rice ascorbate peroxidase gene family encodes functionally diverse isoforms localized in different subcellular compartments. Planta 2006, 224, 300–314. [Google Scholar] [CrossRef]
- Tao, C.; Jin, X.; Zhu, L.; Xie, Q.; Wang, X.; Li, H. Genome-wide investigation and expression profiling of APX gene family in Gossypium hirsutum provide new insights in redox homeostasis maintenance during different fiber development stages. Mol. Gen. Gen. 2018, 293, 685–697. [Google Scholar] [CrossRef]
- Bela, K.; Horváth, E.; Gallé, Á.; Szabados, L.; Tari, I.; Csiszár, J. Plant glutathione peroxidases: Emerging role of the antioxidant enzymes in plant development and stress responses. J. Plant Physiol. 2015, 176, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hu, L.; Wu, H.; Jiang, L.; Liu, S. Genome-wide identification and transcriptional expression analysis of cucumber superoxide dismutase (SOD) family in response to various abiotic stresses. Int. J. Genom. 2017, 2017, 7243973. [Google Scholar] [CrossRef] [PubMed]
- Akbudak, M.A.; Filiz, E.; Vatansever, R.; Kontbay, K. Genome-wide identification and expression profiling of ascorbate peroxidase (APX) and glutathione peroxidase (GPX) genes under drought stress in sorghum (Sorghum bicolor L.). J. Plant Growth Reg. 2018, 37, 925–936. [Google Scholar] [CrossRef]
- Mhamdi, A.; Queval, G.; Chaouch, S.; Vanderauwera, S.; Van Breusegem, F.; Noctor, G. Catalase function in plants: A focus on Arabidopsis mutants as stress-mimic models. J. Exp. Bot. 2010, 61, 4197–4220. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Yang, Y.; Jiang, L.; Liu, S. The catalase gene family in cucumber: Genome-wide identification and organization. Genet. Mol. Biol. 2016, 39, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Caverzan, A.; Passaia, G.; Rosa, S.B.; Ribeiro, C.W.; Lazzarotto, F.; Margis-Pinheiro, M. Plant responses to stresses: Role of ascorbate peroxidase in the antioxidant protection. Genet. Mol. Biol. 2012, 35, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 2012, 217037. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, L.; Ye, S.; Jiang, L.; Liu, S. Genome-wide identification of glutathione peroxidase (GPX) gene family and their response to abiotic stress in cucumber. 3 Biotech. 2018, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bartels, D. Molecular responses to dehydration and desiccation in desiccation-tolerant angiosperm plants. J. Exp. Bot. 2018, 69, 3211–3222. [Google Scholar] [CrossRef]
- Farrant, J.M.; Brandt, W.; Lindsey, G.G. An overview of mechanisms of desiccation tolerance in selected angiosperm resurrection plants. Plant Stress J. 2007, 1, 72–84. [Google Scholar]
- Gechev, T.S.; Dinakar, C.; Benina, M.; Toneva, V.; Bartels, D. Molecular mechanisms of desiccation tolerance in resurrection plants. Cell. Mol. Life Sci. 2012, 69, 3175–3186. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, H.W.; Farrant, J.M. Protection mechanisms against excess light in the resurrection plants Craterostigma wilmsii and Xerophyta viscosa. Plant Growth Reg. 1998, 24, 203–210. [Google Scholar] [CrossRef]
- Farrant, J.M. A comparison of mechanisms of desiccation tolerance among three angiosperm resurrection plant species. Plant Ecol. 2000, 151, 29–39. [Google Scholar] [CrossRef]
- Neale, A.D.; Blomstedt, T.; Bronson, T.N.; Guthridge, L.K.; Evans, J.; Gaff, D.F.; Hamill, J.D. The isolation of genes from the resurrection grass Sporobolus stapfianus which are induced during severe drought stress. Plant Cell Environ. 2000, 23, 265–277. [Google Scholar] [CrossRef]
- Kranner, I.; Beckett, R.P.; Wornik, S.; Zorn, M.; Pfeifhofer, H.W. Revival of a resurrection plant correlates with its antioxidant status. Plant J. 2002, 31, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Collett, H.; Shen, A.; Gardner, M.; Farrant, J.M.; Denby, K.J.; Illing, N. Towards transcript profiling of desiccation tolerance in Xerophyta humilis: Construction of a normalized 11 k X. humilis cDNA set and microarray expression analysis of 424 cDNAs in response to dehydration. Physiol. Plant. 2004, 122, 39–53. [Google Scholar] [CrossRef]
- Gechev, T.S.; Benina, M.; Obata, T.; Tohge, T.; Sujeeth, N.; Minkov, I.; Hille, J.; Temanni, M.R.; Marriott, A.S.; Bergström, E.; et al. Molecular mechanisms of desiccation tolerance in the resurrection glacial relic Haberlea rhodopensis. Cell. Mol. Life Sci. 2013, 70, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Yang, G.; Zhang, L.; Yang, X.; Zhao, S.; Ji, Z.; Zhou, Q.; Hu, M.; Wang, Y.; Chen, M.; et al. The resurrection genome of Boea hygrometrica: A blueprint for survival of dehydration. Proc. Natl. Acad. Sci. USA 2015, 112, 5833–5837. [Google Scholar] [CrossRef]
- VanBuren, R.; Wai, C.M.; Ou, S.; Pardo, J.; Bryant, D.; Jiang, N.; Mockler, T.C.; Edger, P.; Michael, T.P. Extreme haplotype variation in the desiccation-tolerant clubmoss Selaginella lepidophylla. Nat. Commun. 2018, 9, 13. [Google Scholar] [CrossRef]
- Costa, M.C.D.; Artur, M.A.; Maia, J.; Jonkheer, E.; Derks, M.F.; Nijveen, H.; Williams, B.; Mundree, S.G.; Jiménez-Gómez, J.M.; Hesselink, T.; et al. A footprint of desiccation tolerance in the genome of Xerophyta viscosa. Nat. Plants 2017, 3, 17038. [Google Scholar] [CrossRef]
- VanBuren, R.; Bryant, D.; Edger, P.P.; Tang, H.; Burgess, D.; Challabathula, D.; Spittle, K.; Hall, R.; Gu, J.; Lyons, E.; et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 2015, 527, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; Albert, V.A.; Aono, V.A.; Aoyama, T.; Ambrose, B.A.; Ashton, N.W.; et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Ozyigit, I.I.; Filiz, E.; Vatansever, R.; Kurtoglu, K.Y.; Koc, I.; Öztürk, M.X.; Anjum, N.A. Identification and comparative analysis of H2O2-scavenging enzymes (ascorbate peroxidase and glutathione peroxidase) in selected plants employing bioinformatics approaches. Front. Plant Sci. 2016, 7, 301. [Google Scholar] [CrossRef] [PubMed]
- Inzé, A.; Vanderauwera, S.; Hoeberichts, F.A.; Vandorpe, M.; Van Gaever, T.I.; Van Breusegem, F. A subcellular localization compendium of hydrogen peroxide-induced proteins. Plant Cell Environ. 2012, 35, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Henzler, T.; Steudle, E. Transport and metabolic degradation of hydrogen peroxide in Chara corallina: Model calculations and measurements with the pressure probe suggest transport of H2O2 across water channels. J. Exp. Bot. 2000, 51, 2053–2066. [Google Scholar] [CrossRef] [PubMed]
- Asada, K. Ascorbate peroxidase—a hydrogen peroxide-scavenging enzyme in plants. Physiol. Plant. 1992, 85, 235–241. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef]
- Fukao, T.; Xu, K.; Ronald, P.C.; Bailey-Serres, J. A variable cluster of ethylene response factor–like genes regulates metabolic and developmental acclimation responses to submergence in rice. Plant Cell 2006, 18, 2021–2034. [Google Scholar] [CrossRef]
- Rizzon, C.; Ponger, L.; Gaut, B.S. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comp. Biol. 2006, 2, e115. [Google Scholar] [CrossRef]
- Hanada, K.; Zou, C.; Lehti-Shiu, M.D.; Shinozaki, K.; Shiu, S.H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 2008, 148, 993–1003. [Google Scholar] [CrossRef]
- Zou, C.; Lehti-Shiu, M.D.; Thomashow, M.; Shiu, S.H. Evolution of stress-regulated gene expression in duplicate genes of Arabidopsis thaliana. PLoS Genet. 2009, 5, e1000581. [Google Scholar] [CrossRef]
- VanBuren, R.; Pardo, J.; Wai, C.M.; Evans, S.; Bartels, D. Massive tandem proliferation of ELIPs supports convergent evolution of desiccation tolerance across land plants. Plant Physiol. 2019, 179, 1040–1049. [Google Scholar] [CrossRef]
- Hutin, C.; Nussaume, L.; Moise, N.; Moya, I.; Kloppstech, K.; Havaux, M. Early light-induced proteins protect Arabidopsis from photooxidative stress. Proc. Natl. Acad. Sci. USA 2003, 100, 4921–4926. [Google Scholar] [CrossRef]
- Bartels, D.; Hanke, C.; Schneider, K.; Michel, D.; Salamini, F. A desiccation-related Elip-like gene from the resurrection plant Craterostigma plantagineum is regulated by light and ABA. EMBO J. 1992, 11, 2771–2778. [Google Scholar] [CrossRef]
- Alamillo, J.M.; Bartels, D. Effects of desiccation on photosynthesis pigments and the ELIP-like dsp 22 protein complexes in the resurrection plant Craterostigma plantagineum. Plant Sci. 2001, 160, 1161–1170. [Google Scholar] [CrossRef]
- Yobi, A.; Schlauch, K.A.; Tillett, R.L.; Yim, W.C.; Espinoza, C.; Wone, B.W.; Cushman, J.C.; Oliver, M.J. Sporobolus stapfianus: Insights into desiccation tolerance in the resurrection grasses from linking transcriptomics to metabolomics. BMC Plant Biol. 2017, 17, 67. [Google Scholar] [CrossRef]
- Milla, M.A.R.; Maurer, A.; Huete, A.R.; Gustafson, J.P. Glutathione peroxidase genes in Arabidopsis are ubiquitous and regulated by abiotic stresses through diverse signaling pathways. Plant J. 2003, 36, 602–615. [Google Scholar] [CrossRef]
- Chen, M.; Li, K.; Li, H.; Song, C.P.; Miao, Y. The glutathione peroxidase gene family in Gossypium hirsutum: Genome-wide identification, classification, gene expression and functional analysis. Sci. Rep. 2017, 7, 44743. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and con-sequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef]
- Lecharny, A.; Boudet, N.; Gy, I.; Aubourg, S.; Kreis, M. Introns in, introns out in plant gene families: A genomic approach of the dynamics of gene structure. J. Struct. Funct. Genom. 2003, 3, 111–116. [Google Scholar] [CrossRef]
- Bolle, C.; Herrmann, R.G.; Oelmüller, R. Intron sequences are involved in the plastid- and light-dependent expression of the spinach PsaD gene. Plant J. 1996, 10, 919–924. [Google Scholar] [CrossRef]
- Zhan, X.; Qian, B.; Cao, F.; Wu, W.; Yang, L.; Guan, Q.; Gu, X.; Wang, P.; Okusolubo, T.A.; Dunn, S.L.; et al. An Arabidopsis PWI and RRM motif-containing protein is critical for pre-mRNA splicing and ABA responses. Nat. Commun. 2015, 6, 8139. [Google Scholar] [CrossRef]
- Thatcher, S.R.; Danilevskaya, O.N.; Meng, X.; Beatty, M.; Zastrow-Hayes, G.; Harris, C.; Van Allen, B.; Habben, J.; Li, B. Genome-wide analysis of alternative splicing during development and drought stress in maize. Plant Physiol. 2016, 170, 586–599. [Google Scholar] [CrossRef]
- Gelsthorpe, M.; Pulumati, M.; McCallum, C.; Dang-Vu, K.; Tsubota, S.I. The putative cell cycle gene, enhancer of rudimentary, encodes a highly conserved protein found in plants and animals. Gene 1997, 186, 189–195. [Google Scholar] [CrossRef]
- Wojcik, E.; Murphy, A.M.; Fares, H.; Dang-Vu, K.; Tsubota, S.I. Enhancer of rudimentaryp1, e(r)p1, a highly conserved enhancer of the rudimentary gene. Genetics 1994, 138, 1163–1170. [Google Scholar]
- Tsubota, S.I.; Vogel, A.C.; Phillips, A.C.; Ibach, S.M.; Weber, N.K.; Kostrzebski, M.A.; Spencer, S.A. Interactions between enhancer of rudimentary and Notch and deltex reveal a regulatory function of enhancer of rudimentary in the Notch signaling pathway in Drosophila melanogaster. Fly 2011, 5, 275–284. [Google Scholar] [CrossRef][Green Version]
- Karpinski, S.; Escobar, C.; Karpinska, B.; Creissen, G.; Mullineaux, P.M. Photosynthetic electron transport regulates the expression of cytosolic ascorbate peroxidase genes in Arabidopsis during excess light stress. Plant Cell 1997, 9, 627–640. [Google Scholar] [CrossRef]
- Kurepa, J.; Montagu, M.V.; Inzé, D. Expression of sodCp and sodB genes in Nicotiana tabacum: Effects of light and copper excess. J. Exp. Bot. 1997, 48, 2007–2014. [Google Scholar] [CrossRef]
- Karpinski, S.; Reynolds, H.; Karpinska, B.; Wingsle, G.; Creissen, G.; Mullineaux, P. Systemic signaling and acclimation in response to excess excitation energy in Arabidopsis. Science 1999, 284, 654–657. [Google Scholar] [CrossRef]
- Sunkar, R.; Kapoor, A.; Zhu, J.K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef]
- Wang, T.; Chen, L.; Zhao, M.; Tian, Q.; Zhang, W.H. Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genom. 2011, 12, 367. [Google Scholar] [CrossRef]
- Li, T.; Li, H.; Zhang, Y.X.; Liu, J.Y. Identification and analysis of seven H2O2-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica). Nucl. Acids Res. 2011, 39, 2821–2833. [Google Scholar] [CrossRef]
- Jiang, G.; Wang, Z.; Shang, H.; Yang, W.; Hu, Z.; Phillips, J.; Deng, X. Proteome analysis of leaves from the resurrection plant Boea hygrometrica in response to dehydration and rehydration. Planta 2007, 225, 1405–1420. [Google Scholar] [CrossRef]
- Pandey, V.; Ranjan, S.; Deeba, F.; Pandey, A.K.; Singh, R.; Shirke, P.A.; Pathre, U.V. Desiccation-induced physiological and biochemical changes in resurrection plant, Selaginella bryopteris. J. Plant Physiol. 2010, 167, 1351–1359. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive oxygen gene network of plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef]
- Liu, Z.J.; Zhang, X.L.; Bai, J.G.; Suo, B.X.; Xu, P.L.; Wang, L. Exogenous paraquat changes antioxidant enzyme activities and lipid peroxidation in drought-stressed cucumber leaves. Sci. Hort. 2009, 121, 138–143. [Google Scholar] [CrossRef]
- Sandhya, V.S.; Ali, S.Z.; Grover, M.; Reddy, G.; Venkateswarlu, B. Effect of plant growth promoting Pseudomonas spp. on compatible solutes, antioxidant status and plant growth of maize under drought stress. Plant Growth Regul. 2010, 62, 21–30. [Google Scholar] [CrossRef]
- Sharma, P.; Dubey, R.S. Drought induces oxidative stress and enhances the activities of antioxidant enzymes in growing rice seedlings. Plant Growth Regul. 2005, 46, 209–221. [Google Scholar] [CrossRef]
- Pinheiro, H.A.; DaMatta, F.M.; Chaves, A.R.; Fontes, E.P.; Loureiro, M.E. Drought tolerance in relation to protection against oxidative stress in clones of Coffea canephora subjected to long-term drought. Plant Sci. 2004, 167, 1307–1314. [Google Scholar] [CrossRef]
- Miao, Y.; Lv, D.; Wang, P.; Wang, X.C.; Chen, J.; Miao, C.; Song, C.P. An Arabidopsis glutathione peroxidase functions as both a redox transducer and a scavenger in abscisic acid and drought stress responses. Plant Cell. 2006, 18, 2749–2766. [Google Scholar] [CrossRef]
- Zhang, J.; Kirkham, M.B. Enzymatic responses of the ascorbate-glutathione cycle to drought in sorghum and sunflower plants. Plant Sci. 1996, 113, 139–147. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2017, 35, 543–548. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comp. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucl. Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucl. Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucl. Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucl. Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; Dong, Y.; Dijkwel, P.P.; Mueller-Roeber, B.; Gechev, T.S. Genome-Wide Analysis of ROS Antioxidant Genes in Resurrection Species Suggest an Involvement of Distinct ROS Detoxification Systems during Desiccation. Int. J. Mol. Sci. 2019, 20, 3101. https://doi.org/10.3390/ijms20123101
Gupta S, Dong Y, Dijkwel PP, Mueller-Roeber B, Gechev TS. Genome-Wide Analysis of ROS Antioxidant Genes in Resurrection Species Suggest an Involvement of Distinct ROS Detoxification Systems during Desiccation. International Journal of Molecular Sciences. 2019; 20(12):3101. https://doi.org/10.3390/ijms20123101
Chicago/Turabian StyleGupta, Saurabh, Yanni Dong, Paul P. Dijkwel, Bernd Mueller-Roeber, and Tsanko S. Gechev. 2019. "Genome-Wide Analysis of ROS Antioxidant Genes in Resurrection Species Suggest an Involvement of Distinct ROS Detoxification Systems during Desiccation" International Journal of Molecular Sciences 20, no. 12: 3101. https://doi.org/10.3390/ijms20123101
APA StyleGupta, S., Dong, Y., Dijkwel, P. P., Mueller-Roeber, B., & Gechev, T. S. (2019). Genome-Wide Analysis of ROS Antioxidant Genes in Resurrection Species Suggest an Involvement of Distinct ROS Detoxification Systems during Desiccation. International Journal of Molecular Sciences, 20(12), 3101. https://doi.org/10.3390/ijms20123101