Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury

Abstract
1. Introduction
1.1. Cellular Import and Export Mechanisms
1.2. Cisplatin-Induced AKI Mouse Models
1.2.1. Non-Tumour Bearing
1.2.2. Tumour Bearing Models
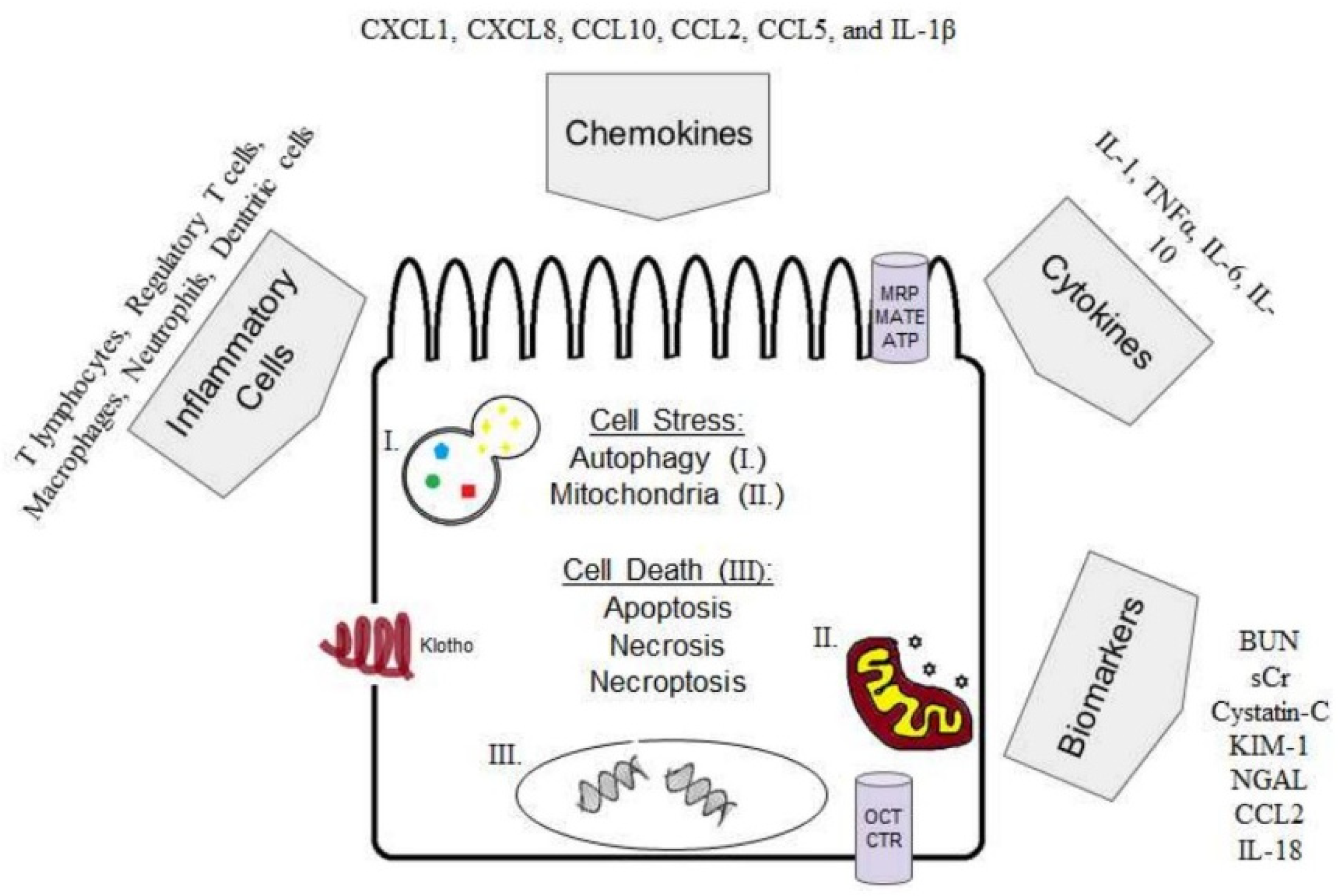
1.3. Cell Stress
1.3.1. Reactive Oxygen Species and Mitochondrial Dysfunction in Cisplatin-Induced AKI
1.3.2. Markers of Oxidative Stress and Mitochondrial Dysfunction
1.4. Cell Death
1.4.1. Apoptosis in Cisplatin-Induced AKI
1.4.2. Necrosis and Necroptosis in Cisplatin-Induced AKI
1.4.3. Methods for Detection of Cell Death and Caveats
1.5. Inflammation in Cisplatin-Induced AKI
1.5.1. Cytokines
1.5.2. Inflammatory Cells
1.6. Autophagy in Cisplatin-Induced AKI
1.7. Klotho in Cisplatin-Induced AKI
1.8. Cisplatin-Induced Chronic Kidney Disease (CKD)
Urinary Exosomes in Cisplatin-Induced AKI
2. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.S.; Kim, H.J.; Shen, A.; Lee, S.B.; Khadka, D.; Pandit, A.; So, H.S. Cisplatin-induced Kidney Dysfunction and Perspectives on Improving Treatment Strategies. Electrolyte Blood Press. 2014, 12, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Shad, F.; Smith, M.C. Acute kidney injury: A guide to diagnosis and management. Am. Fam. Physician 2012, 86, 631–639. [Google Scholar] [PubMed]
- George, B.; Joy, M.S.; Aleksunes, L.M. Urinary protein biomarkers of kidney injury in patients receiving cisplatin chemotherapy. Exp. Biol. Med. (Maywood) 2018, 243, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Harrill, A.H.; Lin, H.; Tobacyk, J.; Seely, J.C. Mouse population-based evaluation of urinary protein and miRNA biomarker performance associated with cisplatin renal injury. Exp. Biol. Med. (Maywood) 2018, 243, 237–247. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kim, T.H.; Won, A.J.; Jung, J.Y.; Kwack, S.J.; Kacew, S.; Chung, K.H.; Lee, B.M.; Kim, H.S. Age-related differences in kidney injury biomarkers induced by cisplatin. Environ. Toxicol. Pharmacol. 2014, 37, 1028–1039. [Google Scholar] [CrossRef]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Guo, C.; Tang, C.; Cai, J.; Dong, Z. Epigenetic Regulation in Kidney Toxicity: Insights from Cisplatin Nephrotoxicity. Semin. Nephrol. 2019, 39, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Schanz, M.; Schricker, S.; Pfister, F.; Alscher, M.D.; Kimmel, M. Renal complications of cancer therapies. Drugs Today (Barc) 2018, 54, 561–575. [Google Scholar] [CrossRef]
- Perse, M.; Veceric-Haler, Z. Cisplatin-Induced Rodent Model of Kidney Injury: Characteristics and Challenges. Biomed. Res. Int. 2018, 2018, 1462802. [Google Scholar] [CrossRef]
- Hultstrom, M.; Becirovic-Agic, M.; Jonsson, S. Comparison of acute kidney injury of different etiology reveals in-common mechanisms of tissue damage. Physiol. Genom. 2018, 50, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.N.; Siskind, L.J. Developing better mouse models to study cisplatin-induced kidney injury. Am. J. Physiol. Ren. Physiol. 2017, 313, F835–F841. [Google Scholar] [CrossRef] [PubMed]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Tang, C.; Ma, Z.; Huang, S.; Dong, Z. DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol. Appl. Pharmacol. 2016, 313, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef]
- Malyszko, J.; Kozlowska, K.; Kozlowski, L.; Malyszko, J. Nephrotoxicity of anticancer treatment. Nephrol. Dial. Transplant. 2017, 32, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Hayati, F.; Hossainzadeh, M.; Shayanpour, S.; Abedi-Gheshlaghi, Z.; Beladi Mousavi, S.S. Prevention of cisplatin nephrotoxicity. J. Nephropharmacol. 2016, 5, 57–60. [Google Scholar] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. Biomed. Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef]
- Dahal, A.; Bellows, B.K.; Sonpavde, G.; Tantravahi, S.K.; Choueiri, T.K.; Galsky, M.D.; Agarwal, N. Incidence of Severe Nephrotoxicity with Cisplatin Based on Renal Function Eligibility Criteria: Indirect Comparison Meta-analysis. Am. J. Clin. Oncol. 2016, 39, 497–506. [Google Scholar] [CrossRef]
- Ciarimboli, G. Membrane transporters as mediators of Cisplatin effects and side effects. Scientifica (Cairo) 2012, 2012, 473829. [Google Scholar] [CrossRef]
- Estrela, G.R.; Wasinski, F.; Felizardo, R.J.F.; Souza, L.L.; Camara, N.O.S.; Bader, M.; Araujo, R.C. MATE-1 modulation by kinin B1 receptor enhances cisplatin efflux from renal cells. Mol. Cell. Biochem. 2017, 428, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Harrach, S.; Ciarimboli, G. Role of transporters in the distribution of platinum-based drugs. Front. Pharmacol. 2015, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, H.; Yamashita, M.; Katsura, H.; Yu, J.; Waki, Y.; Nagata, N.; Sai, Y.; Miyamoto, K. Protecting cisplatin-induced nephrotoxicity with cimetidine does not affect antitumor activity. Biol. Pharm. Bull. 2010, 33, 1867–1871. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Okamoto, K.; Kobayashi, M.; Narumi, K.; Yamada, T.; Iseki, K. Magnesium attenuates cisplatin-induced nephrotoxicity by regulating the expression of renal transporters. Eur. J. Pharmacol. 2017, 811, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Suliman, F.A.; Khodeer, D.M.; Ibrahiem, A.; Mehanna, E.T.; El-Kherbetawy, M.K.; Mohammad, H.M.F.; Zaitone, S.A.; Moustafa, Y.M. Renoprotective effect of the isoflavonoid biochanin A against cisplatin induced acute kidney injury in mice: Effect on inflammatory burden and p53 apoptosis. Int. Immunopharmacol. 2018, 61, 8–19. [Google Scholar] [CrossRef]
- Kumar, G.; Solanki, M.H.; Xue, X.; Mintz, R.; Madankumar, S.; Chatterjee, P.K.; Metz, C.N. Magnesium improves cisplatin-mediated tumor killing while protecting against cisplatin-induced nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2017, 313, F339–F350. [Google Scholar] [CrossRef]
- Solanki, M.H.; Chatterjee, P.K.; Gupta, M.; Xue, X.; Plagov, A.; Metz, M.H.; Mintz, R.; Singhal, P.C.; Metz, C.N. Magnesium protects against cisplatin-induced acute kidney injury by regulating platinum accumulation. Am. J. Physiol. Ren. Physiol. 2014, 307, F369–F384. [Google Scholar] [CrossRef]
- Solanki, M.H.; Chatterjee, P.K.; Xue, X.; Gupta, M.; Rosales, I.; Yeboah, M.M.; Kohn, N.; Metz, C.N. Magnesium protects against cisplatin-induced acute kidney injury without compromising cisplatin-mediated killing of an ovarian tumor xenograft in mice. Am. J. Physiol. Ren. Physiol. 2015, 309, F35–F47. [Google Scholar] [CrossRef]
- Sharp, C.N.; Doll, M.A.; Megyesi, J.; Oropilla, G.B.; Beverly, L.J.; Siskind, L.J. Subclinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2018, 315, F161–F172. [Google Scholar] [CrossRef]
- Kim, Y.K.; Choi, T.R.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S. Beneficial effect of pentoxifylline on cisplatin-induced acute renal failure in rabbits. Ren. Fail. 2003, 25, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Afjal, M.A.; Abdi, S.H.; Sharma, S.; Ahmad, S.; Fatima, M.; Dabeer, S.; Akhter, J.; Raisuddin, S. Anti-inflammatory role of tempol (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl) in nephroprotection. Hum. Exp. Toxicol. 2019, 38, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.M.; Khalaf, M.M.; Sadek, S.A.; Abo-Youssef, A.M. Protective effects of apigenin and myricetin against cisplatin-induced nephrotoxicity in mice. Pharm. Biol. 2017, 55, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Schubert, R.; Sann, J.; Frueh, J.T.; Ullrich, E.; Geiger, H.; Baer, P.C. Tracking of Adipose-Derived Mesenchymal Stromal/Stem Cells in a Model of Cisplatin-Induced Acute Kidney Injury: Comparison of Bioluminescence Imaging versus qRT-PCR. Int. J. Mol. Sci. 2018, 19, 2564. [Google Scholar] [CrossRef] [PubMed]
- Selim, R.E.; Ahmed, H.H.; Abd-Allah, S.H.; Sabry, G.M.; Hassan, R.E.; Khalil, W.K.B.; Abouhashem, N.S. Mesenchymal Stem Cells: A Promising Therapeutic Tool for Acute Kidney Injury. Appl. Biochem. Biotechnol. 2019. [Google Scholar] [CrossRef]
- Nazari Soltan Ahmad, S.; Rashtchizadeh, N.; Argani, H.; Roshangar, L.; Ghorbanihaghjo, A.; Sanajou, D.; Panah, F.; Ashrafi Jigheh, Z.; Dastmalchi, S.; Kalantary-Charvadeh, A. Tangeretin protects renal tubular epithelial cells against experimental cisplatin toxicity. Iran. J. Basic Med. Sci. 2019, 22, 179–186. [Google Scholar] [PubMed]
- Overath, J.M.; Gauer, S.; Obermuller, N.; Schubert, R.; Schafer, R.; Geiger, H.; Baer, P.C. Short-term preconditioning enhances the therapeutic potential of adipose-derived stromal/stem cell-conditioned medium in cisplatin-induced acute kidney injury. Exp. Cell Res. 2016, 342, 175–183. [Google Scholar] [CrossRef]
- Nishihara, K.; Masuda, S.; Shinke, H.; Ozawa, A.; Ichimura, T.; Yonezawa, A.; Nakagawa, S.; Inui, K.; Bonventre, J.V.; Matsubara, K. Urinary chemokine (C-C motif) ligand 2 (monocyte chemotactic protein-1) as a tubular injury marker for early detection of cisplatin-induced nephrotoxicity. Biochem. Pharmacol. 2013, 85, 570–582. [Google Scholar] [CrossRef]
- Oberoi, H.S.; Nukolova, N.V.; Laquer, F.C.; Poluektova, L.Y.; Huang, J.; Alnouti, Y.; Yokohira, M.; Arnold, L.L.; Kabanov, A.V.; Cohen, S.M.; et al. Cisplatin-loaded core cross-linked micelles: Comparative pharmacokinetics, antitumor activity, and toxicity in mice. Int. J. Nanomed. 2012, 7, 2557–2571. [Google Scholar] [CrossRef]
- Sadhukhan, P.; Saha, S.; Dutta, S.; Sil, P.C. Mangiferin Ameliorates Cisplatin Induced Acute Kidney Injury by Upregulating Nrf-2 via the Activation of PI3K and Exhibits Synergistic Anticancer Activity with Cisplatin. Front. Pharmacol. 2018, 9, 638. [Google Scholar] [CrossRef]
- Dupre, T.V.; Doll, M.A.; Shah, P.P.; Sharp, C.N.; Kiefer, A.; Scherzer, M.T.; Saurabh, K.; Saforo, D.; Siow, D.; Casson, L.; et al. Suramin protects from cisplatin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2016, 310, F248–F258. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gui, Y.; Ren, J.; Liu, X.; Feng, Y.; Zeng, Z.; He, W.; Yang, J.; Dai, C. Metformin Protects Against Cisplatin-Induced Tubular Cell Apoptosis and Acute Kidney Injury via AMPKα-regulated Autophagy Induction. Sci. Rep. 2016, 6, 23975. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ravichandran, K.; Ozkok, A.; Wang, Q.; He, Z.; Jani, A.; Ljubanovic, D.; Douglas, I.S.; Edelstein, C.L. The water-soluble triptolide derivative PG490-88 protects against cisplatin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 2014, 349, 518–525. [Google Scholar] [CrossRef]
- Yu, X.; Meng, X.; Xu, M.; Zhang, X.; Zhang, Y.; Ding, G.; Huang, S.; Zhang, A.; Jia, Z. Celastrol ameliorates cisplatin nephrotoxicity by inhibiting NF-kappaB and improving mitochondrial function. EBioMedicine 2018, 36, 266–280. [Google Scholar] [CrossRef]
- Wen, X.; Buckley, B.; McCandlish, E.; Goedken, M.J.; Syed, S.; Pelis, R.; Manautou, J.E.; Aleksunes, L.M. Transgenic expression of the human MRP2 transporter reduces cisplatin accumulation and nephrotoxicity in Mrp2-null mice. Am. J. Pathol. 2014, 184, 1299–1308. [Google Scholar] [CrossRef]
- Miyasato, Y.; Yoshizawa, T.; Sato, Y.; Nakagawa, T.; Miyasato, Y.; Kakizoe, Y.; Kuwabara, T.; Adachi, M.; Ianni, A.; Braun, T.; et al. Sirtuin 7 Deficiency Ameliorates Cisplatin-induced Acute Kidney Injury Through Regulation of the Inflammatory Response. Sci. Rep. 2018, 8, 5927. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Kinoshita, K.; Hino, S.; Yano, T.; Niki, K.; Hirooka, Y.; Kishimoto, K.; Funauchi, M.; Matsumura, I. Signaling Rho-kinase mediates inflammation and apoptosis in T cells and renal tubules in cisplatin nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2015, 308, F899–F909. [Google Scholar] [CrossRef]
- He, Y.X.; Diao, T.T.; Song, S.M.; Wang, C.C.; Wang, Y.; Zhou, C.L.; Bai, Y.B.; Yu, S.S.; Mi, X.; Yang, X.Y.; et al. Wnt4 is significantly upregulated during the early phases of cisplatin-induced acute kidney injury. Sci. Rep. 2018, 8, 10555. [Google Scholar] [CrossRef]
- Kong, M.J.; Bak, S.H.; Han, K.H.; Kim, J.I.; Park, J.W.; Park, K.M. Fragmentation of kidney epithelial cell primary cilia occurs by cisplatin and these cilia fragments are excreted into the urine. Redox Biol. 2019, 20, 38–45. [Google Scholar] [CrossRef]
- Ravichandran, K.; Holditch, S.; Brown, C.N.; Wang, Q.; Ozkok, A.; Weiser-Evans, M.C.; Nemenoff, R.; Miyazaki, M.; Thiessen-Philbrook, H.; Parikh, C.R.; et al. IL-33 deficiency slows cancer growth but does not protect against cisplatin-induced AKI in mice with cancer. Am. J. Physiol. Ren. Physiol. 2018, 314, F356–F366. [Google Scholar] [CrossRef]
- Ma, Q.; Devarajan, S.R.; Devarajan, P. Amelioration of cisplatin-induced acute kidney injury by recombinant neutrophil gelatinase-associated lipocalin. Ren. Fail. 2016, 38, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Soni, H.; Kaminski, D.; Gangaraju, R.; Adebiyi, A. Cisplatin-induced oxidative stress stimulates renal Fas ligand shedding. Ren. Fail. 2018, 40, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Oya, M.; Nangaku, M.; Yasuda, Y.; Komatsu, Y.; Yanagita, M.; Kitagawa, Y.; Kuwano, H.; Nishiyama, H.; Ishioka, C.; et al. Guidelines for treatment of renal injury during cancer chemotherapy 2016. Clin. Exp. Nephrol. 2018, 22, 210–244. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.W.; Yuan, Y.; Chen, J.H.; Lin, W.Q. Kidney disease models: Tools to identify mechanisms and potential therapeutic targets. Zool. Res. 2018, 39, 72–86. [Google Scholar] [PubMed]
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Semin. Nephrol. 2003, 23, 460–464. [Google Scholar] [CrossRef]
- Chen, W.Y.; Hsiao, C.H.; Chen, Y.C.; Ho, C.H.; Wang, J.J.; Hsing, C.H.; Wang, H.Y.; Kan, W.C.; Wu, C.C. Cisplatin Nephrotoxicity Might Have a Sex Difference. An analysis Based on Women’s Sex Hormone Changes. J. Cancer 2017, 8, 3939–3944. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Nguyen, D.X.; Massague, J. Modeling metastasis in the mouse. Curr. Opin. Pharmacol. 2010, 10, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Chiba, S.; Yamashina, T.; Yoshiyama, H.; Jinushi, M. MFG-E8 regulates the immunogenic potential of dendritic cells primed with necrotic cell-mediated inflammatory signals. PLoS ONE 2012, 7, e39607. [Google Scholar] [CrossRef]
- Li, L.; Khan, M.N.; Li, Q.; Chen, X.; Wei, J.; Wang, B.; Cheng, J.W.; Gordon, J.R.; Li, F. G31P, CXCR1/2 inhibitor, with cisplatin inhibits the growth of mice hepatocellular carcinoma and mitigates highdose cisplatin-induced nephrotoxicity. Oncol. Rep. 2015, 33, 751–757. [Google Scholar] [CrossRef]
- Ravichandran, K.; Wang, Q.; Ozkok, A.; Jani, A.; Li, H.; He, Z.; Ljubanovic, D.; Weiser-Evans, M.C.; Nemenoff, R.A.; Edelstein, C.L. CD4 T cell knockout does not protect against kidney injury and worsens cancer. J. Mol. Med. 2016, 94, 443–455. [Google Scholar] [CrossRef]
- Kim, H.; Lee, H.; Lee, G.; Jang, H.; Kim, S.S.; Yoon, H.; Kang, G.H.; Hwang, D.S.; Kim, S.K.; Chung, H.S.; et al. Phospholipase A2 inhibits cisplatin-induced acute kidney injury by modulating regulatory T cells by the CD206 mannose receptor. Kidney Int. 2015, 88, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Trans. Med. 2015, 7, 279ra36. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Shi, Y.; Liu, N.; Xu, L.; Zang, X.; Li, P.; Zhang, J.; Zheng, X.; Qiu, A.; Zhuang, S. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin. Sci. (Lond.) 2018, 132, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Morigi, M.; Rota, C.; Breno, M.; Mele, C.; Noris, M.; Introna, M.; Capelli, C.; Longaretti, L.; Rottoli, D.; et al. Human mesenchymal stromal cells transplanted into mice stimulate renal tubular cells and enhance mitochondrial function. Nat. Commun. 2017, 8, 983. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef]
- Guo, Y.; Ni, J.; Chen, S.; Bai, M.; Lin, J.; Ding, G.; Zhang, Y.; Sun, P.; Jia, Z.; Huang, S.; et al. MicroRNA-709 Mediates Acute Tubular Injury through Effects on Mitochondrial Function. J. Am. Soc. Nephrol. 2018, 29, 449–461. [Google Scholar] [CrossRef]
- Tsushida, K.; Tanabe, K.; Masuda, K.; Tanimura, S.; Miyake, H.; Arata, Y.; Sugiyama, H.; Wada, J. Estrogen-related receptor alpha is essential for maintaining mitochondrial integrity in cisplatin-induced acute kidney injury. Biochem. Biophys. Res. Commun. 2018, 498, 918–924. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Huang, Z.; Li, Q.; Yuan, Y.; Zhang, C.; Wu, L.; Liu, X.; Cao, W.; Guo, H.; Duan, S.; Xu, X.; et al. Renalase attenuates mitochondrial fission in cisplatin-induced acute kidney injury via modulating sirtuin-3. Life Sci. 2019, 222, 78–87. [Google Scholar] [CrossRef]
- Li, Y.; Ye, Z.; Lai, W.; Rao, J.; Huang, W.; Zhang, X.; Yao, Z.; Lou, T. Activation of Sirtuin 3 by Silybin Attenuates Mitochondrial Dysfunction in Cisplatin-induced Acute Kidney Injury. Front. Pharmacol. 2017, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; BHuang, W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Ibrahim, A.; Al-Hizab, F.A.; Abushouk, A.I.; Abdel-Daim, M.M. Nephroprotective Effects of Benzyl Isothiocyanate and Resveratrol Against Cisplatin-Induced Oxidative Stress and Inflammation. Front. Pharmacol. 2018, 9, 1268. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Li, Z.; Li, W.; Liu, Y.; Wang, C.; Lin, H.; Liu, J.; Li, P. Pseudoginsengenin DQ Exhibits Therapeutic Effects in Cisplatin-Induced Acute Kidney Injury via Sirt1/NF-kappaB and Caspase Signaling Pathway without Compromising Its Antitumor Activity in Mice. Molecules 2018, 23, 3038. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.F.; Han, X.Y.; Sun, Y.S.; Zhang, L.X.; Liu, W.; Liu, X.X.; Li, W.; Liu, Y.Y. Kidney Protection Effect of Ginsenoside Re and Its Underlying Mechanisms on Cisplatin-Induced Kidney Injury. Cell. Physiol. Biochem. 2018, 48, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, F.; Cao, X.; Zhai, Z.; GangHuang; Du, X.; Wang, Y.; Zhang, J.; Huang, Y.; Zhao, J.; et al. P2 × 7 receptor blockade protects against cisplatin-induced nephrotoxicity in mice by decreasing the activities of inflammasome components, oxidative stress and caspase-3. Toxicol. Appl. Pharmacol. 2014, 281, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Xu, Y.; Weng, Y.Y.; Fan, X.Y.; Bai, Y.F.; Zheng, X.Y.; Lou, L.J.; Zhang, F. Astilbin ameliorates cisplatin-induced nephrotoxicity through reducing oxidative stress and inflammation. Food Chem. Toxicol. 2018, 114, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Ren, S.; Yan, X.T.; Li, H.P.; Li, W.; Zheng, B.; Wang, Z.; Liu, Y.Y. Improvement of Cisplatin-induced renal dysfunction by Schisandra chinensis stems via anti-inflammation and anti-apoptosis effects. J. Ethnopharmacol. 2018, 217, 228–237. [Google Scholar] [CrossRef]
- Singh, M.P.; AChauhan, K.; Kang, S.C. Morin hydrate ameliorates cisplatin-induced ER stress, inflammation and autophagy in HEK-293 cells and mice kidney via PARP-1 regulation. Int. Immunopharmacol. 2018, 56, 156–167. [Google Scholar] [CrossRef]
- Cao, S.S.; Yan, M.; Hou, Z.Y.; Chen, Y.; Jiang, Y.S.; Fan, X.R.; Fang, P.F.; Zhang, B.K. Danshen modulates Nrf2-mediated signaling pathway in cisplatin-induced renal injury. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Fu, Z.; Zou, Y.; Wen, D.; Ma, H.; Zhou, F.; Chen, Y.; Zhang, M.; Zhang, W. MicroRNA-140-5p attenuated oxidative stress in Cisplatin induced acute kidney injury by activating Nrf2/ARE pathway through a Keap1-independent mechanism. Exp. Cell Res. 2017, 360, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.L.; Wang, Z.; Li, W.; Hou, J.G.; Liu, Y.; Li, X.D.; Li, H.P.; Wang, Y.P. Nephroprotective Effects of Anthocyanin from the Fruits of Panax ginseng (GFA) on Cisplatin-Induced Acute Kidney Injury in Mice. Phytother. Res. 2017, 31, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.N.; Li, Y.Z.; Li, W.; Yan, X.T.; Yang, G.; Zhang, J.; Zhao, L.C.; Yang, L.M. Nephroprotective Effects of Saponins from Leaves of Panax quinquefolius against Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2017, 18, 1407. [Google Scholar] [CrossRef] [PubMed]
- Veceric-Haler, Z.; Erman, A.; Cerar, A.; Motaln, H.; Kolosa, K.; Lah Turnsek, T.; Sodin Semrl, S.; Lakota, K.; Mrak-Poljsak, K.; Skrajnar, S.; et al. Improved Protective Effect of Umbilical Cord Stem Cell Transplantation on Cisplatin-Induced Kidney Injury in Mice Pretreated with Antithymocyte Globulin. Stem Cells Int. 2016, 2016, 3585362. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, T.W.; Park, S.R.; Kim, H.T.; Jung, D.Y.; Ryu, S.Y.; Jung, J.Y. Deletion of NAD(P)H:quinone oxidoreductase 1 represses Mre11-Rad50-Nbs1 complex protein expression in cisplatin-induced nephrotoxicity. Toxicol. Lett. 2016, 243, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Kim, Y.J.; Kim, H.T.; Park, S.R.; Jung, J.Y. beta-Lapachone enhances Mre11-Rad50-Nbs1 complex expression in cisplatin-induced nephrotoxicity. Pharmacol. Rep. 2016, 68, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Tilyek, A.; Chai, C.; Hou, X.; Zhou, B.; Zhang, C.; Cao, Z.; Yu, B. The protective effects of Ribes diacanthum Pall on cisplatin-induced nephrotoxicity in mice. J. Ethnopharmacol. 2016, 178, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.D.; Kumar, J.M.; Sistla, R. Baicalein, a Bioflavonoid, Prevents Cisplatin-Induced Acute Kidney Injury by Up-Regulating Antioxidant Defenses and Down-Regulating the MAPKs and NF-kappaB Pathways. PLoS ONE 2015, 10, e0134139. [Google Scholar] [CrossRef]
- Saifi, M.A.; Sangomla, S.; Khurana, A.; Godugu, C. Protective Effect of Nanoceria on Cisplatin-Induced Nephrotoxicity by Amelioration of Oxidative Stress and Pro-inflammatory Mechanisms. Biol. Trace Elem. Res. 2019, 189, 145–156. [Google Scholar] [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef]
- Valencia, A.; Moran, J. Reactive oxygen species induce different cell death mechanisms in cultured neurons. Free Radic. Biol. Med. 2004, 36, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Meng, X.M.; Ren, G.L.; Gao, L.; Yang, Q.; Li, H.D.; Wu, W.F.; Huang, C.; Zhang, L.; Lv, X.W.; Li, J. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab. Investig. 2018, 98, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.N.; Doll, M.A.; Dupre, T.V.; Shah, P.P.; Subathra, M.; Siow, D.; Arteel, G.E.; Megyesi, J.; Beverly, L.J.; Siskind, L.J. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am. J. Physiol. Ren. Physiol. 2016, 310, F560–F568. [Google Scholar] [CrossRef]
- Sen, Z.; Jie, M.; Jingzhi, Y.; Dongjie, W.; Dongming, Z.; Xiaoguang, C. Total Coumarins from Hydrangea paniculata Protect against Cisplatin-Induced Acute Kidney Damage in Mice by Suppressing Renal Inflammation and Apoptosis. Evid. Based Complement. Altern. Med. 2017, 2017, 5350161. [Google Scholar] [CrossRef]
- Rath, P.C.; Aggarwal, B.B. TNF-induced signaling in apoptosis. J. Clin. Immunol. 1999, 19, 350–364. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Dupre, T.V.; Doll, M.A.; Shah, P.P.; Sharp, C.N.; Siow, D.; Megyesi, J.; Shayman, J.; Bielawska, A.; Bielawski, J.; Beverly, L.J.; et al. Inhibiting glucosylceramide synthase exacerbates cisplatin-induced acute kidney injury. J. Lipid Res. 2017, 58, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kakizoe, Y.; Iwata, Y.; Miyasato, Y.; Mizumoto, T.; Adachi, M.; Izumi, Y.; Kuwabara, T.; Suenaga, N.; Narita, Y.; et al. Doxycycline attenuates cisplatin-induced acute kidney injury through pleiotropic effects. Am. J. Physiol. Ren. Physiol. 2018, 315, F1347–F1357. [Google Scholar] [CrossRef]
- Watanabe, M.; Oe, Y.; Sato, E.; Sekimoto, A.; Sato, H.; Ito, S.; Takahashi, N. Protease-activated receptor 2 exacerbates cisplatin-induced nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2019, 316, F654–F659. [Google Scholar] [CrossRef] [PubMed]
- Soni, H.; Matthews, A.T.; Pallikkuth, S.; Gangaraju, R.; Adebiyi, A. Gamma-secretase inhibitor DAPT mitigates cisplatin-induced acute kidney injury by suppressing Notch1 signaling. J. Cell. Mol. Med. 2019, 23, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hou, J.; Yan, X.; Leng, J.; Li, R.; Zhang, J.; Xing, J.; Chen, C.; Wang, Z.; Li, W. Platycodon grandiflorum Saponins Ameliorate Cisplatin-Induced Acute Nephrotoxicity through the NF-kappaB-Mediated Inflammation and PI3K/Akt/Apoptosis Signaling Pathways. Nutrients 2018, 10, 1328. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, M.; Mou, J.; Zhao, Z.; Yang, J.; Zhu, F.; Pei, G.; Zhu, H.; Wang, Y.; Xu, G.; et al. Pretreatment of Huaiqihuang extractum protects against cisplatin-induced nephrotoxicity. Sci. Rep. 2018, 8, 7333. [Google Scholar] [CrossRef]
- Kim, I.H.; Kwon, M.J.; Jung, J.H.; Nam, T.J. Protein extracted from Porphyra yezoensis prevents cisplatin-induced nephrotoxicity by downregulating the MAPK and NF-kappaB pathways. Int. J. Mol. Med. 2018, 41, 511–520. [Google Scholar]
- Ma, T.; Huang, C.; Xu, Q.; Yang, Y.; Liu, Y.; Meng, X.; Li, J.; Ye, M.; Liang, H. Suppression of BMP-7 by histone deacetylase 2 promoted apoptosis of renal tubular epithelial cells in acute kidney injury. Cell Death Dis. 2017, 8, e3139. [Google Scholar] [CrossRef]
- Huang, Y.C.; Tsai, M.S.; Hsieh, P.C.; Shih, J.H.; Wang, T.S.; Wang, Y.C.; Lin, T.H.; Wang, S.H. Galangin ameliorates cisplatin-induced nephrotoxicity by attenuating oxidative stress, inflammation and cell death in mice through inhibition of ERK and NF-kappaB signaling. Toxicol. Appl. Pharmacol. 2017, 329, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.K.; Kondeti, V.K.; Sharma, I.; Chandel, N.S.; Quaggin, S.E.; Kanwar, Y.S. Beneficial Effects of Myo-Inositol Oxygenase Deficiency in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 1421–1436. [Google Scholar] [CrossRef] [PubMed]
- Potocnjak, I.; Domitrovic, R. Carvacrol attenuates acute kidney injury induced by cisplatin through suppression of ERK and PI3K/Akt activation. Food Chem. Toxicol. 2016, 98, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Huang, C.; Meng, X.; Li, X.; Zhang, Y.; Ji, S.; Li, J.; Ye, M.; Liang, H. A potential adjuvant chemotherapeutics, 18beta-glycyrrhetinic acid, inhibits renal tubular epithelial cells apoptosis via enhancing BMP-7 epigenetically through targeting HDAC2. Sci. Rep. 2016, 6, 25396. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zhang, S.; Su, X.; Qiu, G.; Wu, Z. Protective effects of icariin on cisplatin-induced acute renal injury in mice. Am. J. Transl. Res. 2015, 7, 2105–2114. [Google Scholar] [PubMed]
- Potocnjak, I.; Skoda, M.; Pernjak-Pugel, E.; Persic, M.P.; Domitrovic, R. Oral administration of oleuropein attenuates cisplatin-induced acute renal injury in mice through inhibition of ERK signaling. Mol. Nutr. Food Res. 2016, 60, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kong, L.; Zhang, J.; Yu, G.; Lv, G.; Zhang, F.; Chen, X.; Tian, J.; Fu, F. The pseudoginsenoside F11 ameliorates cisplatin-induced nephrotoxicity without compromising its anti-tumor activity in vivo. Sci. Rep. 2014, 4, 4986. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Liu, H.Z.; Wang, H.B.; Zhong, J.Y.; Yang, C.X.; Zhang, B. Dexmedetomidine protects against cisplatin-induced acute kidney injury in mice through regulating apoptosis and inflammation. Inflamm. Res. 2017, 66, 399–411. [Google Scholar] [CrossRef]
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-kappaB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 2016, 240, 105–113. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.M.; Park, G.; Lee, S.H.; Seo, C.S.; Shin, H.K.; Oh, D.S. Assessing the recovery from prerenal and renal acute kidney injury after treatment with single herbal medicine via activity of the biomarkers HMGB1, NGAL and KIM-1 in kidney proximal tubular cells treated by cisplatin with different doses and exposure times. BMC Complement. Altern. Med. 2017, 17, 544. [Google Scholar] [CrossRef]
- Wei, Z.; He, X.; Kou, J.; Wang, J.; Chen, L.; Yao, M.; Zhou, E.; Fu, Y.; Guo, C.; Yang, Z. Renoprotective mechanisms of morin in cisplatin-induced kidney injury. Int. Immunopharmacol. 2015, 28, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Peyrou, M.; Hanna, P.E.; Cribb, A.E. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol. Sci. 2007, 99, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, K.; Barone, S.; Destefano-Shields, C.; Brooks, M.; Murray-Stewart, T.; Dunworth, M.; Li, W.; Doherty, J.R.; Hall, M.A.; Smith, R.D.; et al. Activation of endoplasmic reticulum stress response by enhanced polyamine catabolism is important in the mediation of cisplatin-induced acute kidney injury. PLoS ONE 2017, 12, e0184570. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef]
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zhen, X.; Zhu, F.; Hu, Z.; Lei, W.; Li, S.; Zha, Y.; Nie, J. Hyperhomocysteinemia Exacerbates Cisplatin-induced Acute Kidney Injury. Int. J. Biol. Sci. 2017, 13, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Pei, L.; Xiao, X.; Wei, Q.; Chen, J.K.; Ding, H.F.; Huang, S.; Fan, G.; Shi, H.; Dong, Z. DNA methylation protects against cisplatin-induced kidney injury by regulating specific genes, including interferon regulatory factor 8. Kidney Int. 2017, 92, 1194–1205. [Google Scholar] [CrossRef]
- Pellegrini, K.L.; Han, T.; Bijol, V.; Saikumar, J.; Craciun, F.L.; Chen, W.W.; Fuscoe, J.C.; Vaidya, V.S. MicroRNA-155 deficient mice experience heightened kidney toxicity when dosed with cisplatin. Toxicol. Sci. 2014, 141, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.A.; Lyon, T.T.; Yin, Y.; Lang, J.E.; Solomon, G.; Annab, L.; Srinivasan, D.G.; Alcorta, D.A.; Barrett, J.C. Induction of apoptosis by c-Fos protein. Mol. Cell. Biol. 1996, 16, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Brines, M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ. 2004, 11 (Suppl. 1), S37–S44. [Google Scholar] [CrossRef]
- Mohamed, H.E.; El-Swefy, S.E.; Mohamed, R.H.; Ghanim, A.M. Effect of erythropoietin therapy on the progression of cisplatin induced renal injury in rats. Exp. Toxicol. Pathol. 2013, 65, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zhuo, L.; Gao, C.; Shi, S.; Wang, N.; Huang, Z.; Li, W.; Hao, L. Erythropoietin protects against cisplatin-induced nephrotoxicity by attenuating endoplasmic reticulum stress-induced apoptosis. J. Nephrol. 2013, 26, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Herzog, C.; Yang, C.; Holmes, A.; Kaushal, G.P. zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am. J. Physiol. Ren. Physiol. 2012, 303, F1239–F1250. [Google Scholar] [CrossRef]
- Gao, L.; Liu, M.M.; Zang, H.M.; Ma, Q.Y.; Yang, Q.; Jiang, L.; Ren, G.L.; Li, H.D.; Wu, W.F.; Wang, J.N.; et al. Restoration of E-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab. Investig. 2018, 98, 911–923. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef]
- Meng, X.M.; Li, H.D.; Wu, W.F.; Ming-Kuen Tang, P.; Ren, G.L.; Gao, L.; Li, X.F.; Yang, Y.; Xu, T.; Ma, T.T.; et al. Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab. Investig. 2018, 98, 79–94. [Google Scholar] [CrossRef]
- Landau, S.I.; Guo, X.; Velazquez, H.; Torres, R.; Olson, E.; Garcia-Milian, R.; Moeckel, G.W.; Desir, G.V.; Safirstein, R. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int. 2019, 95, 797–814. [Google Scholar] [CrossRef]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar] [CrossRef]
- Gobe, G.; Zhang, X.J.; Willgoss, D.A.; Schoch, E.; Hogg, N.A.; Endre, Z.H. Relationship between expression of Bcl-2 genes and growth factors in ischemic acute renal failure in the rat. J. Am. Soc. Nephrol. 2000, 11, 454–467. [Google Scholar] [PubMed]
- Andrade, L.; Vieira, J.M.; Safirstein, R. How cells die counts. Am. J. Kidney Dis. 2000, 36, 662–668. [Google Scholar] [CrossRef]
- Sancho-Martinez, S.M.; Piedrafita, F.J.; Cannata-Andia, J.B.; Lopez-Novoa, J.M.; Lopez-Hernandez, F.J. Necrotic concentrations of cisplatin activate the apoptotic machinery but inhibit effector caspases and interfere with the execution of apoptosis. Toxicol. Sci. 2011, 122, 73–85. [Google Scholar] [CrossRef]
- Dursun, B.; He, Z.; Somerset, H.; Oh, D.J.; Faubel, S.; Edelstein, C.L. Caspases and calpain are independent mediators of cisplatin-induced endothelial cell necrosis. Am. J. Physiol. Ren. Physiol. 2006, 291, F578–F587. [Google Scholar] [CrossRef] [PubMed]
- Yard, B.A.; Daha, M.R.; Kooymans-Couthino, M.; Bruijn, J.A.; Paape, M.E.; Schrama, E.; van Es, L.A.; van der Woude, F.J. IL-1 alpha stimulated TNF alpha production by cultured human proximal tubular epithelial cells. Kidney Int. 1992, 42, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Francis, J.; Majid, D.S. Tumor necrosis factor-alpha induces renal vasoconstriction as well as natriuresis in mice. Am. J. Physiol. Ren. Physiol. 2008, 295, F1836–F1844. [Google Scholar] [CrossRef] [PubMed]
- Privratsky, J.R.; Zhang, J.; Lu, X.; Rudemiller, N.; Wei, Q.; Yu, Y.R.; Gunn, M.D.; Crowley, S.D. Interleukin 1 receptor (IL-1R1) activation exacerbates toxin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2018, 315, F682–F691. [Google Scholar] [CrossRef]
- Liu, Y.; Webb, H.K.; Fukushima, H.; Micheli, J.; Markova, S.; Olson, J.L.; Kroetz, D.L. Attenuation of cisplatin-induced renal injury by inhibition of soluble epoxide hydrolase involves nuclear factor kappaB signaling. J. Pharmacol. Exp. Ther. 2012, 341, 725–734. [Google Scholar] [CrossRef]
- Wang, C.; Dai, H.; Xiong, Z.; Song, Q.; Zou, Z.; Li, M.; Nie, J.; Bai, X.; Chen, Z. Loss of DEPTOR in renal tubules protects against cisplatin-induced acute kidney injury. Cell Death Dis. 2018, 9, 441. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Yoshioka, K.; Tatematsu, S.; Hara, Y.; Minakuchi, H.; Sueyasu, K.; Washida, N.; Tokuyama, H.; Tzukerman, M.; et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J. Biol. Chem. 2010, 285, 13045–13056. [Google Scholar] [CrossRef]
- Li, Z.; Xu, K.; Zhang, N.; Amador, G.; Wang, Y.; Zhao, S.; Li, L.; Qiu, Y.; Wang, Z. Overexpressed SIRT6 attenuates cisplatin-induced acute kidney injury by inhibiting ERK1/2 signaling. Kidney Int. 2018, 93, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am. J. Physiol. Ren. Physiol. 2003, 285, F610–F618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, M.Y.; Seelaender, M.; Castoldi, A.; de Almeida, D.C.; Bacurau, A.V.; Andrade-Oliveira, V.; Enjiu, L.M.; Pisciottano, M.; Hayashida, C.Y.; Hiyane, M.I.; et al. Long-term aerobic exercise protects against cisplatin-induced nephrotoxicity by modulating the expression of IL-6 and HO-1. PLoS ONE 2014, 9, e108543. [Google Scholar] [CrossRef]
- Ribeiro, R.S.; Passos, C.S.; Novaes, A.S.; Maquigussa, E.; Gloria, M.A.; Visona, I.; Ykuta, O.; Oyama, L.M.; Boim, M.A. Precocious obesity predisposes the development of more severe cisplatin-induced acute kidney injury in young adult mice. PLoS ONE 2017, 12, e0174721. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Bolisetty, S.; Agarwal, A. Neutrophils in acute kidney injury: Not neutral any more. Kidney Int. 2009, 75, 674–676. [Google Scholar] [CrossRef]
- Woodfin, A.; Voisin, M.B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.M.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E.; et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011, 12, 761–769. [Google Scholar] [CrossRef]
- Kim, S.C.; Ko, Y.S.; Lee, H.Y.; Kim, M.G.; Jo, S.K.; Cho, W.Y. Blocking junctional adhesion molecule C promotes the recovery of cisplatin-induced acute kidney injury. Korean J. Intern. Med. 2017, 32, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J.; Williams, W.W., Jr.; Colvin, R.B.; Meehan, S.M.; Springer, T.A.; Gutierrez-Ramos, J.C.; Bonventre, J.V. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Investig. 1996, 97, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J.; Meehan, S.M.; Colvin, R.B.; Williams, W.W.; Bonventre, J.V. Protection from toxicant-mediated renal injury in the rat with anti-CD54 antibody. Kidney Int. 1999, 56, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Blumenreich, M.S. The White Blood Cell and Differential Count. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Butterworth: Boston, MA, USA, 1990. [Google Scholar]
- Zhang, J.; Rudemiller, N.P.; Patel, M.B.; Wei, Q.; Karlovich, N.S.; Jeffs, A.D.; Wu, M.; Sparks, M.A.; Privratsky, J.R.; Herrera, M.; et al. Competing Actions of Type 1 Angiotensin II Receptors Expressed on T Lymphocytes and Kidney Epithelium during Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2016, 27, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chien, C.C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774. [Google Scholar] [CrossRef]
- Yang, L.; Brooks, C.R.; Xiao, S.; Sabbisetti, V.; Yeung, M.Y.; Hsiao, L.L.; Ichimura, T.; Kuchroo, V.; Bonventre, J.V. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J. Clin. Investig. 2015, 125, 1620–1636. [Google Scholar] [CrossRef]
- Kinsey, G.R.; Sharma, R.; Okusa, M.D. Regulatory T cells in AKI. J. Am. Soc. Nephrol. 2013, 24, 1720–1726. [Google Scholar] [CrossRef]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef]
- Stremska, M.E.; Jose, S.; Sabapathy, V.; Huang, L.; Bajwa, A.; Kinsey, G.R.; Sharma, P.R.; Mohammad, S.; Rosin, D.L.; Okusa, M.D.; et al. IL233, A Novel IL-2 and IL-33 Hybrid Cytokine, Ameliorates Renal Injury. J. Am. Soc. Nephrol. 2017, 28, 2681–2693. [Google Scholar] [CrossRef]
- Faubel, S.; Ljubanovic, D.; Reznikov, L.; Somerset, H.; Dinarello, C.A.; Edelstein, C.L. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int. 2004, 66, 2202–2213. [Google Scholar] [CrossRef]
- Belliere, J.; Casemayou, A.; Ducasse, L.; Zakaroff-Girard, A.; Martins, F.; Iacovoni, J.S.; Guilbeau-Frugier, C.; Buffin-Meyer, B.; Pipy, B.; Chauveau, D.; et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J. Am. Soc. Nephrol. 2015, 26, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Tadagavadi, R.K.; Reeves, W.B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.H.; Oh, D.J.; Dursun, B.; He, Z.; Hoke, T.S.; Faubel, S.; Edelstein, C.L. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J. Pharmacol. Exp. Ther. 2008, 324, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.D. Mononuclear phagocyte depletion strategies in models of acute kidney disease: What are they trying to tell us? Kidney Int. 2012, 82, 835–837. [Google Scholar] [CrossRef][Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Silva, M.; Cenedeze, M.A.; Perandini, L.A.; Felizardo, R.J.F.; Watanabe, I.K.M.; Agudelo, J.S.H.; Castoldi, A.; Goncalves, G.M.; Origassa, C.S.T.; Semedo, P.; et al. TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin. Sci. (Lond.) 2018, 132, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jia, H.; Zhang, B.; Wang, J.; Ji, C.; Zhu, X.; Yan, Y.; Yin, L.; Yu, J.; Qian, H.; et al. Pre-incubation with hucMSC-exosomes prevents cisplatin-induced nephrotoxicity by activating autophagy. Stem Cell Res. Ther. 2017, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Nie, J.; Zheng, Z.L.; Zhao, J.; Wu, L.M.; Zhu, Y.; Su, Z.Q.; Zheng, G.J.; Feng, B. Renoprotective effect of scutellarin on cisplatin-induced renal injury in mice: Impact on inflammation, apoptosis, and autophagy. Biomed. Pharmacother. 2019, 112, 108647. [Google Scholar] [CrossRef]
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 322. [Google Scholar] [CrossRef]
- Jia, H.; Liu, W.; Zhang, B.; Wang, J.; Wu, P.; Tandra, N.; Liang, Z.; Ji, C.; Yin, L.; Hu, X.; et al. HucMSC exosomes-delivered 14-3-3zeta enhanced autophagy via modulation of ATG16L in preventing cisplatin-induced acute kidney injury. Am. J. Transl. Res. 2018, 10, 101–113. [Google Scholar] [PubMed]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xu, X.; Dong, Z. PRKCD/PKCdelta contributes to nephrotoxicity during cisplatin chemotherapy by suppressing autophagy. Autophagy 2017, 13, 631–632. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, D.; Pan, J.; Xiang, X.; Liu, Y.; Dong, G.; Livingston, M.J.; Chen, J.K.; Yin, X.M.; Dong, Z. Protein Kinase Cdelta Suppresses Autophagy to Induce Kidney Cell Apoptosis in Cisplatin Nephrotoxicity. J. Am. Soc. Nephrol. 2017, 28, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-o, M.; et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010, 24, 3438–3450. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef]
- Seo, M.Y.; Yang, J.; Lee, J.Y.; Kim, K.; Kim, S.C.; Chang, H.; Won, N.H.; Kim, M.G.; Jo, S.K.; Cho, W.; et al. Renal Klotho expression in patients with acute kidney injury is associated with the severity of the injury. Korean J. Intern. Med. 2015, 30, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Case, J.; Khan, S.; Khalid, R.; Khan, A. Epidemiology of acute kidney injury in the intensive care unit. Crit. Care Res. Pract. 2013, 2013, 479730. [Google Scholar] [CrossRef] [PubMed]
- Silver, S.A.; Harel, Z.; McArthur, E.; Nash, D.M.; Acedillo, R.; Kitchlu, A.; Garg, A.X.; Chertow, G.M.; Bell, C.M.; Wald, R. Causes of Death after a Hospitalization with AKI. J. Am. Soc. Nephrol. 2018, 29, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Bucaloiu, I.D.; Kirchner, H.L.; Norfolk, E.R.; Hartle, J.E., 2nd; Perkins, R.M. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012, 81, 477–485. [Google Scholar] [CrossRef]
- Hsu, C.Y. Yes, AKI truly leads to CKD. J. Am. Soc. Nephrol. 2012, 23, 967–969. [Google Scholar] [CrossRef]
- Cerda, J.; Lameire, N.; Eggers, P.; Pannu, N.; Uchino, S.; Wang, H.; Bagga, A.; Levin, A. Epidemiology of acute kidney injury. Clin. J. Am. Soc. Nephrol. 2008, 3, 881–886. [Google Scholar] [CrossRef]
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Negishi, K.; Sugaya, T.; Nangaku, M.; Noiri, E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016, 89, 374–385. [Google Scholar] [CrossRef]
- Torres, R.; Velazquez, H.; Chang, J.J.; Levene, M.J.; Moeckel, G.; Desir, G.V.; Safirstein, R. Three-Dimensional Morphology by Multiphoton Microscopy with Clearing in a Model of Cisplatin-Induced CKD. J. Am. Soc. Nephrol. 2016, 27, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.; Romay, C.; Borrego, A.; Hernandez, F.; Merino, N.; Zamora, Z.; Rojas, E. Lipid peroxides and antioxidant enzymes in cisplatin-induced chronic nephrotoxicity in rats. Mediat. Inflamm. 2005, 2005, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Dalton, R.N. Serum creatinine and glomerular filtration rate: Perception and reality. Clin. Chem. 2010, 56, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Lacquaniti , A.; Coppolino, G.; Donato, V.; Campo, S.; Fazio, M.R.; Nicocia, G.; Buemi, M. Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, O.M. Phosphate and Klotho. Kidney Int. Suppl. 2011, 121, S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Razzaque, M.S. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010, 24, 3562–3571. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; McMillan, K.L.; Wu, J.; Gillings, N.; Flores, B.; Moe, O.W.; Hu, M.C. Cisplatin nephrotoxicity as a model of chronic kidney disease. Lab. Investig. 2018, 98, 1105–1121. [Google Scholar] [CrossRef]
- Edelstein, C.L. Biomarkers in Acute Kidney Injury. In Biomarkers of Kidney Disease; Edelstein, C.L., Ed.; Elsevier: London, UK, 2017; pp. 241–303. [Google Scholar]
- Zhou, H.; Cheruvanky, A.; Hu, X.; Matsumoto, T.; Hiramatsu, N.; Cho, M.E.; Berger, A.; Leelahavanichkul, A.; Doi, K.; Chawla, L.S.; et al. Urinary exosomal transcription factors, a new class of biomarkers for renal disease. Kidney Int. 2008, 74, 613–621. [Google Scholar] [CrossRef]
- Sonoda, H.; Oshikawa-Hori, S.; Ikeda, M. An Early Decrease in Release of Aquaporin-2 in Urinary Extracellular Vesicles After Cisplatin Treatment in Rats. Cells 2019, 8, 139. [Google Scholar] [CrossRef]
- Zhou, H.; Pisitkun, T.; Aponte, A.; Yuen, P.S.; Hoffert, J.D.; Yasuda, H.; Hu, X.; Chawla, L.; Shen, R.F.; Knepper, M.A.; et al. Exosomal Fetuin-A identified by proteomics: A novel urinary biomarker for detecting acute kidney injury. Kidney Int. 2006, 70, 1847–1857. [Google Scholar] [CrossRef]
- Bulacio, R.P.; Anzai, N.; Ouchi, M.; Torres, A.M. Organic Anion Transporter 5 (Oat5) Urinary Excretion Is a Specific Biomarker of Kidney Injury: Evaluation of Urinary Excretion of Exosomal Oat5 after N-Acetylcysteine Prevention of Cisplatin Induced Nephrotoxicity. Chem. Res. Toxicol. 2015, 28, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.; Segarra, A.B.; Montoro-Molina, S.; de Gracia, M.D.; Osuna, A.; O’Valle, F.; Gomez-Guzman, M.; Vargas, F.; Wangensteen, R. Glutamyl aminopeptidase in microvesicular and exosomal fractions of urine is related with renal dysfunction in cisplatin-treated rats. PLoS ONE 2017, 12, e0175462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, H.; Xu, W.; Wang, B.; Wu, H.; Tao, Y.; Zhang, B.; Wang, M.; Mao, F.; Yan, Y.; et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res. Ther. 2013, 4, 34. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type | Model | |||||
|---|---|---|---|---|---|---|
| Low Dose, Short-Term | Low Dose, Long-Term | High Dose, Short-Term | ||||
| Cisplatin dosage | 5–15 mg/kg | 5–15 mg/kg | 20–30 mg/kg | |||
| Study duration | <7 Days | >7 Days | <7 Days | |||
| Routine AKI biomarker | BUN [33] (40–60 mg/dL) SCr [34] (0.1–0.5 mg/dL) | ** Cystatin-C [35] (1–1.3 µg/mL) KIM-1 [36] (10–12 fold increase) NGAL [37] (0.3–1.3 µg/mL) CCL2 [38] (0.5–10 fold increase mRNA) | BUN [39] (30–150 mg/dL) SCr [40] (0.5–3 mg/dL) | KIM-1 [41] (0.05–0.1 mg/mL) NGAL [41] (20–40 mg/mL) | BUN [42] (50–200 mg/dL) SCr [43] (0.5–3 mg/dL) | * Cystatin-C [44] (14–18 ng/mL) KIM-1 [45] 50–100 fold increased) NGAL/SCr [46] (6–10 µg/mg) CCL2 [46] (10–30 fold increase mRNA) IL-18 [47] (20–25 pg/mg) |
| Timing of detection | >48 h | >72 h | >48 h | >72 h | >48 h | >72 h |
| Novel AKI biomarker | miRNAs [5] (10 fold increase): miR-130a, 151–3p, 218, 320, 680, 138, 152, 221, 328, 685 | Urinary Wnt4 [48] (0.1–0.7 µg/mg) Urinary ARL13B [49] (15–45 fold increase) | N/A | Renal IL-33 [50] (0.3–1.0 µg/mg) | N/A | Urinary NGAL [51] (40–120 units/g SCr) Urinary FasL [52] (45–150 pg/mL) Renal IL-33 [50] (0.3–1.0 µg/mg) |
| Timing of detection | <24 h | >24 h | N/A | >24 h | N/A | >24 h |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. https://doi.org/10.3390/ijms20123011
Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. International Journal of Molecular Sciences. 2019; 20(12):3011. https://doi.org/10.3390/ijms20123011
Chicago/Turabian StyleHolditch, Sara J., Carolyn N. Brown, Andrew M. Lombardi, Khoa N. Nguyen, and Charles L. Edelstein. 2019. "Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury" International Journal of Molecular Sciences 20, no. 12: 3011. https://doi.org/10.3390/ijms20123011
APA StyleHolditch, S. J., Brown, C. N., Lombardi, A. M., Nguyen, K. N., & Edelstein, C. L. (2019). Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. International Journal of Molecular Sciences, 20(12), 3011. https://doi.org/10.3390/ijms20123011