Potential Mechanisms of T Cell-Mediated and Eosinophil-Independent Bronchial Hyperresponsiveness


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Th2 Cell-Mediated Eosinophilic Asthma
3. Pharmacological Therapies for Th2 Cell-Mediated Asthma
4. Th1 and Th17 Cell-Mediated Neutrophilic Asthma
5. Th9 Cells
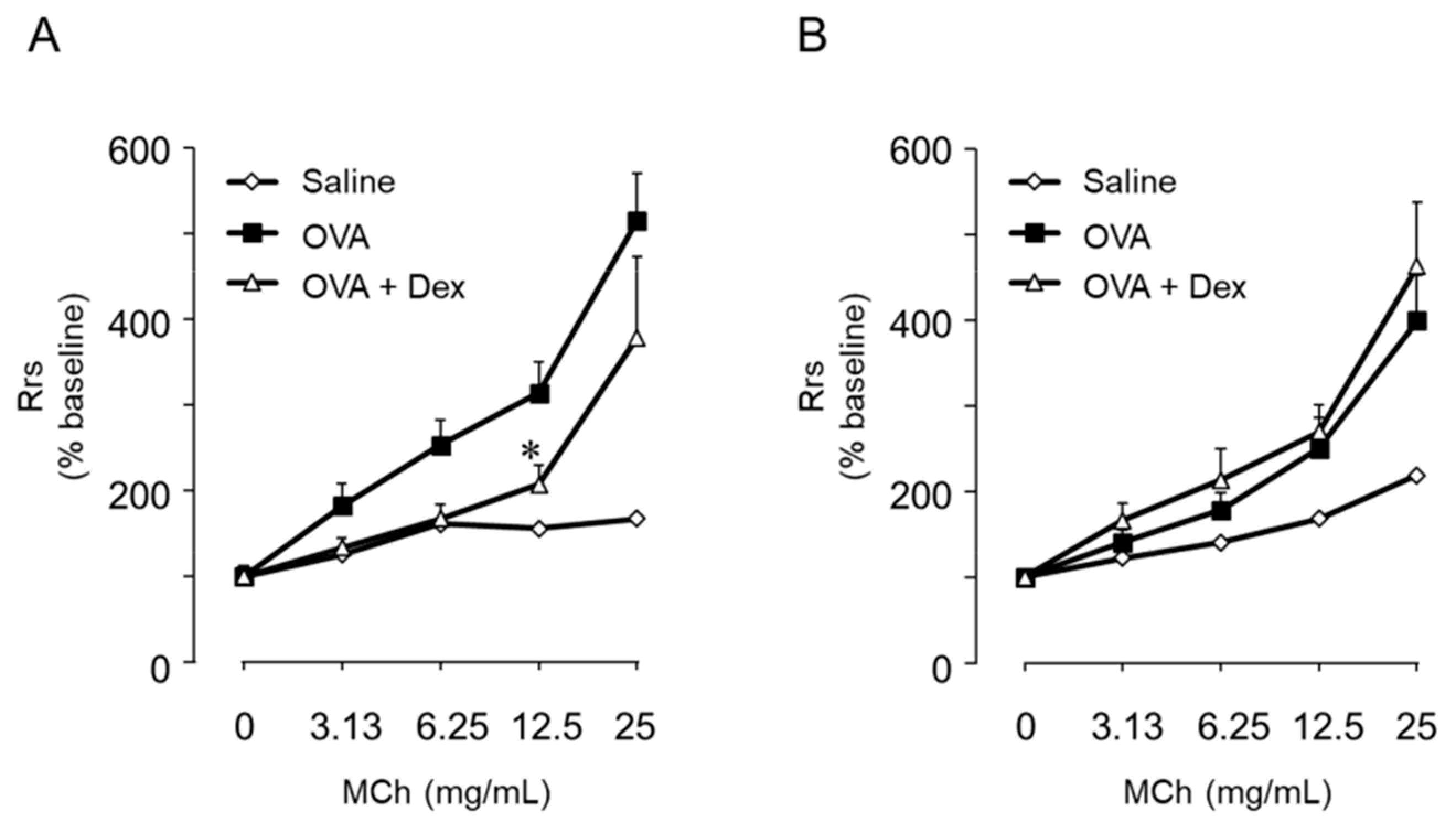
6. Th9 Cells Induce BHR Accompanied by but Not Dependent on Eosinophil Infiltration
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef]
- Wenzel, S.E. Asthma: Defining of the persistent adult phenotypes. Lancet 2006, 368, 804–813. [Google Scholar] [CrossRef]
- Lötvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F., Jr.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 2011, 127, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Haldar, P.; Pavord, I.D.; Shaw, D.E.; Berry, M.A.; Thomas, M.; Brightling, C.E.; Wardlaw, A.J.; Green, R.H. Cluster analysis and clinical asthma phenotypes. Am. J. Respir. Crit. Care Med. 2008, 178, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A.; Bachert, C.; Cingi, C.; Dykewicz, M.S.; Hellings, P.W.; Naclerio, R.M.; Schleimer, R.P.; Ledford, D. Endotypes and phenotypes of chronic rhinosinusitis: A PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 2013, 131, 1479–1490. [Google Scholar] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Robinson, D.S.; Hamid, Q.; Ying, S.; Tsicopoulos, A.; Barkans, J.; Bentley, A.M.; Corrigan, C.; Durham, S.R.; Kay, A.B. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N. Engl. J. Med. 1992, 326, 298–304. [Google Scholar] [CrossRef]
- Van Reijsen, F.C.; Bruijnzeel-Koomen, C.A.; Kalthoff, F.S.; Maggi, E.; Romagnani, S.; Westland, J.K.; Mudde, G.C. Skin-derived aeroallergen-specific T-cell clones of Th2 phenotype in patients with atopic dermatitis. J. Allergy Clin. Immunol. 1992, 90, 184–193. [Google Scholar] [CrossRef]
- Wills-Karp, M. Immunological basis of antigen-induced airways hyperresponsiveness. Annu. Rev. Immunol. 1999, 17, 255–281. [Google Scholar] [CrossRef]
- Kay, A.B. Asthma and inflammation. J. Allergy Clin. Immunol. 1991, 87, 893–910. [Google Scholar] [CrossRef]
- Bochner, B.S.; Undem, B.J.; Lichtenstein, L.M. Immunological aspects of allergic asthma. Annu. Rev. Immunol. 1994, 12, 295–335. [Google Scholar] [CrossRef] [PubMed]
- Snapper, C.M.; Finkelman, F.D.; Paul, W.E. Differential regulation of IgG1 and IgE synthesis by interleukin 4. J. Exp. Med. 1988, 167, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Sellge, G.; Lorentz, A.; Sebald, W.; Raab, R.; Manns, M.P. IL-4 enhances proliferation and mediator release in mature human mast cells. Proc. Natl. Acad. Sci. USA 1999, 96, 8080–8085. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Paul, W.E.; Davis, M.M.; Fazekas de St Groth, B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J. Exp. Med. 1992, 176, 1091–1098. [Google Scholar] [CrossRef]
- Hsieh, C.S.; Heimberger, A.B.; Gold, J.S.; O’Garra, A.K.; Murphy, M. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an αβ T-cell-receptor transgenic system. Proc. Natl. Acad. Sci. USA 1992, 89, 6065–6069. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L.; Weinberg, A.D.; English, M.; Huston, G. IL-4 directs the development of Th2-like helper effectors. J. Immunol. 1990, 145, 3796–3808. [Google Scholar]
- Brusselle, G.G.; Kips, J.C.; Tavernier, J.H.; van der Heyden, J.G.; Cuvelier, C.A.; Pauwels, R.A.; Bluethmann, H. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin. Exp. Allergy. 1994, 24, 73–80. [Google Scholar] [CrossRef]
- Emson, C.L.; Bell, S.E.; Jones, A.; Wisden, W.; McKenzie, A.N. Interleukin (IL)-4-independent induction of immunoglobulin (Ig)E, and perturbation of T cell development in transgenic mice expressing IL-13. J. Exp. Med. 1998, 188, 399–404. [Google Scholar] [CrossRef]
- Wills-Karp, M.; Luyimbazi, J.; Xu, X.; Schofield, B.; Neben, T.Y.; Karp, C.L.; Donaldson, D.D. Interleukin-13: Central mediator of allergic asthma. Science 1998, 282, 2258–2261. [Google Scholar] [CrossRef]
- Grünig, G.; Warnock, M.; Wakil, A.E.; Venkayya, R.; Brombacher, F.; Rennick, D.M.; Sheppard, D.; Mohrs, M.; Donaldson, D.D.; Locksley, R.M.; et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 1998, 282, 2261–2263. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Homer, R.J.; Wang, Z.; Chen, Q.; Geba, G.P.; Wang, J.; Zhang, Y.; Elias, J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Investig. 1999, 103, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.C.; McKenzie, A.N.; Koskinen, A.M.; Yang, M.; Mattes, J.; Foster, P.S. Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J. Immunol. 2000, 165, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Suresh, V.; Mih, J.D.; George, S.C. Measurement of IL-13–Induced iNOS-Derived Gas Phase Nitric Oxide in Human Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2007, 37, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Chibana, K.; Trudeau, J.B.; Mustovich, A.T.; Hu, H.; Zhao, J.; Balzar, S.; Chu, H.W.; Wenzel, S.E. IL-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synthase as compared with effects on arginases in human primary bronchial epithelial cells. Clin. Exp. Allergy 2008, 38, 936–946. [Google Scholar] [CrossRef]
- Walter, D.M.; McIntire, J.J.; Berry, G.; McKenzie, A.N.; Donaldson, D.D.; DeKruyff, R.H.; Umetsu, D.T. Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J. Immunol. 2001, 167, 4668–4675. [Google Scholar] [CrossRef]
- Chiba, Y.; Nakazawa, S.; Todoroki, M.; Shinozaki, K.; Sakai, H.; Misawa, M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am. J. Respir. Cell Mol. Biol. 2009, 40, 159–167. [Google Scholar] [CrossRef]
- Kinashi, T.; Harada, N.; Severinson, E.; Tanabe, T.; Sideras, P.; Konishi, M.; Azuma, C.; Tominaga, A.; Bergstedt-Lindqvist, S.; Takahashi, M.; et al. Cloning of complementary DNA encoding T-cell replacing factor and identity with B-cell growth factor II. Nature 1986, 324, 70–73. [Google Scholar] [CrossRef]
- Hamelmann, E.; Cieslewicz, G.; Schwarze, J.; Ishizuka, T.; Joetham, A.; Heusser, C.; Gelfand, E.W. Anti-IL-5 but not anti-IgE prevents airway inflammation and airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 1999, 160, 934–941. [Google Scholar] [CrossRef]
- Kumar, R.K.; Herbert, C.; Webb, D.C.; Li, L.; Foster, P.S. Effects of anticytokine therapy in a mouse model of chronic asthma. Am. J. Respir. Crit. Care Med. 2004, 170, 1043–1048. [Google Scholar] [CrossRef]
- Leckie, M.J.; ten Brinke, A.; Khan, J.; Diamant, Z.; O’Connor, B.J.; Walls, C.M.; Mathur, A.K.; Cowley, H.C.; Chung, K.F.; Djukanovic, R.; et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 2000, 356, 2144–2148. [Google Scholar] [CrossRef]
- Bousquet, J.; Chanez, P.; Lacoste, J.Y.; Barneon, G.; Ghavanian, N.; Enander, I.; Venge, P.; Ahlstedt, S.; Simony-Lafontaine, J.; Godard, P.; et al. Eosinophilic inflammation in asthma. N. Engl. J. Med. 1990, 323, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Taille, C.; Laveneziana, P.; Bourdin, A.; Chanez, P.; Humbert, M. Anti-interleukin-5 therapy in severe asthma. Eur. Respir. Rev. 2013, 22, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Price, D.B.; Rigazio, A.; Campbell, J.D.; Bleecker, E.R.; Corrigan, C.J.; Thomas, M.; Wenzel, S.E.; Wilson, A.M.; Small, M.B.; Gopalan, G.; et al. Blood eosinophil count and prospective annual asthma disease burden: A UK cohort study. Lancet Respir. Med. 2015, 3, 849–858. [Google Scholar] [CrossRef]
- McBrien, C.N.; Menzies-Gow, A. The Biology of Eosinophils and Their Role in Asthma. Front. Med. 2017, 4, 93. [Google Scholar]
- Humbles, A.A.; Lloyd, C.M.; McMillan, S.J.; Friend, D.S.; Xanthou, G.; McKenna, E.E.; Ghiran, S.; Gerard, N.P.; Yu, C.; Orkin, S.H.; et al. A critical role for eosinophils in allergic airways remodeling. Science 2004, 305, 1776–1779. [Google Scholar] [CrossRef]
- Lee, J.J.; Dimina, D.; Macias, M.P.; Ochkur, S.I.; McGarry, M.P.; O’Neill, K.R.; Protheroe, C.; Pero, R.; Nguyen, T.; Cormier, S.A.; et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science 2004, 305, 1773–1776. [Google Scholar] [CrossRef]
- Saeki, M.; Kaminuma, O.; Nishimura, T.; Kitamura, N.; Mori, A.; Hiroi, T. Th9 cells elicit eosinophil-independent bronchial hyperresponsiveness in mice. Allergol. Int. 2016, 65S, 24–29. [Google Scholar] [CrossRef]
- Walsh, E.R.; Sahu, N.; Kearley, J.; Benjamin, E.; Kang, B.H.; Humbles, A.; August, A. Strain-specific requirement for eosinophils in the recruitment of T cells to the lung during the development of allergic asthma. J. Exp. Med. 2008, 205, 1285–1292. [Google Scholar] [CrossRef]
- 2018 GINA Report, Global Strategy for Asthma Management and Prevention. Available online: https://ginasthma.org/wp-content/uploads/2018/04/wms-GINA-2018-report-V1.3-002.pdf (accessed on 25 May 2019).
- Doi, S.; Gemou-Engesaeth, V.; Kay, A.B.; Corrigan, C.J. Polymerase chain reaction quantification of cytokine messenger RNA expression in peripheral blood mononuclear cells of patients with acute exacerbations of asthma: Effect of glucocorticoid therapy. Clin. Exp. Allergy 1994, 24, 854–867. [Google Scholar] [CrossRef]
- Gemou-Engesaeth, V.; Bush, A.; Kay, A.B.; Hamid, Q.; Corrigan, C.J. Inhaled Glucocorticoid Therapy of Childhood Asthma Is Associated with Reduced Peripheral Blood T Cell Activation and ‘Th2-Type’ Cytokine mRNA Expression. Pediatrics 1997, 99, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Gemou-Engesaeth, V.; Fagerhol, M.K.; Toda, M.; Hamid, Q.; Halvorsen, S.; Groegaard, J.B.; Corrigan, C.J. Expression of activation markers and cytokine mRNA by peripheral blood CD4 and CD8 T cells in atopic and nonatopic childhood asthma: Effect of inhaled glucocorticoid therapy. Pediatrics 2002, 109, E24. [Google Scholar] [CrossRef] [PubMed]
- Wallin, A.; Sandström, T.; Cioppa, G.D.; Holgate, S.; Wilson, S. The effects of regular inhaled formoterol and budesonide on preformed Th-2 cytokines in mild asthmatics. Respir. Med. 2002, 96, 1021–1025. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barrat, F.J.; Cua, D.J.; Boonstra, A.; Richards, D.F.; Crain, C.; Savelkoul, H.F.; de Waal-Malefyt, R.; Coffman, R.L.; Hawrylowicz, C.M.; O’Garra, A. In vitro generation of interleukin 10-producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 2002, 195, 603–616. [Google Scholar] [CrossRef] [PubMed]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef] [PubMed]
- Milgrom, H.; Berger, W.; Nayak, A.; Gupta, N.; Pollard, S.; McAlary, M.; Taylor, A.F.; Rohane, P. Treatment of childhood asthma with anti-immunoglobulin E antibody (omalizumab). Pediatrics 2001, 108, E36. [Google Scholar] [CrossRef] [PubMed]
- Buh, R.; Solèr, M.; Matz, J.; Townley, R.; O’Brien, J.; Noga, O.; Champain, K.; Fox, H.; Thirlwell, J.; Della Cioppa, G. Omalizumab provides long-term control in patients with moderate-to-severe allergic asthma. Eur. Respir. J. 2002, 20, 73–78. [Google Scholar]
- Bousquet, J.; Cabrera, P.; Berkman, N.; Buhl, R.; Holgate, S.; Wenzel, S.; Fox, H.; Hedgecock, S.; Blogg, M.; Cioppa, G.D. The effect of treatment with omalizumab, an anti-IgE antibody, on asthma exacerbations and emergency medical visits in patients with severe persistent asthma. Allergy 2005, 60, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Djukanovic, R.; Casale, T.; Bousquet, J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases: An update on anti-inflammatory activity and clinical efficacy. Clin. Exp. Allergy 2005, 35, 408–416. [Google Scholar] [CrossRef]
- Dahlén, S.E.; Hedqvist, P.; Hammarström, S.; Samuelsson, B. Leukotrienes are potent constrictors of human bronchi. Nature 1980, 288, 484–486. [Google Scholar] [CrossRef]
- Dahlén, S.E.; Hansson, G.; Hedqvist, P.; Björck, T.; Granström, E.; Dahlén, B. Allergen challenge of lung tissue from asthmatics elicits bronchial contraction that correlates with the release of leukotrienes C4, D4, and E4. Proc. Natl. Acad. Sci. USA 1983, 80, 1712–1716. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Henderson, W.R., Jr. Leukotrienes. N. Engl. J. Med. 2007, 357, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.W.; Muccitelli, R.M.; Tucker, S.S.; Vickery-Clark, L.M.; Wilson, K.A.; Gleason, J.G.; Hall, R.F.; Wasserman, M.A.; Torphy, T.J. Pharmacologic profile of SK&F 104353: A novel, potent and selective peptidoleukotriene receptor antagonist in guinea pig and human airways. J. Pharmacol. Exp. Ther. 1987, 243, 474–481. [Google Scholar] [PubMed]
- Watanabe-Kohno, S.; Yasui, K.; Nabe, T.; Yamamura, H.; Horiba, M.; Ohata, K. Significant role of peptide leukotrienes (p-LTs) in the antigen-induced contractions of human and guinea pig lung parenchymas and bronchi or tracheas in vitro. Jpn. J. Pharmacol. 1992, 60, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kohrogi, H.; Honda, I.; Kawano, O.; Sugimoto, M.; Araki, S.; Ando, M.A. Novel leukotriene antagonist, ONO-1078, inhibits and reverses human bronchial contraction induced by leukotrienes C4 and D4 and antigen in vitro. Am. Rev. Respir. Dis. 1992, 146, 923–929. [Google Scholar] [CrossRef]
- Knorr, B.; Matz, J.; Bernstein, J.A.; Nguyen, H.; Seidenberg, B.C.; Reiss, T.F.; Becker, A. Montelukast for chronic asthma in 6- to 14-year-old children: A randomized, double-blind trial. Pediatric Montelukast Study Group. JAMA 1998, 279, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Reiss, T.F.; Chervinsky, P.; Dockhorn, R.J.; Shingo, S.; Seidenberg, B.; Edwards, T.B. Montelukast, a once-daily leukotriene receptor antagonist, in the treatment of chronic asthma: A multicenter, randomized, double-blind trial. Montelukast Clinical Research Study Group. Arch. Intern. Med. 1998, 158, 1213–1220. [Google Scholar] [CrossRef]
- Israel, E.; Cohn, J.; Dube, L.; Drazen, J.M. Effect of treatment with zileuton, a 5-lipoxygenase inhibitor, in patients with asthma. A randomized controlled trial. Zileuton Clinical Trial Group. JAMA 1996, 275, 931–936. [Google Scholar] [CrossRef]
- Nelson, H.; Kemp, J.; Berger, W.; Corren, J.; Casale, T.; Dube, L.; Walton-Bowen, K.; LaVallee, N.; Stepanians, M. Efficacy of zileuton controlled-release tablets administered twice daily in the treatment of moderate persistent asthma: A 3-month randomized controlled study. Ann. Allergy Asthma Immunol. 2007, 99, 178–184. [Google Scholar] [CrossRef]
- Shiota, Y.; Arikita, H.; Horita, N.; Hiyama, J.; Ono, T.; Yamakido, M. Intracellular IL-5 and T-lymphocyte subsets in atopic and nonatopic bronchial asthma. J. Allergy Clin. Immunol. 2002, 109, 294–298. [Google Scholar] [CrossRef]
- Hamid, Q.; Azzawi, M.; Ying, S.; Moqbel, R.; Wardlaw, A.J.; Corrigan, C.J.; Bradley, B.; Durham, S.R.; Collins, J.V.; Jeffery, P.K. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J. Clin. Investig. 1991, 87, 1541–1546. [Google Scholar] [CrossRef]
- Truyen, E.; Coteur, L.; Dilissen, E.; Overbergh, L.; Dupont, L.J.; Ceuppens, J.L.; Bullens, D.M. Evaluation of airway inflammation by quantitative Th1/Th2 cytokine mRNA measurement in sputum of asthma patients. Thorax 2006, 61, 202–208. [Google Scholar] [CrossRef]
- Walker, C.; Bode, E.; Boer, L.; Hansel, T.T.; Blaser, K.; Virchow, J.C., Jr. Allergic and nonallergic asthmatics have distinct patterns of T-cell activation and cytokine production in peripheral blood and bronchoalveolar lavage. Am. Rev. Respir. Dis. 1992, 146, 109–115. [Google Scholar] [CrossRef]
- Walker, C.; Bauer, W.; Braun, R.K.; Menz, G.; Braun, P.; Schwarz, F.; Hansel, T.T.; Villiger, B. Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am. J. Respir. Crit. Care Med. 1994, 150, 1038–1048. [Google Scholar] [CrossRef]
- Nagai, H.; Yamaguchi, S.; Inagaki, N.; Tsuruoka, N.; Hitoshi, Y.; Takatsu, K. Effect of anti-IL-5 monoclonal antibody on allergic bronchial eosinophilia and airway hyperresponsiveness in mice. Life Sci. 1993, PL243–PL247. [Google Scholar] [CrossRef]
- Kung, T.T.; Stelts, D.M.; Zurcher, J.A.; Adams, G.K., 3rd; Egan, R.W.; Kreutner, W.; Watnick, A.S.; Jones, H.; Chapman, R.W. Involvement of IL-5 in a murine model of allergic pulmonary inflammation: Prophylactic and therapeutic effect of an anti-IL-5 antibody. Am. J. Respir. Cell Mol. Biol. 1995, 13, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Flood-Page, P.; Swenson, C.; Faiferman, I.; Matthews, J.; Williams, M.; Brannick, L.; Robinson, D.; Wenzel, S.; Busse, W.; Hansel, T.T.; et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.; Chupp, G.; Bardin, P.; Bourdin, A.; Garcia, G.; Hartley, B.; Yancey, S.; Humbert, M. The role of mepolizumab in atopic and nonatopic severe asthma with persistent eosinophilia. Eur. Respir. J. 2014, 44, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Pizzichini, M.M.; Kjarsgaard, M.; Inman, M.D.; Efthimiadis, A.; Pizzichini, E.; Hargreave, F.E.; O’Byrne, P.M. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N. Engl. J. Med. 2009, 360, 985–993. [Google Scholar] [CrossRef]
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.N.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): A multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659. [Google Scholar] [CrossRef]
- Bel, E.H.; Wenzel, S.E.; Thompson, P.J.; Prazma, C.M.; Keene, O.N.; Yancey, S.W.; Ortega, H.G.; Pavord, I.D.; SIRIUS Investigators. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.Y.; Crocker, I.C.; Koenig, G.; Romero, F.A.; Townley, R.G. Anti-interleukin-4 inhibits immunoglobulin E production in a murine model of atopic asthma. J. Asthma 1997, 34, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Borish, L.C.; Nelson, H.S.; Lanz, M.J.; Claussen, L.; Whitmore, J.B.; Agosti, J.M.; Garrison, L. Interleukin-4 receptor in moderate atopic asthma. A phase I/II randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 1999, 160, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A. Therapies for allergic inflammation: Refining strategies to induce tolerance. Nat. Med. 2012, 18, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.K.; Blackburn, M.N.; Brigham-Burke, M.; Dede, K.; Al-Mahdi, N.; Zia-Amirhosseini, P.; Cook, R.M. Preclinical efficacy and safety of pascolizumab (SB 240683): A humanized anti-interleukin-4 antibody with therapeutic potential in asthma. Clin. Exp. Immunol. 2002, 130, 93–100. [Google Scholar] [CrossRef]
- Tomlinson, K.L.; Davies, G.C.; Sutton, D.J.; Palframan, R.T. Neutralisation of Interleukin-13 in Mice Prevents Airway Pathology Caused by Chronic Exposure to House Dust Mite. PLoS One 2010, 5, e13136. [Google Scholar]
- Corren, J.; Lemanske, R.F.; Hanania, N.A.; Korenblat, P.E.; Parsey, M.V.; Arron, J.R.; Harris, J.M.; Scheerens, H.; Wu, L.C.; Su, Z.; et al. Lebrikizumab treatment in adults with asthma. N. Engl. J. Med. 2011, 365, 1088–1198. [Google Scholar] [CrossRef]
- Noonan, M.; Korenblat, P.; Mosesova, S.; Scheerens, H.; Arron, J.R.; Zheng, Y.; Putnam, W.S.; Parsey, M.V.; Bohen, S.P.; Matthews, J.G. Dose-ranging study of lebrikizumab in asthmatic patients not receiving inhaled steroids. J. Allergy Clin. Immunol. 2013, 132, 567–574.e12. [Google Scholar] [CrossRef]
- Scheerens, H.; Arron, J.R.; Zheng, Y.; Putnam, W.S.; Erickson, R.W.; Choy, D.F.; Harris, J.M.; Lee, J.; Jarjour, N.N.; Matthews, J.G. The effects of lebrikizumab in patients with mild asthma following whole lung allergen challenge. Clin. Exp. Allergy 2014, 44, 38–46. [Google Scholar] [CrossRef]
- Hanania, N.A.; Noonan, M.; Corren, J.; Korenblat, P.; Zheng, Y.; Fischer, S.K.; Cheu, M.; Putnam, W.S.; Murray, E.; Scheerens, H.; et al. Lebrikizumab in moderate-to-severe asthma: Pooled data from two randomised placebo-controlled studies. Thorax 2015, 70, 748–756. [Google Scholar] [CrossRef]
- U.S. National Institutes of Health. LAVOLTA I Study Record. Available online: https://clinicaltrials.gov/ct2/show/NCT01868061 (accessed on 25 May 2019).
- U.S. National Institutes of Health. LAVOLTA II Study Record. Available online: https://clinicaltrials.gov/ct2/show/NCT01867125 (accessed on 25 May 2019).
- Oh, C.K.; Geba, G.P.; Molfino, N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur. Respir. Rev. 2010, 19, 46–54. [Google Scholar] [CrossRef]
- Wenzel, S.; Ford, L.; Pearlman, D.; Spector, S.; Sher, L.; Skobieranda, F.; Wang, L.; Kirkesseli, S.; Rocklin, R.; Bock, B.; et al. Dupilumab in persistent asthma with elevated eosinophil levels. N. Engl. J. Med. 2013, 368, 2455–2466. [Google Scholar] [CrossRef] [PubMed]
- Thaçi, D.; Simpson, E.L.; Beck, L.A.; Bieber, T.; Blauvelt, A.; Papp, K.; Soong, W.; Worm, M.; Szepietowski, J.C.; Sofen, H.; et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: A randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet 2016, 387, 40–52. [Google Scholar] [CrossRef]
- Castro, M.; Corren, J.; Pavord, I.D.; Maspero, J.; Wenzel, S.; Rabe, K.F.; Busse, W.W.; Ford, L.; Sher, L.; FitzGerald, J.M.; et al. Dupilumab efficacy and safety in moderateto-severe uncontrolled asthma. N. Engl. J. Med. 2018, 378, 2486–2496. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Nair, P.; Brusselle, G.; Maspero, J.F.; Castro, M.; Sher, L.; Zhu, H.; Hamilton, J.D.; Swanson, B.N.; Khan, A.; et al. Efficacy and Safety of Dupilumab in Glucocorticoid-Dependent Severe Asthma. N. Engl. J. Med. 2018, 378, 2475–2485. [Google Scholar] [CrossRef] [PubMed]
- FDA approves asthma indication for Dupixent (dupilumab). Available online: http://www.news.sanofi.us/2018-10-19-FDA-approves-asthsma-indication-for-Dupixent-R-dupilumab (accessed on 18 June 2019).
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Gavett, S.H.; O’Hearn, D.J.; Li, X.; Huang, S.K.; Finkelman, F.D.; Wills-Karp, M. Interleukin 12 inhibits antigen-induced airway hyperresponsiveness, inflammation, and Th2 cytokine expression in mice. J. Exp. Med. 1995, 182, 1527–1536. [Google Scholar] [CrossRef]
- Kitagaki, K.; Jain, V.V.; Businga, T.R.; Hussain, I.; Kline, J.N. Immunomodulatory effects of CpG oligodeoxynucleotides on established Th2 responses. Clin. Diagn. Lab. Immunol. 2002, 9, 1260–1269. [Google Scholar] [CrossRef]
- Bortolatto, J.; Borducchi, E.; Rodriguez, D.; Keller, A.C.; Faquim-Mauro, E.; Bortoluci, K.R.; Mucida, D.; Gomes, E.; Christ, A.; Schnyder-Candrian, S.; et al. Toll-like receptor 4 agonists adsorbed to aluminium hydroxide adjuvant attenuate ovalbumin-specific allergic airway disease: Role of MyD88 adaptor molecule and interleukin-12/interferon-gamma axis. Clin. Exp. Allergy 2008, 38, 1668–1679. [Google Scholar] [CrossRef]
- Hansen, G.; Berry, G.; DeKruyff, R.H.; Umetsu, D.T. Allergen-specific Th1 cells fail to counterbalance Th2 cell–induced airway hyperreactivity but cause severe airway inflammation. J. Clin. Investig. 1999, 103, 175–183. [Google Scholar] [CrossRef]
- Li, J.J.; Wang, W.; Baines, K.J.; Bowden, N.A.; Hansbro, P.M.; Gibson, P.G.; Kumar, R.K.; Foster, P.S.; Yang, M. IL-27/IFN-γ induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J. Immunol. 2010, 185, 4401–4409. [Google Scholar] [CrossRef] [PubMed]
- Bryan, S.A.; O’Connor, B.J.; Matti, S.; Leckie, M.J.; Kanabar, V.; Khan, J.; Warrington, S.J.; Renzetti, L.; Rames, A.; Bock, J.A.; et al. Effects of recombinant human interleukin-12 on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 2000, 356, 2149–2153. [Google Scholar] [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Tanaka, Y.; Tsuji, T.; Jinushi, T.; Hoshino, A.; Asakura, Y.; Mita, Y.; Watanabe, K.; Nakaike, S.; Togashi, Y.; et al. A critical role for mouse CXC chemokine(s) in pulmonary neutrophilia during Th type 1-dependent airway inflammation. J. Immunol. 2001, 167, 2349–2353. [Google Scholar] [CrossRef] [PubMed]
- Liles, W.C.; Dale, D.C.; Klebanoff, S.J. Glucocorticoids inhibit apoptosis of human neutrophils. Blood 1995, 86, 3181–3188. [Google Scholar] [PubMed]
- Saffar, A.S.; Ashdown, H.; Gounni, A.S. The molecular mechanisms of glucocorticoids-mediated neutrophil survival. Curr. Drug Targets 2011, 12, 556–562. [Google Scholar] [CrossRef]
- Chesné, J.; Braza, F.; Mahay, G.; Brouard, S.; Aronica, M.; Magnan, A. IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med. 2014, 190, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Molet, S.; Hamid, Q.; Davoine, F.; Nutku, E.; Taha, R.; Page, N.; Olivenstein, R.; Elias, J.; Chakir, J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J. Allergy Clin. Immunol. 2001, 108, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Kokubu, F.; Kuga, H.; Matsukura, S.; Hoshino, H.; Ieki, K.; Imai, T.; Adachi, M.; Huang, S.K. Modulation of bronchial epithelial cells by IL-17. J. Allergy Clin. Immunol. 2001, 108, 804–809. [Google Scholar] [CrossRef]
- Rahman, M.S.; Yamasaki, A.; Yang, J.; Shan, L.; Halayko, A.J.; Gounni, A.S. IL-17A induces eotaxin-1/CC chemokine ligand 11 expression in human airway smooth muscle cells: Role of MAPK (Erk1/2, JNK, and p38) pathways. J. Immunol. 2006, 177, 4064–4071. [Google Scholar] [CrossRef] [PubMed]
- Al-Alwan, L.A.; Chang, Y.; Baglole, C.J.; Risse, P.A.; Halayko, A.J.; Martin, J.G.; Eidelman, D.H.; Hamid, Q. Autocrine-regulated airway smooth muscle cell migration is dependent on IL-17-induced growth-related oncogenes. J. Allergy Clin. Immunol. 2012, 130, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Al-Alwan, L.; Risse, P.A.; Halayko, A.J.; Martin, J.G.; Baglole, C.J.; Eidelman, D.H.; Hamid, Q. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J. 2012, 26, 5152–5160. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Melton, A.C.; Chen, C.; Engler, M.B.; Huang, K.E.; Ren, X.; Wang, Y.; Bernstein, X.; Li, J.T.; Atabai, K.; et al. IL-17A produced by αβ T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 2012, 18, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.W.; Holgate, S.; Kerwin, E.; Chon, Y.; Feng, J.; Lin, J.; Lin, S.L. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves New Psoriasis Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-psoriasis-drug (accessed on 25 May 2019).
- Faulkner, H.; Humphreys, N.; Renauld, J.C.; Van Snick, J.; Grencis, R. Interleukin-9 is involved in host protective immunity to intestinal nematode infection. Eur. J. Immunol. 1997, 27, 2536–2540. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Staudt, V.; Bothur, E.; Klein, M.; Lingnau, K.; Reuter, S.; Grebe, N.; Gerlitzki, B.; Hoffmann, M.; Ulges, A.; Taube, C.; et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 2010, 33, 192–202. [Google Scholar] [CrossRef]
- Goswami, R.; Jabeen, R.; Yagi, R.; Pham, D.; Zhu, J.; Goenka, S.; Kaplan, M.H. STAT6-dependent regulation of Th9 development. J. Immunol. 2012, 188, 968–975. [Google Scholar] [CrossRef]
- Chang, H.C.; Sehra, S.; Goswami, R.; Yao, W.; Yu, Q.; Stritesky, G.L.; Jabeen, R.; McKinley, C.; Ahyi, A.N.; Han, L.; et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. 2010, 11, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, R.; Goswami, R.; Awe, O.; Kulkarni, A.; Nguyen, E.T.; Attenasio, A.; Walsh, D.; Olson, M.R.; Kim, M.H.; Tepper, R.S.; et al. Th9 cell development requires a BATF-regulated transcriptional network. J. Clin. Invest. 2013, 123, 4641–4653. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.R.; Verdan, F.F.; Hufford, M.M.; Dent, A.L.; Kaplan, M.H. STAT3 Impairs STAT5 Activation in the Development of IL-9-Secreting T Cells. J. Immunol. 2016, 196, 3297–3304. [Google Scholar] [CrossRef] [PubMed]
- Mantel, P.Y.; Kuipers, H.; Boyman, O.; Rhyner, C.; Ouaked, N.; Rückert, B.; Karagiannidis, C.; Lambrecht, B.N.; Hendriks, R.W.; Crameri, R.; et al. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 2007, 12, e329. [Google Scholar]
- Jäger, A.; Dardalhon, V.; Sobel, R.A.; Bettelli, E.; Kuchroo, V.K. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 2009, 183, 7169–7177. [Google Scholar] [CrossRef]
- Zhou, Y.; Sonobe, Y.; Akahori, T.; Jin, S.; Kawanokuchi, J.; Noda, M.; Iwakura, Y.; Mizuno, T.; Suzumura, A. IL-9 promotes Th17 cell migration into the central nervous system via CC chemokine ligand-20 produced by astrocytes. J. Immunol. 2011, 186, 4415–4421. [Google Scholar] [CrossRef] [PubMed]
- Purwar, R.; Schlapbach, C.; Xiao, S.; Kang, H.S.; Elyaman, W.; Jiang, X.; Jetten, A.M.; Khoury, S.J.; Fuhlbrigge, R.C.; Kuchroo, V.K.; et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012, 18, 1248–1253. [Google Scholar] [CrossRef]
- Lu, Y.; Hong, S.; Li, H.; Park, J.; Hong, B.; Wang, L.; Zheng, Y.; Liu, Z.; Xu, J.; He, J.; et al. Th9 cells promote antitumor immune responses in vivo. J. Clin. Investig. 2012, 122, 4160–4171. [Google Scholar] [CrossRef]
- Ye, Z.J.; Yuan, M.L.; Zhou, Q.; Du, R.H.; Yang, W.B.; Xiong, X.Z.; Zhang, J.C.; Wu, C.; Qin, S.M.; Shi, H.Z. Differentiation and recruitment of Th9 cells stimulated by pleural mesothelial cells in human Mycobacterium tuberculosis infection. PLoS One 2012, 7, e31710. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Q.; Xue, G.; Bi, E.; Ma, X.; Wang, A.; Qian, J.; Dong, C.; Yi, Q. Th9 Cells Represent a Unique Subset of CD4+ T Cells Endowed with the Ability to Eradicate Advanced Tumors. Cancer Cell 2018, 33, 1048–1060.e7. [Google Scholar] [CrossRef]
- Rivera Vargas, T.; Humblin, E.; Végran, F.; Ghiringhelli, F.; Apetoh, L. Th9 cells in anti-tumor immunity. Semin. Immunopathol. 2017, 39, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, T.; Kaminuma, O.; Yamada, J.; Kitamura, N.; Abe, A.; Kobayashi, N.; Suko, M.; Mori, A. Eosinophils are required for the induction of bronchial hyperresponsiveness in a Th transfer model of BALB/c background. Int. Arch. Allergy Immunol. 2010, 152S1, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Fan, Y.; Li, J.; Zhang, X.; Lou, X.; Dou, Y.; Shi, X.; Lan, P.; Xiao, Y.; Minze, L.; et al. Guidance of super-enhancers in regulation of IL-9 induction and airway inflammation. J. Exp. Med. 2018, 215, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Kerzerho, J.; Maazi, H.; Speak, A.O.; Szely, N.; Lombardi, V.; Khoo, B.; Geryak, S.; Lam, J.; Soroosh, P.; Van Snick, J.; et al. Programmed cell death ligand 2 regulates TH9 differentiation and induction of chronic airway hyperreactivity. J. Allergy Clin. Immunol. 2013, 131, e1–e2. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, Y.; Li, J.; Li, S.; Zhang, Y.; Shen, J.; Tan, W.; Wu, C. Detection of IL-9 producing T cells in the PBMCs of allergic asthmatic patients. BMC Immunol. 2017, 18, 38. [Google Scholar] [CrossRef]
- Hoppenot, D.; Malakauskas, K.; Lavinskienė, S.; Bajoriūnienė, I.; Kalinauskaitė, V.; Sakalauskas, R. Peripheral blood Th9 cells and eosinophil apoptosis in ashma patients. Medicina 2015, 51, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.K.; Leigh, R.; McLaurin, K.K.; Kim, K.; Hultquist, M.; Molfino, N.A. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir. Res. 2013, 14, 93. [Google Scholar] [CrossRef]
- Saeki, M.; Kaminuma, O.; Nishimura, T.; Kitamura, N.; Mori, A.; Hiroi, T. Th9 cells induce steroid-resistant bronchial hyperresponsiveness in mice. Allergol. Int. 2017, 66S, 35–40. [Google Scholar] [CrossRef]
- Percopo, C.M.; Brenner, T.A.; Ma, M.; Kraemer, L.S.; Hakeem, R.M.; Lee, J.J.; Rosenberg, H.F. SiglecF+Gr1hi eosinophils are a distinct subpopulation within the lungs of allergen-challenged mice. J. Leukoc. Biol. 2017, 101, 321–328. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef]
- Abe, A.; Koyama, S.; Ohtomo, T.; Kitamura, N.; Kaminuma, O.; Mori, A. Murine T cell-derived contractile activity for bronchial smooth muscle cells. Int. Arch. Allergy Immunol. 2012, 158, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, T.; Kaminuma, O.; Kitamura, N.; Suko, M.; Kobayashi, N.; Mori, A. Murine Th clones confer late asthmatic response upon antigen challenge. Int. Arch. Allergy Immunol. 2009, 149, 2–6. [Google Scholar] [CrossRef]
- Pelaia, G.; Renda, T.; Gallelli, L.; Vatrella, A.; Busceti, M.T.; Agati, S.; Caputi, M.; Cazzola, M.; Maselli, R.; Marsico, S.A. Molecular mechanisms underlying airway smooth muscle contraction and proliferation: Implications for asthma. Respir. Med. 2008, 102, 117311–117381. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.J.; Killian, K. Airway smooth muscle as a target of asthma therapy: History and new directions. Respir. Res. 2006, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Chiba, Y.; Matsusue, K.; Hattori, Y.; Maitani, Y.; Sakai, H.; Kimura, S.; Misawa, M. The proximal STAT6 and NF-κB sites are responsible for IL-13- and TNF-alpha-induced RhoA transcriptions in human bronchial smooth muscle cells. Pharmacol. Res. 2010, 61, 466–472. [Google Scholar] [CrossRef]
- Chen, H.; Tliba, O.; Van Besien, C.R.; Panettieri, R.A., Jr.; Amrani, Y. TNF-alpha modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J. Appl. Physiol. 2003, 95, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kaminuma, O.; Saeki, M.; Kitamura, N.; Matsuoka, K.; Yonekawa, H.; Mori, A.; Hiroi, T. Essential Contribution of CD4+ T Cells to Antigen-Induced Nasal Hyperresponsiveness in Experimental Allergic Rhinitis. PLoS ONE 2016, 11, e0146686. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeki, M.; Nishimura, T.; Kitamura, N.; Hiroi, T.; Mori, A.; Kaminuma, O. Potential Mechanisms of T Cell-Mediated and Eosinophil-Independent Bronchial Hyperresponsiveness. Int. J. Mol. Sci. 2019, 20, 2980. https://doi.org/10.3390/ijms20122980
Saeki M, Nishimura T, Kitamura N, Hiroi T, Mori A, Kaminuma O. Potential Mechanisms of T Cell-Mediated and Eosinophil-Independent Bronchial Hyperresponsiveness. International Journal of Molecular Sciences. 2019; 20(12):2980. https://doi.org/10.3390/ijms20122980
Chicago/Turabian StyleSaeki, Mayumi, Tomoe Nishimura, Noriko Kitamura, Takachika Hiroi, Akio Mori, and Osamu Kaminuma. 2019. "Potential Mechanisms of T Cell-Mediated and Eosinophil-Independent Bronchial Hyperresponsiveness" International Journal of Molecular Sciences 20, no. 12: 2980. https://doi.org/10.3390/ijms20122980
APA StyleSaeki, M., Nishimura, T., Kitamura, N., Hiroi, T., Mori, A., & Kaminuma, O. (2019). Potential Mechanisms of T Cell-Mediated and Eosinophil-Independent Bronchial Hyperresponsiveness. International Journal of Molecular Sciences, 20(12), 2980. https://doi.org/10.3390/ijms20122980