The Pleiotropic Role of Retinoic Acid/Retinoic Acid Receptors Signaling: From Vitamin A Metabolism to Gene Rearrangements in Acute Promyelocytic Leukemia



Abstract
1. Metabolism of Vitamin A
2. Retinoid Receptors
3. RA and RAR Signaling during Embryonic Development
4. The Pleiotropic Roles of RA/RAR Signaling: From the Immune System to the Bone Remodeling
5. RA/RAR Signaling Involvement in Apoptosis
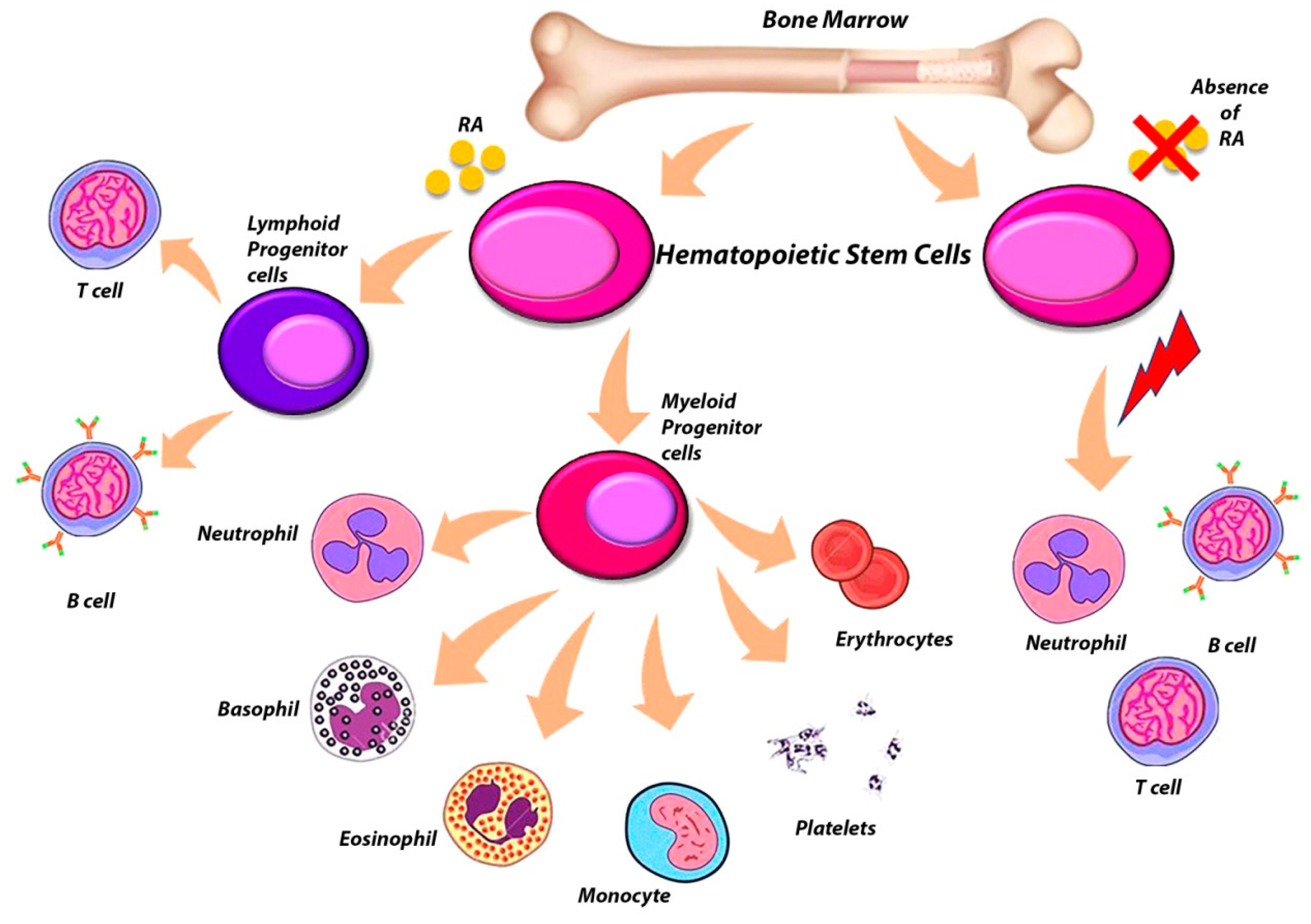
6. The Complex Role of RARs in the Regulation of Hematopoietic Stem Cells (HSCs)
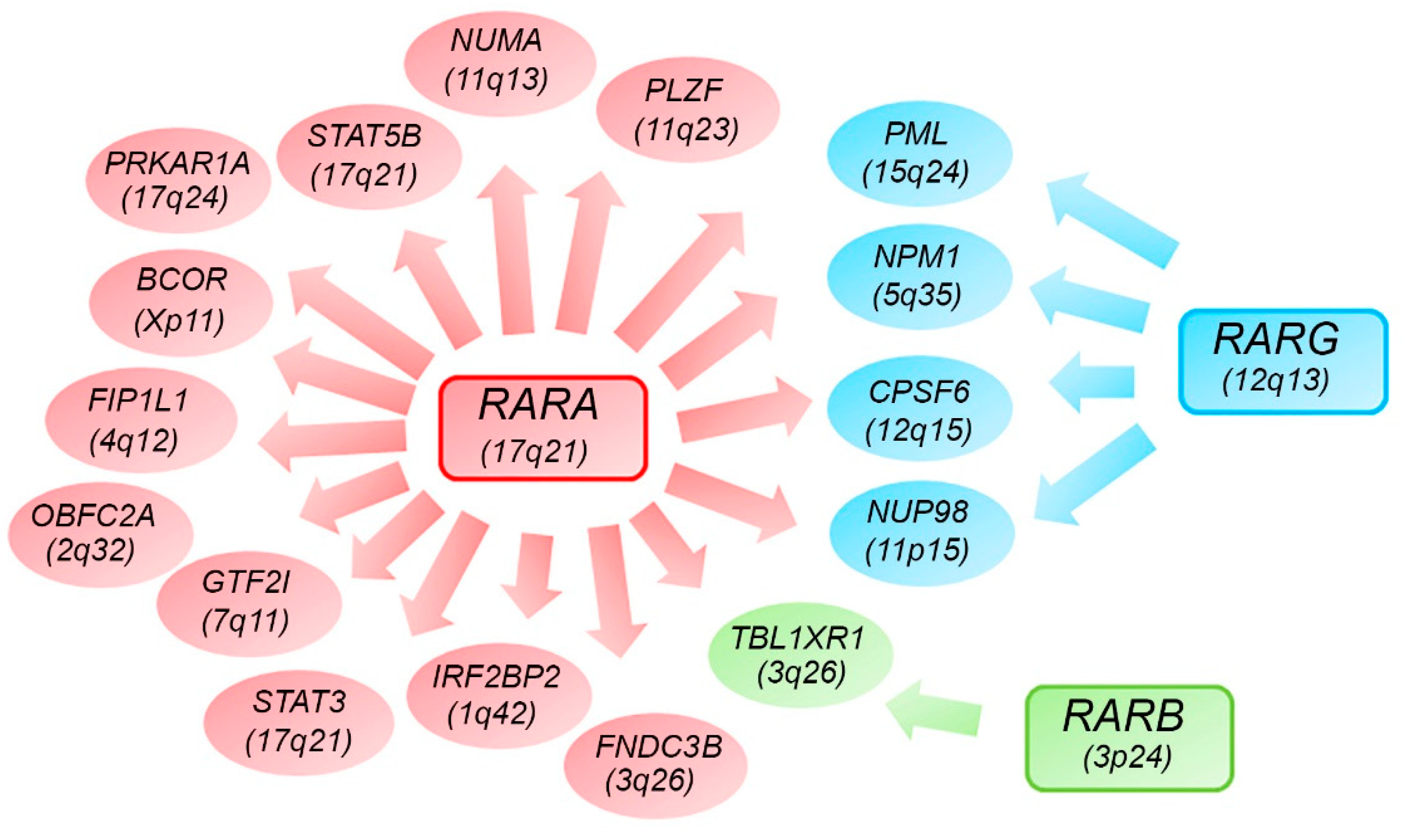
7. Alternative RARs-Rearrangements Resemble Acute Promyelocitic Leukemia (APL)
8. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| APL | acute promyelocytic leukemia |
| AML | acute myeloid leukemia |
| RBPs | retinol-binding proteins |
| ADHs | alcohol dehydrogenases |
| RAL | retinaldehyde |
| RA | retinoic acid |
| RALDHs | retinaldehyde dehydrogenases |
| CRABPs | cellular retinoic acid-binding proteins |
| RARs | retinoic acid receptors |
| PPARs | peroxisome proliferator–activated receptors |
| ATRA | all-trans retinoic acid |
| RXRs | retinoid X receptors |
| RAREs | retinoic acid response elements |
| HATs | histone acetyl transferases |
| NCoA3 | nuclear receptor coactivator 3 |
| ACTR | activator of the thyroid and RA receptor |
| HDACs | histone deacetylases |
| NCoR | nuclear receptor corepressor |
| ROR | retinoid-related orphan receptors |
| ROREs | ROR response elements |
| PPREs | peroxisome proliferator response elements |
| GluR1 | glutamate receptor 1 |
| CREB | cyclic AMP response element-binding protein |
| HSCs | hematopoietic stem cells |
| SLOs | secondary lymphoid organs |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| Treg | regulatory T cell |
| FoxP3 | forkhead-box-P3 transcription factor |
| iTreg | induced Treg |
| TGF-β | transforming growth factor-β |
| tTreg | Treg generated in the thymus |
| APCs | antigen-presenting cell |
| Ig | immunoglobulin |
| MAPK | mitogen-activated protein kinase |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| ILCs | innate lymphoid cells |
| PTH | parathyroid hormone |
| RANKL | nuclear factor kappa-β ligand |
| OPG | osteoprotegerin |
| BMP2 | bone morphogenetic protein 2 |
| BM | bone marrow |
| MSCs | mesenchymal stem cells |
| ZA | zoledronic acid |
| PML | promyelocytic leukemia |
| RARA | retinoic acid receptor-α |
| RARB | retinoic acid receptor-β |
| RARG | retinoic acid receptor-γ |
| NUP98 | nucleoporin 98 gene |
| NPCs | nuclear pore complex |
| CPSF6 | polyadenylation specific factor 6 |
| OS | overall survival |
| LFS | leukemia-free survival |
References
- Harrison, E.H. Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 70–77. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A Metabolism: An Update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.W.; Huang, H.S.; Shiratori, T. Tissue distribution and metabolism of newly absorbed vitamin a in the rat. J. Lipid Res. 1965, 6, 390–396. [Google Scholar] [PubMed]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sandell, L.L.; Trainor, P.A.; Koentgen, F.; Duester, G. Alcohol and aldehyde dehydrogenases: Retinoid metabolic effects in mouse knockout models. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ruuska, S.E.; Levinthal, D.J.; Noy, N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 1999, 274, 23695–23698. [Google Scholar] [CrossRef]
- Larange, A.; Cheroutre, H. Retinoic Acid and Retinoic Acid Receptors as Pleiotropic Modulators of the Immune System. Annu. Rev. Immunol. 2016, 34, 369–394. [Google Scholar] [CrossRef]
- Henning, P.; Conaway, H.H.; Lerner, U.H. Retinoid Receptors in Bone and Their Role in Bone Remodeling. Front. Endocrinol. 2015, 6, 31. [Google Scholar] [CrossRef]
- Hale, F. The Relation of Vitamin a to Anophthalmos in Pigs. Am. J. Ophthalmol. 1935, 18, 1087–1093. [Google Scholar] [CrossRef]
- Wilson, J.G.; Warkany, J. Aortic-arch and cardiac anomalies in the offspring of vitamin A deficient rats. Am. J. Anat. 1949, 85, 113–155. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.; Lohnes, D.; Decimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar] [PubMed]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar] [PubMed]
- Clagett-Dame, M.; DeLuca, H.F. The role of vitamin a in mammalian reproduction and embryonic development. Annu. Rev. Nutr. 2002, 22, 347–381. [Google Scholar] [CrossRef]
- Collins, M.D.; Mao, G.E. Teratology of Retinoids. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 399–430. [Google Scholar] [CrossRef]
- Stephensen, C.B. Vitamin A, infection, and immune function. Annu. Rev. Nutr. 2001, 21, 167–192. [Google Scholar] [CrossRef]
- Underwood, B.A.; Arthur, P. The contribution of vitamin A to public health. FASEB J. 1996, 10, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Rochette-Egly, C.; Germain, P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, nrs.07005. [Google Scholar] [CrossRef]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Krust, A.; Mendelsohn, C.; Garnier, J.M.; Zelent, A.; Leroy, P.; Staub, A.; Chambon, P. Murine isoforms of retinoic acid receptor gamma with specific patterns of expression. Proc. Natl. Acad. Sci. USA 1990, 87, 2700–2704. [Google Scholar] [CrossRef]
- Leroy, P.; Krust, A.; Zelent, A.; Mendelsohn, C.; Garnier, J.M.; Kastner, P.; Dierich, A.; Chambon, P. Multiple isoforms of the mouse retinoic acid receptor alpha are generated by alternative splicing and differential induction by retinoic acid. EMBO J. 1991, 10, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zelent, A.; Mendelsohn, C.; Kastner, P.; Krust, A.; Garnier, J.M.; Ruffenach, F.; Leroy, P.; Chambon, P. Differentially expressed isoforms of the mouse retinoic acid receptor beta generated by usage of two promoters and alternative splicing. EMBO J. 1991, 10, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Linney, E. The mouse retinoid-X receptor-gamma gene: Genomic organization and evidence for functional isoforms. Mol. Endocrinol. 1993, 7, 651–658. [Google Scholar] [PubMed]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin A and retinoid signaling: Genomic and nongenomic effects. J. Lipid Res. 2013, 54, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Mic, F.A.; Molotkov, A.; Benbrook, D.M.; Duester, G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 7135–7140. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, R.J.; Schiltz, R.L.; Chakravarti, D.; Nash, A.; Nagy, L.; Privalsky, M.L.; Nakatani, Y.; Evans, R.M. Nuclear Receptor Coactivator ACTR Is a Novel Histone Acetyltransferase and Forms a Multimeric Activation Complex with P/CAF and CBP/p300. Cell 1997, 90, 569–580. [Google Scholar] [CrossRef]
- Farboud, B.; Hauksdottir, H.; Wu, Y.; Privalsky, M.L. Isotype-restricted corepressor recruitment: A constitutively closed helix 12 conformation in retinoic acid receptors beta and gamma interferes with corepressor recruitment and prevents transcriptional repression. Mol. Cell. Biol. 2003, 23, 2844–2858. [Google Scholar] [CrossRef]
- Beard, R.L.; Duong, T.T.; Teng, M.; Klein, E.S.; Standevan, A.M.; Chandraratna, R.A.S. Synthesis and biological activity of retinoic acid receptor-α specific amides. Bioorg. Med. Chem. Lett. 2002, 12, 3145–3148. [Google Scholar] [CrossRef]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing Effects of Retinoic Acid on Cell Growth Result from Alternate Activation of Two Different Nuclear Receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef]
- Stehlin-Gaon, C.; Willmann, D.; Zeyer, D.; Sanglier, S.; Van Dorsselaer, A.; Renaud, J.-P.; Moras, D.; Schüle, R. All-trans retinoic acid is a ligand for the orphan nuclear receptor RORβ. Nat. Struct. Mol. Biol. 2003, 10, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M. Retinoid-Related Orphan Receptors (RORs): Critical Roles in Development, Immunity, Circadian Rhythm, and Cellular Metabolism. Nucl. Recept. Signal. 2009, 7, nrs.07003. [Google Scholar] [CrossRef] [PubMed]
- Giguère, V.; McBroom, L.D.; Flock, G. Determinants of target gene specificity for ROR alpha 1: Monomeric DNA binding by an orphan nuclear receptor. Mol. Cell. Biol. 1995, 15, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Giguère, V.; Tini, M.; Flock, G.; Ong, E.; Evans, R.M.; Otulakowski, G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994, 8, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Cañón, E.; Cosgaya, J.M.; Scsucova, S.; Aranda, A. Rapid effects of retinoic acid on CREB and ERK phosphorylation in neuronal cells. Mol. Biol. Cell 2004, 15, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Napoli, J.L. All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J. 2008, 22, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Onisko, B.; Napoli, J.L. The nuclear transcription factor RARalpha associates with neuronal RNA granules and suppresses translation. J. Biol. Chem. 2008, 283, 20841–20847. [Google Scholar] [CrossRef]
- Poon, M.M.; Chen, L. Retinoic acid-gated sequence-specific translational control by RARalpha. Proc. Natl. Acad. Sci. USA 2008, 105, 20303–20308. [Google Scholar] [CrossRef]
- Aggarwal, S.; Kim, S.-W.; Cheon, K.; Tabassam, F.H.; Yoon, J.-H.; Koo, J.S. Nonclassical Action of Retinoic Acid on the Activation of the cAMP Response Element-binding Protein in Normal Human Bronchial Epithelial Cells. Mol. Biol. Cell 2006, 17, 566–575. [Google Scholar] [CrossRef]
- Ross, S.A.; McCaffery, P.J.; Drager, U.C.; De Luca, L.M. Retinoids in Embryonal Development. Physiol. Rev. 2000, 80, 1021–1054. [Google Scholar] [CrossRef]
- Duester, G. Retinoic Acid Synthesis and Signaling during Early Organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Bain, G.; Ray, W.J.; Yao, M.; Gottlieb, D.I. Retinoic Acid Promotes Neural and Represses Mesodermal Gene Expression in Mouse Embryonic Stem Cells in Culture. Biochem. Biophys. Res. Commun. 1996, 223, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, C.; Asada, N.; Frenette, P.S. The hematopoietic stem cell niche: From embryo to adult. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Chanda, B.; Ditadi, A.; Iscove, N.N.; Keller, G. Retinoic Acid Signaling Is Essential for Embryonic Hematopoietic Stem Cell Development. Cell 2013, 155, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Ghyselinck, N.B.; Dupé, V.; Dierich, A.; Messaddeq, N.; Garnier, J.M.; Rochette-Egly, C.; Chambon, P.; Mark, M. Role of the retinoic acid receptor beta (RARbeta) during mouse development. Int. J. Dev. Biol. 1997, 41, 425–447. [Google Scholar] [PubMed]
- Sitnik, K.M.; Kotarsky, K.; White, A.J.; Jenkinson, W.E.; Anderson, G.; Agace, W.W. Mesenchymal cells regulate retinoic acid receptor-dependent cortical thymic epithelial cell homeostasis. J. Immunol. 2012, 188, 4801–4809. [Google Scholar] [CrossRef] [PubMed]
- Van de Pavert, S.A.; Ferreira, M.; Domingues, R.G.; Ribeiro, H.; Molenaar, R.; Moreira-Santos, L.; Almeida, F.F.; Ibiza, S.; Barbosa, I.; Goverse, G.; et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 2014, 508, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Grace, C.S.; Mikkola, H.K.A.; Dou, D.R.; Calvanese, V.; Ronn, R.E.; Purton, L.E. Protagonist or antagonist? The complex roles of retinoids in the regulation of hematopoietic stem cells and their specification from pluripotent stem cells. Exp. Hematol. 2018, 65, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.-X.; Liu, Y.; Yang, J.-F.; Wang, G.-Y.; Mu, L.; Zhang, T.-S.; Xie, X.-L.; Wang, J.-H.; Liu, Y.-M.; Kong, Q.-F.; et al. All-trans-retinoic acid ameliorates experimental allergic encephalomyelitis by affecting dendritic cell and monocyte development. Immunology 2013, 138, 333–345. [Google Scholar] [CrossRef]
- Denning, T.L.; Wang, Y.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17–producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Cannons, J.L.; Grainger, J.R.; Dos Santos, L.M.; Hand, T.W.; Naik, S.; Wohlfert, E.A.; Chou, D.B.; Oldenhove, G.; Robinson, M.; et al. Essential Role for Retinoic Acid in the Promotion of CD4+ T Cell Effector Responses via Retinoic Acid Receptor Alpha. Immunity 2011, 34, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kong, N.; Wang, J.; Fan, H.; Zou, H.; Horwitz, D.; Brand, D.; Liu, Z.; Zheng, S.G. Cutting edge: All-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J. Immunol. 2010, 185, 2675–2679. [Google Scholar] [CrossRef] [PubMed]
- Van, Y.-H.; Lee, W.-H.; Ortiz, S.; Lee, M.-H.; Qin, H.-J.; Liu, C.-P. All-trans Retinoic Acid Inhibits Type 1 Diabetes by T Regulatory (Treg)-Dependent Suppression of Interferon-γ–Producing T-cells Without Affecting Th17 Cells. Diabetes 2009, 58, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Jin, H.; Korn, T.; Liu, S.M.; Oukka, M.; Lim, B.; Kuchroo, V.K. Retinoic Acid Increases Foxp3+ Regulatory T Cells and Inhibits Development of Th17 Cells by Enhancing TGF-β-Driven Smad3 Signaling and Inhibiting IL-6 and IL-23 Receptor Expression. J. Immunol. 2008, 181, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- La, P.; Morgan, T.A.; Sykes, S.M.; Mao, H.; Schnepp, R.W.; Petersen, C.D.; Hua, X. Fusion proteins of retinoid receptors antagonize TGF-β-induced growth inhibition of lung epithelial cells. Oncogene 2003, 22, 198–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Benson, M.J.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007, 204, 1765–1774. [Google Scholar] [CrossRef]
- Sun, C.-M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef]
- Wiedermann, U.; Hanson, L.A.; Kahu, H.; Dahlgren, U.I. Aberrant T-cell function in vitro and impaired T-cell dependent antibody response in vivo in vitamin A-deficient rats. Immunology 1993, 80, 581–586. [Google Scholar]
- Cantorna, M.T.; Nashold, F.E.; Hayes2, C.E. In Vitamin A Deficiency Multiple Mechanisms Establish a Regulatory T Helper Cell Imbalance with Excess Th1 and Insufficient Th2 Function. J. Immunol. 1994, 152, 1515–1522. [Google Scholar] [PubMed]
- Racke, M.K.; Burnett, D.; Pak, S.H.; Albert, P.S.; Cannella, B.; Raine, C.S.; McFarlin, D.E.; Scott, D.E. Retinoid treatment of experimental allergic encephalomyelitis. IL-4 production correlates with improved disease course. J. Immunol. 1995, 154, 450–458. [Google Scholar] [PubMed]
- Brown, C.C.; Esterhazy, D.; Sarde, A.; London, M.; Pullabhatla, V.; Osma-Garcia, I.; al-Bader, R.; Ortiz, C.; Elgueta, R.; Arno, M.; et al. Retinoic acid is essential for th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015, 42, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Na, S.Y.; Kang, B.Y.; Chung, S.W.; Han, S.J.; Ma, X.; Trinchieri, G.; Im, S.Y.; Lee, J.W.; Kim, T.S. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFκB. J. Biol. Chem. 1999, 274, 7674–7680. [Google Scholar] [CrossRef] [PubMed]
- Austenaa, L.M.; Ross, A.C. Potentiation of interferon-gamma-stimulated nitric oxide production by retinoic acid in RAW 264.7 cells. J. Leukoc. Biol. 2001, 70, 121–129. [Google Scholar]
- Hoag, K.A.; Nashold, F.E.; Goverman, J.; Hayes, C.E. Retinoic Acid Enhances the T Helper 2 Cell Development That Is Essential for Robust Antibody Responses through Its Action on Antigen-Presenting Cells. J. Nutr. 2002, 132, 3736–3739. [Google Scholar] [CrossRef] [PubMed]
- Pasatiempo, A.M.; Kinoshita, M.; Taylor, C.E.; Ross, A.C. Antibody production in vitamin A-depleted rats is impaired after immunization with bacterial polysaccharide or protein antigens. FASEB J. 1990, 4, 2518–2527. [Google Scholar] [CrossRef]
- Ertesvag, A.; Aasheim, H.C.; Naderi, S.; Blomhoff, H.K. Vitamin A potentiates CpG-mediated memory B-cell proliferation and differentiation: Involvement of early activation of p38MAPK. Blood 2007, 109, 3865–3872. [Google Scholar] [CrossRef]
- Guidoboni, M.; Zancai, P.; Cariati, R.; Rizzo, S.; Dal Col, J.; Pavan, A.; Gloghini, A.; Spina, M.; Cuneo, A.; Pomponi, F.; et al. Retinoic acid inhibits the proliferative response induced by CD40 activation and interleukin-4 in mantle cell lymphoma. Cancer Res. 2005, 65, 587–595. [Google Scholar]
- Naderi, S.; Blomhoff, H.K. Retinoic acid prevents phosphorylation of pRB in normal human B lymphocytes: Regulation of cyclin E, cyclin A, and p21(Cip1). Blood 1999, 94, 1348–1358. [Google Scholar]
- Boller, S.; Grosschedl, R. The regulatory network of B-cell differentiation: A focused view of early B-cell factor 1 function. Immunol. Rev. 2014, 261, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Sirisinha, S.; Darip, M.D.; Moongkarndi, P.; Ongsakul, M.; Lamb, A.J. Lamb Impaired local immune response in vitamin A-deficient rats. Clin. Exp. Immunol. 1980, 40, 127–135. [Google Scholar]
- Gangopadhyay, N.N.; Moldoveanu, Z.; Stephensen, C.B. Vitamin A deficiency has different effects on immunoglobulin A production and transport during influenza A infection in BALB/c mice. J. Nutr. 1996, 126, 2960–2967. [Google Scholar] [CrossRef] [PubMed]
- Bjersing, J.L.; Telemo, E.; Dahlgren, U.; Hanson, L.Å. Loss of ileal IgA+ plasma cells and of CD4+ lymphocytes in ileal Peyer’s patches of vitamin A deficient rats. Clin. Exp. Immunol. 2002, 130, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; von Andrian, U.H. Differentiation and homing of IgA-secreting cells. Mucosal Immunol. 2008, 1, 96–109. [Google Scholar] [CrossRef]
- Tokuyama, H.; Tokuyama, Y. The regulatory effects of all-trans-retinoic acid on isotype switching: Retinoic acid induces IgA switch rearrangement in cooperation with IL-5 and inhibits IgG1 switching. Cell. Immunol. 1999, 192, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Worm, M.; Krah, J.M.; Manz, R.A.; Henz, B.M. Retinoic acid inhibits CD40 + interleukin-4-mediated IgE production in vitro. Blood 1998, 92, 1713–1720. [Google Scholar]
- Ruiter, B.; Patil, S.U.; Shreffler, W.G. Vitamins A and D have antagonistic effects on expression of effector cytokines and gut-homing integrin in human innate lymphoid cells. Clin. Exp. Allergy 2015, 45, 1214–1225. [Google Scholar] [CrossRef]
- Kim, M.H.; Taparowsky, E.J.; Kim, C.H. RetinOic Acid Differentially Regulates The Migration Of Innate Lymphoid Cell Subsets To The Gut. Immunity 2015, 43, 107–119. [Google Scholar] [CrossRef]
- McCarthy, N.E.; Bashir, Z.; Vossenkämper, A.; Hedin, C.R.; Giles, E.M.; Bhattacharjee, S.; Brown, S.G.; Sanders, T.J.; Whelan, K.; MacDonald, T.T.; et al. Proinflammatory Vδ2+ T cells populate the human intestinal mucosa and enhance IFN-γ production by colonic αβ T cells. J. Immunol. 2013, 191, 2752–2763. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S.; Park, O. Liver natural killer and natural killer T cells: Immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 2009, 86, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-K.; Hou, W.-S. Retinoic acid modulates interferon-γ production by hepatic natural killer T cells via phosphatase 2A and the extracellular signal-regulated kinase pathway. J. Interferon Cytokine Res. 2015, 35, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Kneissel, M.; Studer, A.; Cortesi, R.; Šuša, M. Retinoid-induced bone thinning is caused by subperiosteal osteoclast activity in adult rodents. Bone 2005, 36, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Lind, T.; Lind, P.M.; Jacobson, A.; Hu, L.; Sundqvist, A.; Risteli, J.; Yebra-Rodriguez, A.; Rodriguez-Navarro, A.; Andersson, G.; Melhus, H. High dietary intake of retinol leads to bone marrow hypoxia and diaphyseal endosteal mineralization in rats. Bone 2011, 48, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Raisz, L.G. Bone Resorption in Tissue Culture. Factors Influencing the Response to Parathyroid Hormone. J. Clin. Investig. 1965, 44, 103–116. [Google Scholar] [CrossRef]
- Conaway, H.H.; Pirhayati, A.; Persson, E.; Pettersson, U.; Svensson, O.; Lindholm, C.; Henning, P.; Tuckermann, J.; Lerner, U.H. Retinoids stimulate periosteal bone resorption by enhancing the protein RANKL, a response inhibited by monomeric glucocorticoid receptor. J. Biol. Chem. 2011, 286, 31425–31436. [Google Scholar] [CrossRef] [PubMed]
- Thavarajah, M.; Evans, D.B.; Kanis, J.A. 1,25(OH)2D3 induces differentiation of osteoclast-like cells from human bone marrow cultures. Biochem. Biophys. Res. Commun. 1991, 176, 1189–1195. [Google Scholar] [CrossRef]
- Hata, K.; Kukita, T.; Akamine, A.; Kukita, A.; Kurisu, K. Trypsinized osteoclast-like multinucleated cells formed in rat bone marrow cultures efficiently form resorption lacunae on dentine. Bone 1992, 13, 139–146. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Shidoji, Y.; Muto, Y.; Ohishi, N.; Yagi, K.; Ikegami, S.; Shinki, T.; Udagawa, N.; Suda, T.; et al. Effects of Geranylgeranoic Acid in Bone: Induction of Osteoblast Differentiation and Inhibition of Osteoclast Formation. J. Bone Miner. Res. 2002, 17, 91–100. [Google Scholar] [CrossRef]
- Martin, S.J.; Bradley, J.G.; Cotter, T.G. HL-60 cells induced to differentiate towards neutrophils subsequently die via apoptosis. Clin. Exp. Immunol. 2008, 79, 448–453. [Google Scholar] [CrossRef]
- Yang, Y.; Vacchio, M.S.; Ashwell, J.D. 9-cis-retinoic acid inhibits activation-driven T-cell apoptosis: Implications for retinoid X receptor involvement in thymocyte development. Proc. Natl. Acad. Sci. USA 1993, 90, 6170–6174. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.J.; Noy, N. Suppression of Mammary Carcinoma Growth by Retinoic Acid: Proapoptotic Genes Are Targets for Retinoic Acid Receptor and Cellular Retinoic Acid–Binding Protein II Signaling. Cancer Res. 2005, 65, 8193–8199. [Google Scholar] [CrossRef] [PubMed]
- Mrass, P.; Rendl, M.; Mildner, M.; Gruber, F.; Lengauer, B.; Ballaun, C.; Eckhart, L.; Tschachler, E. Retinoic Acid Increases the Expression of p53 and Proapoptotic Caspases and Sensitizes Keratinocytes to Apoptosis. Cancer Res. 2004, 64, 6542–6548. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, H.; Nakamura, Y.; Ozaki, T.; Nakanishi, H.; Ohira, M.; Isogai, E.; Kageyama, H.; Imaizumi, M.; Nakagawara, A. Bcl-2 is a key regulator for the retinoic acid-induced apoptotic cell death in neuroblastoma. Oncogene 2006, 25, 5046–5055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rosdahl, I. Expression profiles of p53, p21, bax and bcl-2 proteins in all-trans-retinoic acid treated primary and metastatic melanoma cells. Int. J. Oncol. 2004, 25, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Mäntymaa, P.; Säily, M.; Savolainen, E.-R.; Vähäkangas, K.; Koistinen, P. p53 Pathway in Apoptosis Induced by All-Trans-Retinoic Acid in Acute Myeloblastic Leukaemia Cells. Acta Haematol. 2000, 103, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Orr, B.; White, K.; Belogortseva, N.; Niles, R.; Boskovic, G.; Nguyen, H.; Dykes, A.; Park, M. Chmp 1A is a mediator of the anti-proliferative effects of All-trans Retinoic Acid in human pancreatic cancer cells. Mol. Cancer 2009, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Thin, T.H.; Li, L.; Chung, T.-K.; Sun, H.; Taneja, R. Stra13 is induced by genotoxic stress and regulates ionizing-radiation-induced apoptosis. EMBO Rep. 2007, 8, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Hormi-Carver, K.; Feagins, L.A.; Spechler, S.J.; Souza, R.F. All trans-retinoic acid induces apoptosis via p38 and caspase pathways in metaplastic Barrett’s cells. Am. J. Physiol. Liver Physiol. 2007, 292, G18–G27. [Google Scholar] [CrossRef]
- Altucci, L.; Rossin, A.; Raffelsberger, W.; Reitmair, A.; Chomienne, C.; Gronemeyer, H. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat. Med. 2001, 7, 680–686. [Google Scholar] [CrossRef]
- Clarke, N.; Jimenez-Lara, A.M.; Voltz, E.; Gronemeyer, H. Tumor suppressor IRF-1 mediates retinoid and interferon anticancer signaling to death ligand TRAIL. EMBO J. 2004, 23, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.K.; Aggarwal, B.B. All-trans-retinoic acid upregulates TNF receptors and potentiates TNF-induced activation of nuclear factors-κB, activated protein-1 and apoptosis in human lung cancer cells. Oncogene 2000, 19, 2110–2119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Witcher, M.; Ross, D.T.; Rousseau, C.; Deluca, L.; Miller, W.H. Synergy between all-trans retinoic acid and tumor necrosis factor pathways in acute leukemia cells. Blood 2003, 102, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) Apoptosis Systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Bovenzi, V.; Lê, N.L.; Côte, S.; Sinnett, D.; Momparler, L.F.; Momparler, R.L. DNA methylation of retinoic acid receptor beta in breast cancer and possible therapeutic role of 5-aza-2′-deoxycytidine. Anti-Cancer Drugs 1999, 10, 471–476. [Google Scholar] [CrossRef]
- Côté, S.; Sinnett, D.; Momparler, R.L. Demethylation by 5-aza-2′-deoxycytidine of specific 5-methylcytosine sites in the promoter region of the retinoic acid receptor beta gene in human colon carcinoma cells. Anti-Cancer Drugs 1998, 9, 743–750. [Google Scholar] [PubMed]
- Nakayama, T.; Watanabe, M.; Yamanaka, M.; Hirokawa, Y.; Suzuki, H.; Ito, H.; Yatani, R.; Shiraishi, T. The Role of Epigenetic Modifications in Retinoic Acid Receptor β2 Gene Expression in Human Prostate Cancers. Lab. Investig. 2001, 81, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, M.Z.; Liao, J.; Yang, G.-Y.; Nie, Y.; Song, Y.; So, C.; Xu, X.; Wang, L.-D.; Yang, C.S. Hypermethylation-Associated Inactivation of Retinoic Acid Receptor β in Human Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. 2003, 9, 5257–5263. [Google Scholar] [PubMed]
- Castillo, L.; Milano, G.; Santini, J.; Demard, F.; Pierrefite, V. Analysis of retinoic acid receptor beta expression in normal and malignant laryngeal mucosa by a sensitive and routine applicable reverse transcription-polymerase chain reaction enzyme-linked immunosorbent assay method. Clin. Cancer Res. 1997, 3, 2137–2142. [Google Scholar]
- Picard, E.; Seguin, C.; Monhoven, N.; Rochette-Egly, C.; Siat, J.; Borrelly, J.; Martinet, Y.; Martinet, N.; Vignaud, J.M. Expression of Retinoid Receptor Genes and Proteins in Non-Small-Cell Lung Cancer. JNCI J. Natl. Cancer Inst. 1999, 91, 1059–1066. [Google Scholar] [CrossRef]
- Widschwendter, M.; Berger, J.; Daxenbichler, G.; Müller-Holzner, E.; Widschwendter, A.; Mayr, A.; Marth, C.; Zeimet, A.G. Loss of Retinoic Acid Receptor β Expression in Breast Cancer and Morphologically Normal Adjacent Tissue but not in the Normal Breast Tissue Distant from the Cancer. Cancer Res. 1997, 57, 4158–4161. [Google Scholar] [CrossRef]
- Lotan, R.; Xu, X.-C.; Lippman, S.M.; Ro, J.Y.; Lee, J.S.; Lee, J.J.; Hong, W.K. Suppression of Retinoic Acid Receptor–β in Premalignant Oral Lesions and Its Up-Regulation by Isotretinoin. N. Engl. J. Med. 1995, 332, 1405–1411. [Google Scholar] [CrossRef]
- Xu, X.-C. Tumor-suppressive activity of retinoic acid receptor-beta in cancer. Cancer Lett. 2007, 253, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Henion, P.D.; Weston, J.A. Retinoic acid selectively promotes the survival and proliferation of neurogenic precursors in cultured neural crest cell populations. Dev. Biol. 1994, 161, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Lie, D.C.; DeCicco, K.L.; Shi, Y.; DeLuca, L.M.; Gage, F.H.; Evans, R.M. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2006, 103, 3902–3907. [Google Scholar] [CrossRef]
- Plum, L.A.; Parada, L.F.; Tsoulfas, P.; Clagett-Dame, M. Retinoic acid combined with neurotrophin-3 enhances the survival and neurite outgrowth of embryonic sympathetic neurons. Exp. Biol. Med. 2001, 226, 766–775. [Google Scholar] [CrossRef]
- Rodriguez-Tébar, A.; Rohrer, H. Retinoic acid induces NGF-dependent survival response and high-affinity NGF receptors in immature chick sympathetic neurons. Development 1991, 112, 813–820. [Google Scholar]
- Sorg, O.; Iran, C.; Carraux, P.; Grand, D.; Hügin, A.; Didierjean, L.; Saurat, J.-H. Spectral Properties of Topical Retinoids Prevent DNA Damage and Apoptosis After Acute UV-B Exposure in Hairless Mice. Photochem. Photobiol. 2007, 81, 830–836. [Google Scholar] [CrossRef]
- Kholodenko, R.; Kholodenko, I.; Sorokin, V.; Tolmazova, A.; Sazonova, O.; Buzdin, A. Anti-apoptotic effect of retinoic acid on retinal progenitor cells mediated by a protein kinase A-dependent mechanism. Cell Res. 2007, 17, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, R.; Baker, K.M.; Pan, J. All-trans retinoic acid prevents angiotensin II- and mechanical stretch-induced reactive oxygen species generation and cardiomyocyte apoptosis. J. Cell. Physiol. 2008, 215, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Besnard, V.; Nabeyrat, E.; Henrion-Caude, A.; Chadelat, K.; Perin, L.; Le Bouc, Y.; Clement, A. Protective role of retinoic acid from antiproliferative action of TNF-α on lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L863–L871. [Google Scholar] [CrossRef] [PubMed]
- Manor, D.; Shmidt, E.N.; Budhu, A.; Flesken-Nikitin, A.; Zgola, M.; Page, R.; Nikitin, A.Y.; Noy, N. Mammary carcinoma suppression by cellular retinoic acid binding protein-II. Cancer Res. 2003, 63, 4426–4433. [Google Scholar]
- Tan, N.S.; Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor-β as a target for wound healing drugs. Expert Opin. Ther. Targets 2004, 8, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Michalik, L.; Noy, N.; Yasmin, R.; Pacot, C.; Heim, M.; Flühmann, B.; Desvergne, B.; Wahli, W. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 2001, 15, 3263–3277. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Smith, P.; Beesley, C.; Igarashi, M.; Fujii, H.; Forootan, S.; Adamson, J.; Foster, C.; Morgan, E. Expression of cutaneous fatty acid-binding protein (C-FABP) in prostate cancer: Potential prognostic marker and target for tumourigenicity-suppression. Int. J. Oncol. 2008, 32, 767–775. [Google Scholar] [CrossRef][Green Version]
- Schug, T.T.; Berry, D.C.; Toshkov, I.A.; Cheng, L.; Nikitin, A.Y.; Noy, N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proc. Natl. Acad. Sci. USA 2008, 105, 7546–7551. [Google Scholar] [CrossRef] [PubMed]
- Green, A.C.; Rudolph-Stringer, V.; Straszkowski, L.; Tjin, G.; Crimeen-Irwin, B.; Walia, M.; Martin, T.J.; Sims, N.A.; Purton, L.E. Retinoic Acid Receptor γ Activity in Mesenchymal Stem Cells Regulates Endochondral Bone, Angiogenesis, and B Lymphopoiesis. J. Bone Miner. Res. 2018, 33, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Breitman, T.; Collins, S.; Keene, B. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood 1981, 57, 1000–1004. [Google Scholar] [PubMed]
- Purton, L.E.; Dworkin, S.; Olsen, G.H.; Walkley, C.R.; Fabb, S.A.; Collins, S.J.; Chambon, P. RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med. 2006, 203, 1283–1293. [Google Scholar] [CrossRef]
- Cabezas-Wallscheid, N.; Buettner, F.; Sommerkamp, P.; Klimmeck, D.; Ladel, L.; Thalheimer, F.B.; Pastor-Flores, D.; Roma, L.P.; Renders, S.; Zeisberger, P.; et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 2017, 169, 807–823.e19. [Google Scholar] [CrossRef]
- Luo, J.; Pasceri, P.; Conlon, R.A.; Rossant, J.; Giguère, V. Mice lacking all isoforms of retinoic acid receptor β develop normally and are susceptible to the teratogenic effects of retinoic acid. Mech. Dev. 1995, 53, 61–71. [Google Scholar] [CrossRef]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor γ in the mouse. Cell 1993, 73, 643–658. [Google Scholar] [CrossRef]
- Lufkin, T.; Lohnes, D.; Mark, M.; Dierich, A.; Gorry, P.; Gaub, M.P.; LeMeur, M.; Chambon, P. High postnatal lethality and testis degeneration in retinoic acid receptor alpha mutant mice. Proc. Natl. Acad. Sci. USA 1993, 90, 7225–7229. [Google Scholar] [CrossRef] [PubMed]
- Walkley, C.R.; Olsen, G.H.; Dworkin, S.; Fabb, S.A.; Swann, J.; McArthur, G.A.; Westmoreland, S.V.; Chambon, P.; Scadden, D.T.; Purton, L.E. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007, 129, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Dewamitta, S.R.; Joseph, C.; Purton, L.E.; Walkley, C.R. Erythroid-extrinsic regulation of normal erythropoiesis by retinoic acid receptors. Br. J. Haematol. 2014, 164, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Lawrence, H.J.; Waltzinger, C.; Ghyselinck, N.B.; Chambon, P.; Chan, S. Positive and negative regulation of granulopoiesis by endogenous RARalpha. Blood 2001, 97, 1314–1320. [Google Scholar] [CrossRef]
- Green, A.C.; Poulton, I.J.; Vrahnas, C.; Häusler, K.D.; Walkley, C.R.; Wu, J.Y.; Martin, T.J.; Gillespie, M.T.; Chandraratna, R.A.S.; Quinn, J.M.W.; et al. RARγ is a negative regulator of osteoclastogenesis. J. Steroid Biochem. Mol. Biol. 2015, 150, 46–53. [Google Scholar] [CrossRef]
- Joseph, C.; Nota, C.; Fletcher, J.L.; Maluenda, A.C.; Green, A.C.; Purton, L.E. Retinoic Acid Receptor γ Regulates B and T Lymphopoiesis via Nestin-Expressing Cells in the Bone Marrow and Thymic Microenvironments. J. Immunol. 2016, 196, 2132–2144. [Google Scholar] [CrossRef]
- Cabezas-Wallscheid, N.; Klimmeck, D.; Hansson, J.; Lipka, D.B.; Reyes, A.; Wang, Q.; Weichenhan, D.; Lier, A.; Von Paleske, L.; Renders, S.; et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 2014, 15, 507–522. [Google Scholar] [CrossRef]
- De Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RARα fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef]
- Degos, L.; Wang, Z.Y. All trans retinoic acid in acute promyelocytic leukemia. Oncogene 2001, 20, 7140–7145. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [PubMed]
- Purton, L.E.; Bernstein, I.D.; Collins, S.J. All-trans retinoic acid delays the differentiation of primitive hematopoietic precursors (lin-c-kit+Sca-1(+)) while enhancing the terminal maturation of committed granulocyte/monocyte progenitors. Blood 1999, 94, 483–495. [Google Scholar] [PubMed]
- Walkley, C.R.; Purton, L.E.; Snelling, H.J.; Yuan, Y.D.; Nakajima, H.; Chambon, P.; Chandraratna, R.A.S.; McArthur, G.A. Identification of the molecular requirements for an RARα-mediated cell cycle arrest during granulocytic differentiation. Blood 2004, 103, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; So, C.W.E. Transcriptional and epigenetic networks in haematological malignancy. FEBS Lett. 2011, 585, 2100–2111. [Google Scholar] [CrossRef]
- Such, E.; Cervera, J.; Valencia, A.; Barragán, E.; Ibañez, M.; Luna, I.; Fuster, O.; Perez-Sirvent, M.L.; Senent, L.; Sempere, A.; et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood 2011, 117, 242–245. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Chen, Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar] [CrossRef]
- De Thé, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef]
- Platzbecker, U.; Avvisati, G.; Cicconi, L.; Thiede, C.; Paoloni, F.; Vignetti, M.; Ferrara, F.; Divona, M.; Albano, F.; Efficace, F.; et al. Improved outcomes with retinoic acid and arsenic trioxide compared with retinoic acid and chemotherapy in non-high-risk acute promyelocytic leukemia: Final results of the randomized Italian-German APL0406 trial. J. Clin. Oncol. 2017, 35, 605–612. [Google Scholar] [CrossRef]
- Cicconi, L.; Divona, M.; Ciardi, C.; Ottone, T.; Ferrantini, A.; Lavorgna, S.; Alfonso, V.; Paoloni, F.; Piciocchi, A.; Avvisati, G.; et al. PML-RARα kinetics and impact of FLT3-ITD mutations in newly diagnosed acute promyelocytic leukaemia treated with ATRA and ATO or ATRA and chemotherapy. Leukemia 2016, 30, 1987–1992. [Google Scholar] [CrossRef]
- La Starza, R.; Brandimarte, L.; Pierini, V.; Nofrini, V.; Gorello, P.; Crescenzi, B.; Berchicci, L.; Matteucci, C.; Romoli, S.; Beacci, D.; et al. A NUP98-positive acute myeloid leukemia with a t(11;12)(p15;q13) without HOXC cluster gene involvement. Cancer Genet. Cytogenet. 2009, 193, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Romana, S.; Radford-Weiss, I.; Abdelali, R.B.; Schluth, C.; Petit, A.; Dastugue, N.; Talmant, P.; Bilhou-Nabera, C.; Mugneret, F.; Lafage-Pochitaloff, M.; et al. NUP98 rearrangements in hematopoietic malignancies: A study of the Groupe Francophone de Cytogénétique Hématologique. Leukemia 2006, 20, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Hollink, I.H.I.M.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T.C.J.M.; Pratcorona, M.; Abbas, S.; Kuipers, J.E.; van Galen, J.F.; Beverloo, H.B.; Sonneveld, E.; Kaspers, G.-J.J.L.; et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011, 118, 3645–3656. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.H.; Aplan, P.D. NUP98 gene fusions in hematologic malignancies. Leukemia 2001, 15, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Hussey, D.J.; Dobrovic, A. Recurrent coiled-coil motifs in NUP98 fusion partners provide a clue to leukemogenesis. Blood 2002, 99, 1097–1098. [Google Scholar] [CrossRef]
- Zhang, X.; Li, F.; Wang, J.; Suo, S.; Ling, Q.; Yu, W.; Jin, J. RARγ-rearrangements resemble acute promyelocytic leukemia and benefit from 3 + 7 regimen. Leuk. Lymphoma 2019, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.J.; Zeisig, B.B.; Li, S.; Liu, W.; Chu, H.; Song, Y.; Giordano, A.; Schwaller, J.; Gronemeyer, H.; Dong, S.; et al. Critical role of retinoid/rexinoid signaling in mediating transformation and therapeutic response of NUP98-RARG leukemia. Leukemia 2015, 29, 1153–1162. [Google Scholar] [CrossRef]
- Such, E.; Cordón, L.; Sempere, A.; Villamón, E.; Ibañez, M.; Luna, I.; Gómez-Seguí, I.; López-Pavía, M.; Alonso, C.; Lo-Coco, F.; et al. In vitro all-trans retinoic acid sensitivity of acute myeloid leukemia blasts with NUP98/RARG fusion gene. Ann. Hematol. 2014, 93, 1931–1933. [Google Scholar] [CrossRef]
- Qin, Y.-Z.; Huang, X.-J.; Zhu, H.-H. Identification of a novel CPSF6-RARG fusion transcript in acute myeloid leukemia resembling acute promyelocytic leukemia. Leukemia 2018, 32, 2285–2287. [Google Scholar] [CrossRef]
- Liu, T.; Wen, L.; Yuan, H.; Wang, Y.; Yao, L.; Xu, Y.; Cen, J.; Ruan, C.; Wu, D.; Chen, S. Identification of novel recurrent CPSF6-RARG fusions in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood 2018, 131, 1870–1873. [Google Scholar] [CrossRef]
- Miller, C.A.; Tricarico, C.; Skidmore, Z.L.; Uy, G.L.; Lee, Y.-S.; Hassan, A.; O’Laughlin, M.D.; Schmidt, H.; Tian, L.; Duncavage, E.J.; et al. A case of acute myeloid leukemia with promyelocytic features characterized by expression of a novel RARG-CPSF6 fusion. Blood Adv. 2018, 2, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-S.; Do, Y.R.; Ki, C.-S.; Lee, C.; Kim, D.-H.; Lee, W.; Ryoo, N.-H.; Jeon, D.-S. Identification of a novel PML-RARG fusion in acute promyelocytic leukemia. Leukemia 2017, 31, 1992–1995. [Google Scholar] [CrossRef] [PubMed]
- Coccaro, N.; Zagaria, A.; Orsini, P.; Anelli, L.; Tota, G.; Casieri, P.; Impera, L.; Minervini, A.; Minervini, C.F.; Cumbo, C.; et al. RARA and RARG gene downregulation associated with EZH2 mutation in acute promyelocytic-like morphology leukemia. Hum. Pathol. 2018, 80, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Osumi, T.; Tsujimoto, S.-I.; Tamura, M.; Uchiyama, M.; Nakabayashi, K.; Okamura, K.; Yoshida, M.; Tomizawa, D.; Watanabe, A.; Takahashi, H.; et al. Recurrent RARB translocations in acute promyelocytic leukemia lacking RARA translocation. Cancer Res. 2018, 78, 4452–4458. [Google Scholar] [CrossRef] [PubMed]
- Campregher, P.V.; da Sliva Halley, N.; Vieira, G.A.; Fernandes, J.F.; Velloso, E.D.R.P.; Ali, S.; Mughal, T.; Miller, V.; Mangueira, C.L.P.; Odone, V.; et al. Identification of a novel fusion TBL1XR1–PDGFRB in a patient with acute myeloid leukemia harboring the DEK–NUP214 fusion and clinical response to dasatinib. Leuk. Lymphoma 2017, 58, 2969–2972. [Google Scholar] [CrossRef]
- Wu, X.; Zhan, Y.; Li, X.; Wei, J.; Santiago, L.; Daniels, G.; Deng, F.; Zhong, X.; Chiriboga, L.; Basch, R.; et al. Nuclear TBLR1 as an ER corepressor promotes cell proliferation, migration and invasion in breast and ovarian cancer. Am. J. Cancer Res. 2016, 6, 2351–2360. [Google Scholar]
- Wen, L.; Xu, Y.; Yao, L.; Wang, N.; Wang, Q.; Liu, T.; Pan, J.; Cen, J.; Zhou, H.; Miao, M.; et al. Clinical and molecular features of acute promyelocytic leukemia with variant retinoid acid receptor fusions. Haematologica 2019, 104, e195–e199. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Hematopoietic Cell Population | Bone Marrow Phenotype | Bone Marrow Defects | Reference | |
|---|---|---|---|---|
| Rara−/− mice | normal granulocyte population normal HSCs numbers and function | normal development | manifold defects in several tissues high post-natal mortality | [97,132,133] |
| Rarb−/− mice | normal development | normal development | none | [131] |
| Rarg−/− mice | ↑ granulocytes in PB, BM and spleen several defects both in erythropoiesis and B lymphopoiesis | ↑osteoclastogenesis ↓ trabecular bone mass | manifold defects in several tissues high post-natal mortality | [132,133,134,135,137] |
| Nestin-Cre: Rarg Δ/Δ mice | impaired BM B lymphopoiesis and thymic T lymphopoiesis normal HSCs numbers and function | NA | / | [102] |
| Prrx1:RargΔ/Δ female mice | ↑ BM granulocytes | ↑ trabecular osteoblasts | enhanced BM vascularization: ↑ expression of Vegf-a | [127] |
| Prrx1:RargΔ/Δ male mice | ↑ pro-B and pre-B lymphocytes in BM unchanged number of splenic and mature PB B lymphocytes | ↑ trabecular osteoclasts | enhanced BM vascularization: ↑ expression of Vegf-a | [127] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conserva, M.R.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. The Pleiotropic Role of Retinoic Acid/Retinoic Acid Receptors Signaling: From Vitamin A Metabolism to Gene Rearrangements in Acute Promyelocytic Leukemia. Int. J. Mol. Sci. 2019, 20, 2921. https://doi.org/10.3390/ijms20122921
Conserva MR, Anelli L, Zagaria A, Specchia G, Albano F. The Pleiotropic Role of Retinoic Acid/Retinoic Acid Receptors Signaling: From Vitamin A Metabolism to Gene Rearrangements in Acute Promyelocytic Leukemia. International Journal of Molecular Sciences. 2019; 20(12):2921. https://doi.org/10.3390/ijms20122921
Chicago/Turabian StyleConserva, Maria Rosa, Luisa Anelli, Antonella Zagaria, Giorgina Specchia, and Francesco Albano. 2019. "The Pleiotropic Role of Retinoic Acid/Retinoic Acid Receptors Signaling: From Vitamin A Metabolism to Gene Rearrangements in Acute Promyelocytic Leukemia" International Journal of Molecular Sciences 20, no. 12: 2921. https://doi.org/10.3390/ijms20122921
APA StyleConserva, M. R., Anelli, L., Zagaria, A., Specchia, G., & Albano, F. (2019). The Pleiotropic Role of Retinoic Acid/Retinoic Acid Receptors Signaling: From Vitamin A Metabolism to Gene Rearrangements in Acute Promyelocytic Leukemia. International Journal of Molecular Sciences, 20(12), 2921. https://doi.org/10.3390/ijms20122921