Repression of TERRA Expression by Subtelomeric DNA Methylation Is Dependent on NRF1 Binding

Abstract
1. Introduction
2. Results
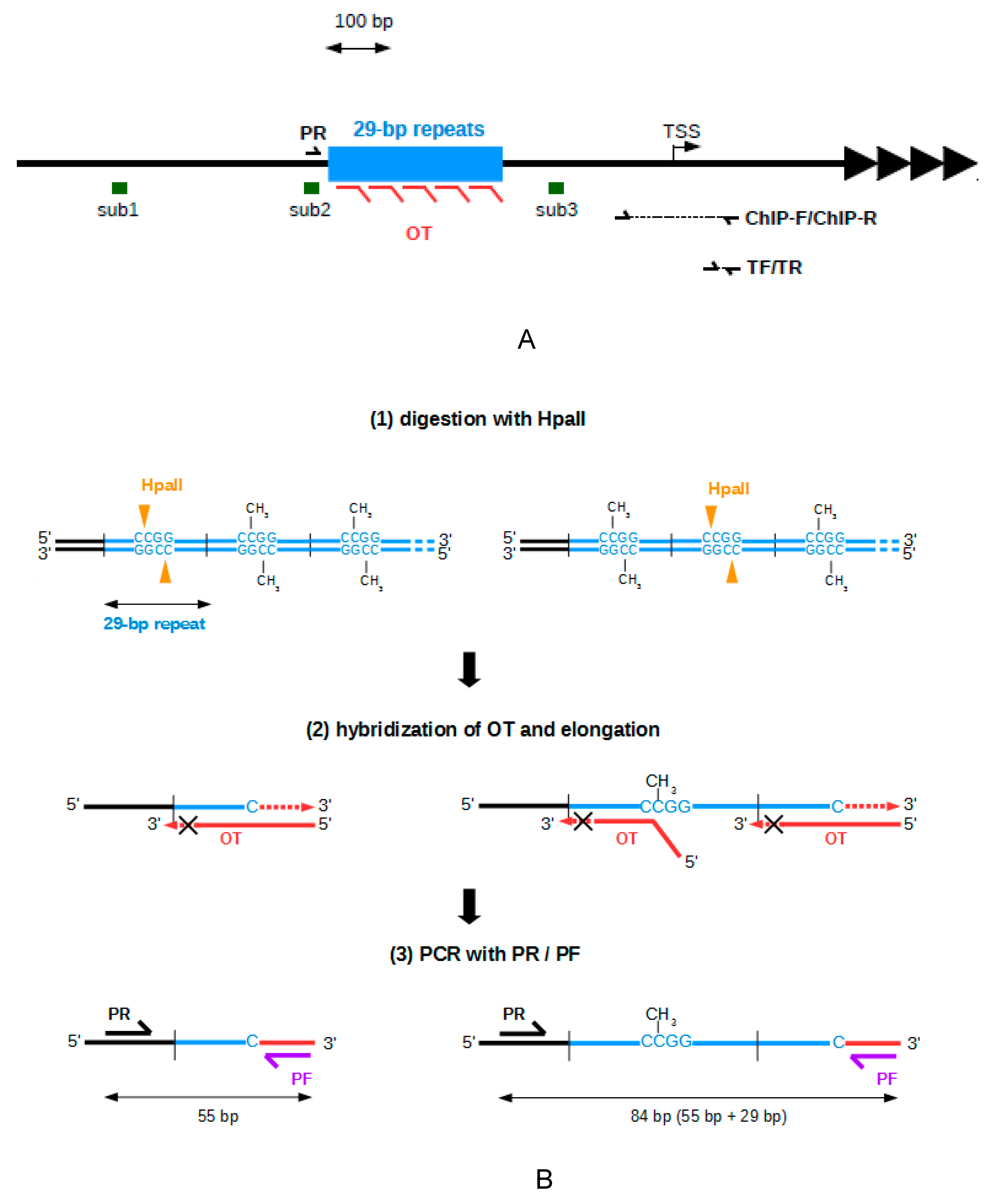
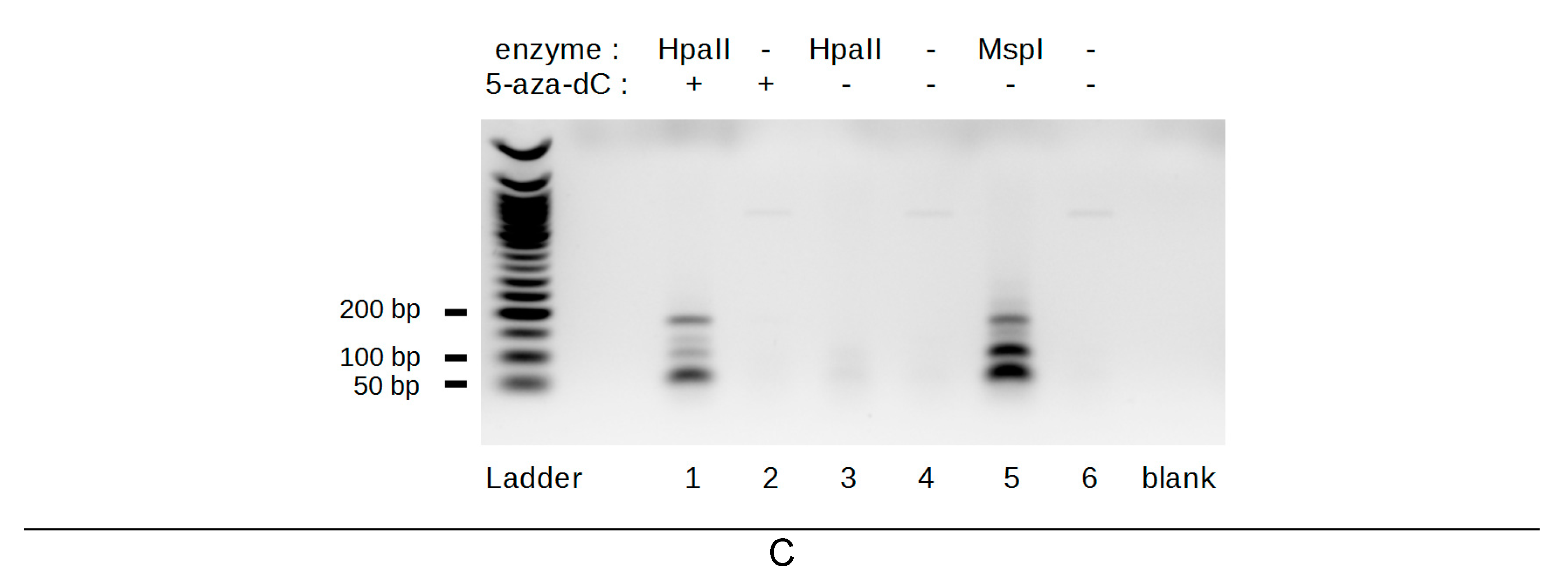
2.1. Development of A Method to Analyze The Methylation of The 29 bp Repeats
2.2. Telomerase-Positive and alternative lengthening of telomeres (ALT) Cells Are Differently Methylated at The Subtelomeric 29 bp Repeat
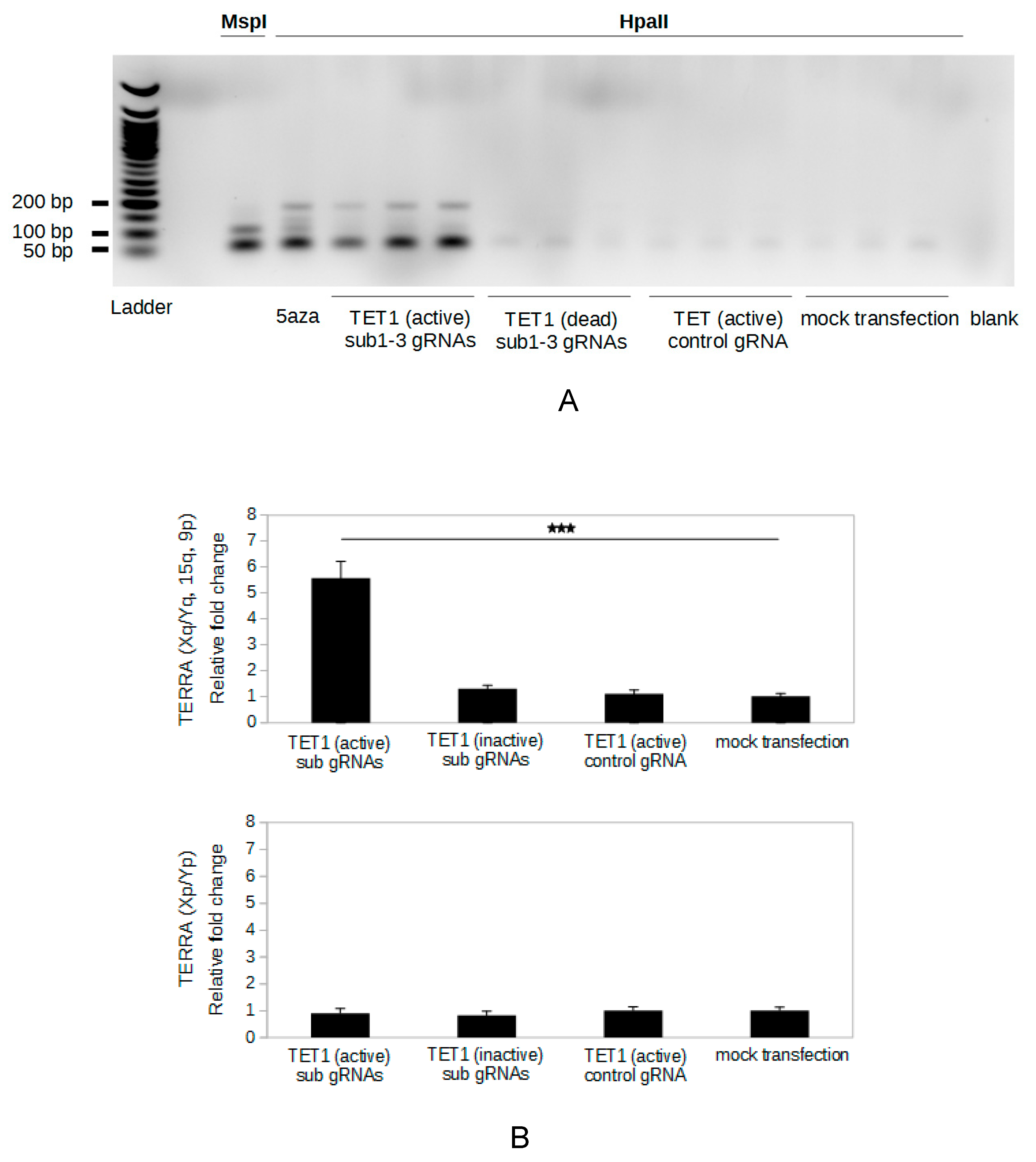
2.3. Specific Demethylation of The Subtelomeric CpG Islands Is Induced by The CRISPR-dCas9-TET1 System
2.4. Targeted Demethylation of The Subtelomeric CpG Islands Increases TERRA Expression
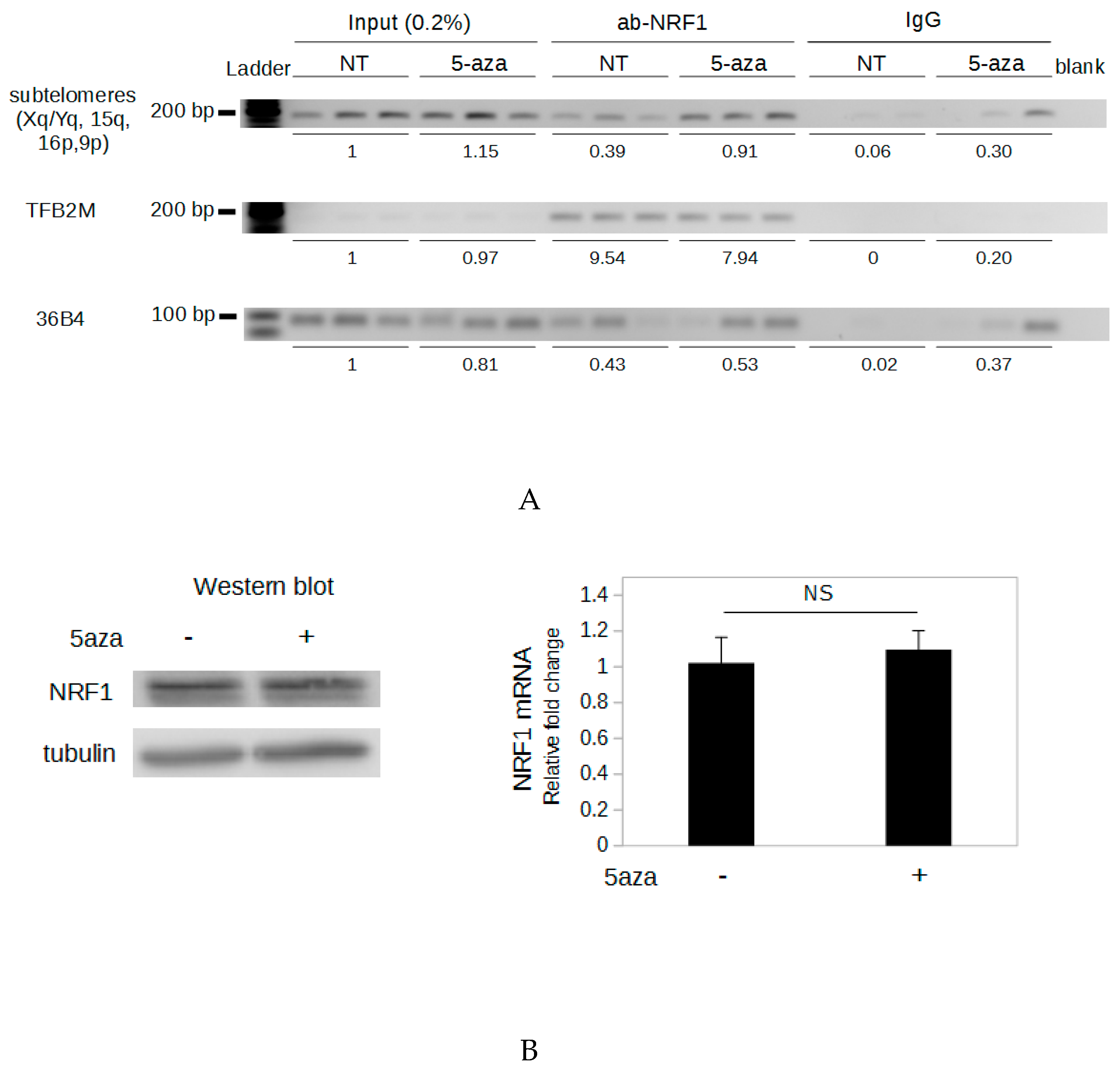
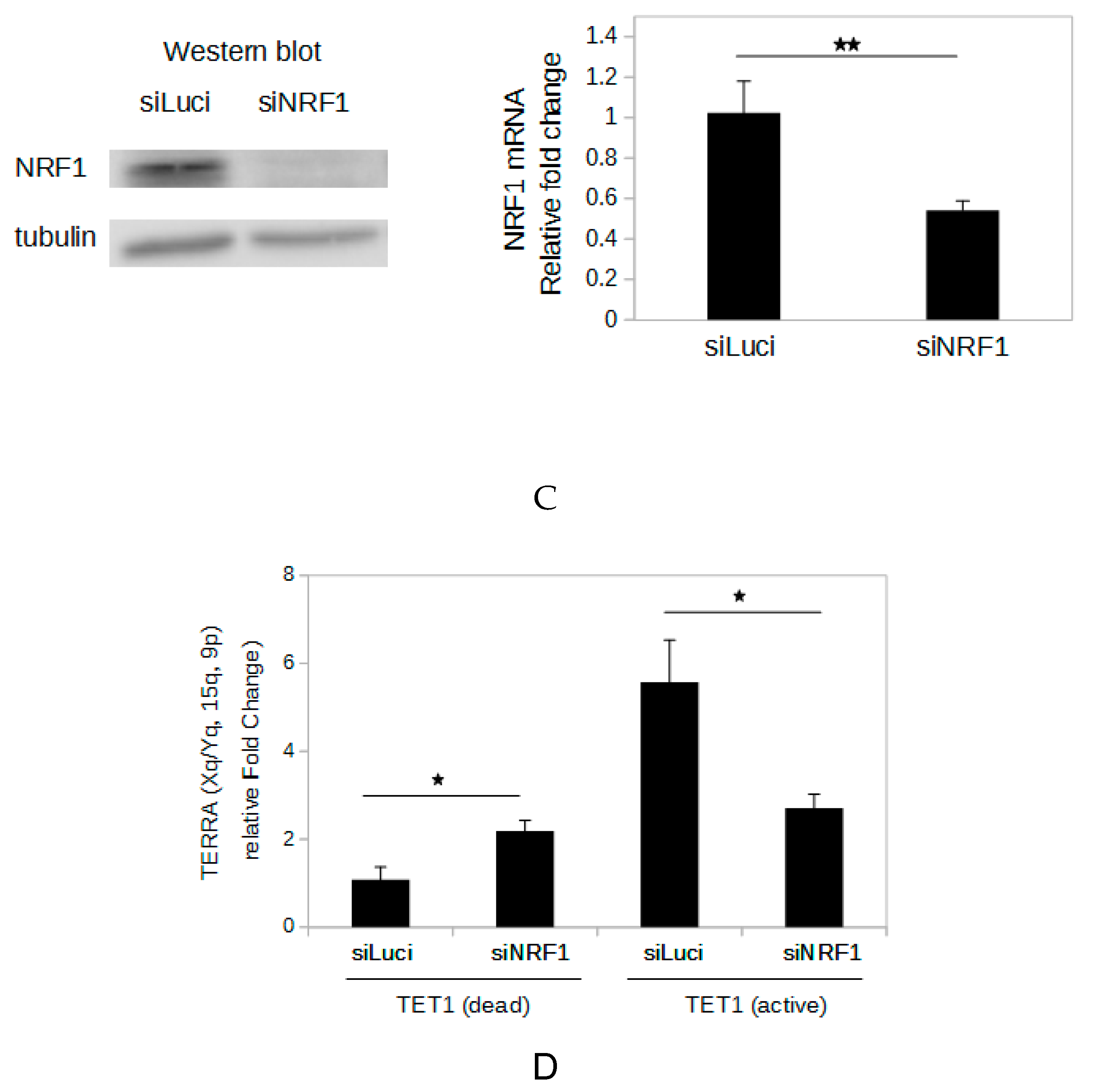
2.5. Demethylation of The Subtelomeric CpG Islands Induces An Increase in NRF1 Binding
2.6. NRF1 Is Involved in The DNA Demethylation-Dependent Up-Regulation of TERRA
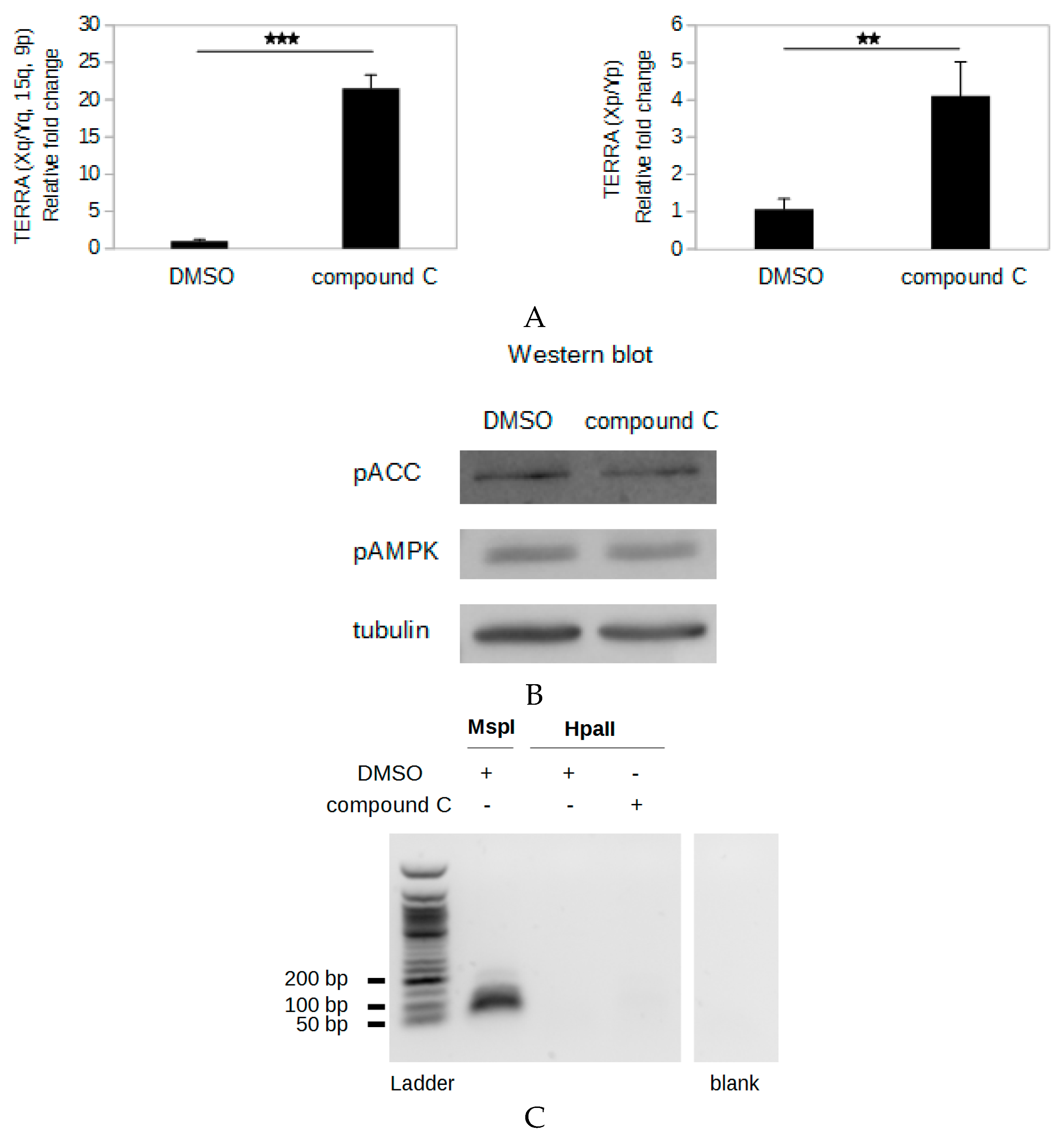
2.7. Compound C Induces An AMPK Inhibition-Independent up-Regulation of TERRA
3. Discussion
4. Materials and Methods
4.1. Construction of Plasmids
4.2. Cell Culture, Transfections, and Treatments
4.3. DNA Methylation Analysis
4.4. RNA and DNA Extraction
4.5. Quantitative RT-PCR Analysis
4.6. Protein Extraction and Western Blot Analysis
4.7. ChIP Assay and Semi-Quantitative PCR Analysis
Supplementary Materials
Author Contributions
Acknowledgements
Conflicts of Interest
References
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Riethman, H.; Ambrosini, A.; Paul, S. Human subtelomere structure and variation. Chromosome Res. 2005, 13, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, N.; Van Beneden, A.; Decottignies, A. Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1α. Nat. Struct. Mol. Biol. 2012, 19, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.J.; López-Silanes, I.; Megías, D.F.; Fraga, M.; Castells-García, Á.; Blasco, M.A. TERRA recruitment of polycomb to telomeres is essential for histone trymethylation marks at telomeric heterochromatin. Nat. Commun. 2018, 9, 1548. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.J.; López de Silanes, I.; Graña, O.; Blasco, M.A. Telomeric RNAs are essential to maintain telomeres. Nat. Commun. 2016, 7, 12534. [Google Scholar] [CrossRef]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef]
- Kreilmeier, T.; Mejri, D.; Hauck, M.; Kleiter, M.; Holzmann, K. Telomere Transcripts Target Telomerase in Human Cancer Cells. Genes (Basel) 2016, 7, 46. [Google Scholar] [CrossRef]
- Nergadze, S.G.; Farnung, B.O.; Wischnewski, H.; Khoriauli, L.; Vitelli, V.; Chawla, R.; Giulotto, E.; Azzalin, C.M. CpG-island promoters drive transcription of human telomeres. RNA 2009, 15, 2186–2194. [Google Scholar] [CrossRef]
- Farnung, B.O.; Brun, C.M.; Arora, R.; Lorenzi, L.E.; Azzalin, C.M. Telomerase Efficiently Elongates Highly Transcribing Telomeres in Human Cancer Cells. PLoS ONE 2012, 7, e35714. [Google Scholar] [CrossRef] [PubMed]
- Yehezkel, S.; Segev, Y.; Viegas-Péquignot, E.; Skorecki, K.; Selig, S. Hypomethylation of subtelomeric regions in ICF syndrome is associated with abnormally short telomeres and enhanced transcription from telomeric regions. Hum. Mol. Genet. 2008, 17, 2776–2789. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S.; et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Moarii, M.; Boeva, V.; Vert, J.-P.; Reyal, F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genom. 2015, 16, 873. [Google Scholar] [CrossRef] [PubMed]
- Guillaumet-Adkins, A.; Richter, J.; Odero, M.D.; Sandoval, J.; Agirre, X.; Catala, A.; Esteller, M.; Prósper, F.; Calasanz, M.J.; Buño, I.; et al. Hypermethylation of the alternative AWT1 promoter in hematological malignancies is a highly specific marker for acute myeloid leukemias despite high expression levels. J. Hematol. Oncol. 2014, 7, 4. [Google Scholar] [CrossRef]
- Sproul, D.; Nestor, C.; Culley, J.; Dickson, J.H.; Dixon, J.M.; Harrison, D.J.; Meehan, R.R.; Sims, A.H.; Ramsahoye, B.H. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4364–4369. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Domcke, S.; Bardet, A.F.; Adrian Ginno, P.; Hartl, D.; Burger, L.; Schübeler, D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015, 528, 575. [Google Scholar] [CrossRef]
- Li, W.; Li, X.; Wang, W.; Li, X.; Tan, Y.; Yi, M.; Yang, J.; McCarthy, J.B.; Xiong, W.; Wu, M.; et al. NOR1 is an HSF1- and NRF1-regulated putative tumor suppressor inactivated by promoter hypermethylation in nasopharyngeal carcinoma. Carcinogenesis 2011, 32, 1305–1314. [Google Scholar] [CrossRef]
- Lioznova, A.V.; Khamis, A.M.; Artemov, A.V.; Besedina, E.; Ramensky, V.; Bajic, V.B.; Kulakovskiy, I.V.; Medvedeva, Y.A. CpG traffic lights are markers of regulatory regions in human genome. BMC Genom. 2019, 20, 102. [Google Scholar] [CrossRef] [PubMed]
- Diman, A.; Boros, J.; Poulain, F.; Rodriguez, J.; Purnelle, M.; Episkopou, H.; Bertrand, L.; Francaux, M.; Deldicque, L.; Decottignies, A. Nuclear respiratory factor 1 and endurance exercise promote human telomere transcription. Sci. Adv. 2016, 2, e1600031. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tang, C.; Wang, Q.; Su, J.; Ni, T.; Yang, W.; Wang, Y.; Chen, W.; Liu, X.; Wang, S.; et al. NRF1 coordinates with DNA methylation to regulate spermatogenesis. FASEB J. 2017, 31, 4959–4970. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Zhang, X.; Vertino, P.M.; Cheng, X. The Mechanisms of Generation, Recognition, and Erasure of DNA 5-Methylcytosine and Thymine Oxidations. J. Biol. Chem. 2015, 290, 20723–20733. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Z.; Stong, N.; Plasschaert, R.; Moczan, A.; Chen, H.-S.; Hu, S.; Wikramasinghe, P.; Davuluri, R.V.; Bartolomei, M.S.; et al. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J. 2012, 31, 4165–4178. [Google Scholar] [CrossRef]
- Roach, H.I.; Hashimoto, K. PCR-Based Methods to Determine DNA Methylation Status at Specific CpG Sites Using Methylation-Sensitive Restriction Enzymes; Scion Publishing Limited: Banbury, UK, 2007; pp. 279–292. [Google Scholar]
- Ng, L.J.; Cropley, J.E.; Pickett, H.A.; Reddel, R.R.; Suter, C.M. Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res. 2009, 37, 1152–1159. [Google Scholar] [CrossRef]
- Tilman, G.; Loriot, A.; Van Beneden, A.; Arnoult, N.; Londoño-Vallejo, J.A.; De Smet, C.; Decottignies, A. Subtelomeric DNA hypomethylation is not required for telomeric sister chromatid exchanges in ALT cells. Oncogene 2009, 28, 1682–1693. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of Mitochondrial Transcription Specificity Factors (TFB1M and TFB2M) by Nuclear Respiratory Factors (NRF-1 and NRF-2) and PGC-1 Family Coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef]
- ENCSR550RTN—ENCODE. Available online: https://www.encodeproject.org/experiments/ENCSR550RTN/ (accessed on 13 March 2019).
- Scarpulla, R.C. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Et Biophys. Acta (Bba) Gene Struct. Expr. 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Thijssen, P.E.; Tobi, E.W.; Balog, J.; Schouten, S.G.; Kremer, D.; El Bouazzaoui, F.; Henneman, P.; Putter, H.; Eline Slagboom, P.; Heijmans, B.T.; et al. Chromatin remodeling of human subtelomeres and TERRA promoters upon cellular senescence: Commonalities and differences between chromosomes. Epigenetics 2013, 8, 512–521. [Google Scholar] [CrossRef][Green Version]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121. [Google Scholar] [CrossRef]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef]
- Lei, Y.; Huang, Y.-H.; Goodell, M.A. DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. 2018, 19, 187. [Google Scholar] [CrossRef]
- Beishline, K.; Vladimirova, O.; Tutton, S.; Wang, Z.; Deng, Z.; Lieberman, P.M. CTCF driven TERRA transcription facilitates completion of telomere DNA replication. Nat. Commun. 2017, 8, 2114. [Google Scholar] [CrossRef]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef]
- Rodriguez, C.; Borgel, J.; Court, F.; Cathala, G.; Forné, T.; Piette, J. CTCF is a DNA methylation-sensitive positive regulator of the INK/ARF locus. Biochem. Biophys. Res. Commun. 2010, 392, 129–134. [Google Scholar] [CrossRef]
- Kang, J.Y.; Song, S.H.; Yun, J.; Jeon, M.S.; Kim, H.P.; Han, S.W.; Kim, T.Y. Disruption of CTCF/cohesin-mediated high-order chromatin structures by DNA methylation downregulates PTGS2 expression. Oncogene 2015, 34, 5677–5684. [Google Scholar] [CrossRef]
- Maurano, M.T.; Wang, H.; John, S.; Shafer, A.; Canfield, T.; Lee, K.; Stamatoyannopoulos, J.A. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep. 2015, 12, 1184–1195. [Google Scholar] [CrossRef]
- Hashimoto, H.; Wang, D.; Horton, J.R.; Zhang, X.; Corces, V.G.; Cheng, X. Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol. Cell 2017, 66, 711–720.e3. [Google Scholar] [CrossRef]
- Choi, Y.S.; Kim, S.; Kyu Lee, H.; Lee, K.-U.; Pak, Y.K. In vitro methylation of nuclear respiratory factor-1 binding site suppresses the promoter activity of mitochondrial transcription factor A. Biochem. Biophys. Res. Commun. 2004, 314, 118–122. [Google Scholar] [CrossRef]
- Vera, E.; Canela, A.; Fraga, M.F.; Esteller, M.; Blasco, M.A. Epigenetic regulation of telomeres in human cancer. Oncogene 2008, 27, 6817–6833. [Google Scholar] [CrossRef]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010, 38, 5797–5806. [Google Scholar] [CrossRef]
- Sagie, S.; Toubiana, S.; Hartono, S.R.; Katzir, H.; Tzur-Gilat, A.; Havazelet, S.; Francastel, C.; Velasco, G.; Chédin, F.; Selig, S. Telomeres in ICF syndrome cells are vulnerable to DNA damage due to elevated DNA:RNA hybrids. Nat. Commun. 2017, 8, 14015. [Google Scholar] [CrossRef]
- Satoh, J.; Kawana, N.; Yamamoto, Y. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul. Syst. Biol. 2013, 7, GRSB.S13204. [Google Scholar] [CrossRef]
- Cam, H.; Balciunaite, E.; Blais, A.; Spektor, A.; Scarpulla, R.C.; Young, R.; Kluger, Y.; Dynlacht, B.D. A Common Set of Gene Regulatory Networks Links Metabolism and Growth Inhibition. Mol. Cell 2004, 16, 399–411. [Google Scholar] [CrossRef]
- de Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol. Cell. Biol. 1990, 10, 518–527. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Cui, Y.; Milton, J.R.; Li, J.; Irudayaraj, J. Selective increase in subtelomeric DNA methylation: An epigenetic biomarker for malignant glioma. Clin. Epigenet. 2015, 7, 107. [Google Scholar] [CrossRef]
- Oh, B.-K.; Um, T.-H.; Choi, G.H.; Park, Y.N. Frequent changes in subtelomeric DNA methylation patterns and its relevance to telomere regulation during human hepatocarcinogenesis. Int. J. Cancer 2011, 128, 857–868. [Google Scholar] [CrossRef]
- Feldmann, A.; Ivanek, R.; Murr, R.; Gaidatzis, D.; Burger, L.; Schübeler, D. Transcription Factor Occupancy Can Mediate Active Turnover of DNA Methylation at Regulatory Regions. PLoS Genet. 2013, 9, e1003994. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; Tiwari, V.K.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490. [Google Scholar] [CrossRef]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schübeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar] [CrossRef]
- Dubois-Chevalier, J.; Oger, F.; Dehondt, H.; Firmin, F.F.; Gheeraert, C.; Staels, B.; Lefebvre, P.; Eeckhoute, J. A dynamic CTCF chromatin binding landscape promotes DNA hydroxymethylation and transcriptional induction of adipocyte differentiation. Nucleic Acids Res. 2014, 42, 10943–10959. [Google Scholar] [CrossRef]
- Gebhard, C.; Benner, C.; Ehrich, M.; Schwarzfischer, L.; Schilling, E.; Klug, M.; Dietmaier, W.; Thiede, C.; Holler, E.; Andreesen, R.; et al. General Transcription Factor Binding at CpG Islands in Normal Cells Correlates with Resistance to De novo DNA Methylation in Cancer Cells. Cancer Res. 2010, 70, 1398–1407. [Google Scholar] [CrossRef]
- Vucicevic, L.; Misirkic, M.; Kristina, J.; Vilimanovich, U.; Sudar, E.; Isenovic, E.; Prica, M.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Vladimir, B.; et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011, 7, 40–50. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK Inhibitor Compound C Is a Potent AMPK-Independent Antiglioma Agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef]
- Cheruiyot, A.; Li, S.; Nickless, A.; Roth, R.; Fitzpatrick, J.A.J.; You, Z. Compound C inhibits nonsense-mediated RNA decay independently of AMPK. PLoS ONE 2018, 13, e0204978. [Google Scholar] [CrossRef]
- Dasgupta, B.; Seibel, W. Compound C/Dorsomorphin: Its Use and Misuse as an AMPK Inhibitor. In AMPK: Methods and Protocols; Neumann, D., Viollet, B., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 195–202. ISBN 978-1-4939-7598-3. [Google Scholar]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; Mclauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Feretzaki, M.; Lingner, J. A practical qPCR approach to detect TERRA, the elusive telomeric repeat-containing RNA. Methods 2017, 114, 39–45. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence (5′-3′) |
|---|---|
| assemb-TET1-F | CGGACCGGTGGCGGTGGCGGAGGGGCTAGCAGATCCGAACTGCCCACCTGCAGC |
| assemb-TET1-R | ATGGCTGATTATGATCTAGAGTCGCGGCCGCTCAGACCCAATGGTTATAGGG |
| TET1-mut-R | TTGTGAATGGCCCTGTAGGGATGAGCACAGAAGTCC |
| TET1-mut-F | CTGTGCTCATCCCTACAGGGCCATTCACAACATGAATAATGGAAG |
| TF | GCAGCCATGAATAATCAAGGT |
| TR | TTCCGCACTGAACCGCTCTAA |
| ChIP-F | GCTCTAACTGGTCTCTGACCT |
| ChIP-R | GTTCTGCTCAGCACAGACCTG |
| TFB2M-R | ACGGTCCACTCACAATCCTC |
| TFB2M-F | CCCACGTGGAACATTTTCTG |
| 36B4-F | CAGCAAGTGGGAAGGTGTAATCC |
| 36B4-R | CCCATTCTATCATCAACGGGTACAA |
| Xp-R | TCTTCTTTCTGGTGGGGTTG |
| Xp-F | GGGGTCCCTTTCCATACTGT |
| GAPDH-R | GAAGGTGAAGGTCGGAGTCAAC |
| GAPDH-F | CAGAGTTAAAAGCAGCCCTGGT |
| OT | GTTAGATCCCAGGCGTAGAACAGGCGCAGGCGCAGAGATddC |
| PR | GGACGCGCTAGCATGTGT |
| PF | GTTAGATCCCAGGCGTAGAACAG |
| sub1 | GCAGGGCTCTCTTGCTTAGAG |
| sub2 | CCTGCGCCACGCCTCCACCCC |
| sub3 | GTCCTCTGCACAGATTTCGG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Berre, G.; Hossard, V.; Riou, J.-F.; Guieysse-Peugeot, A.-L. Repression of TERRA Expression by Subtelomeric DNA Methylation Is Dependent on NRF1 Binding. Int. J. Mol. Sci. 2019, 20, 2791. https://doi.org/10.3390/ijms20112791
Le Berre G, Hossard V, Riou J-F, Guieysse-Peugeot A-L. Repression of TERRA Expression by Subtelomeric DNA Methylation Is Dependent on NRF1 Binding. International Journal of Molecular Sciences. 2019; 20(11):2791. https://doi.org/10.3390/ijms20112791
Chicago/Turabian StyleLe Berre, Gabriel, Virginie Hossard, Jean-Francois Riou, and Anne-Laure Guieysse-Peugeot. 2019. "Repression of TERRA Expression by Subtelomeric DNA Methylation Is Dependent on NRF1 Binding" International Journal of Molecular Sciences 20, no. 11: 2791. https://doi.org/10.3390/ijms20112791
APA StyleLe Berre, G., Hossard, V., Riou, J.-F., & Guieysse-Peugeot, A.-L. (2019). Repression of TERRA Expression by Subtelomeric DNA Methylation Is Dependent on NRF1 Binding. International Journal of Molecular Sciences, 20(11), 2791. https://doi.org/10.3390/ijms20112791