A Structure-Based Drug Discovery Paradigm


Abstract
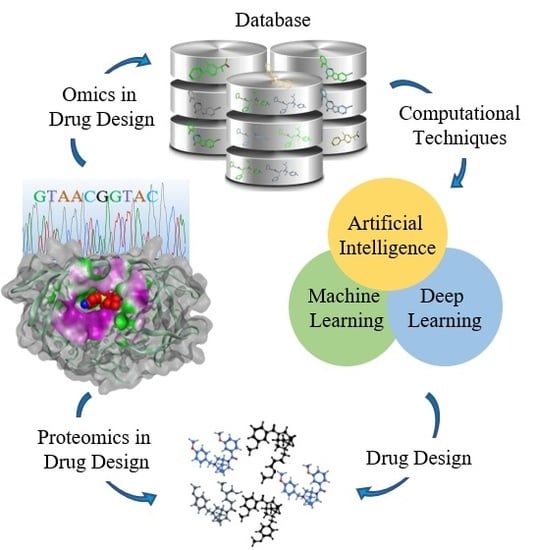
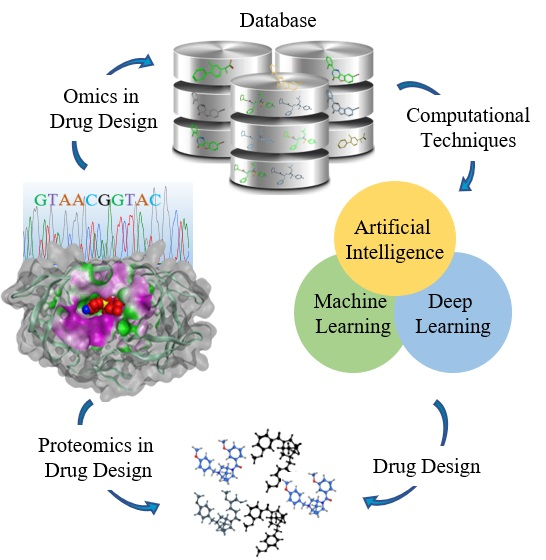
1. Introduction
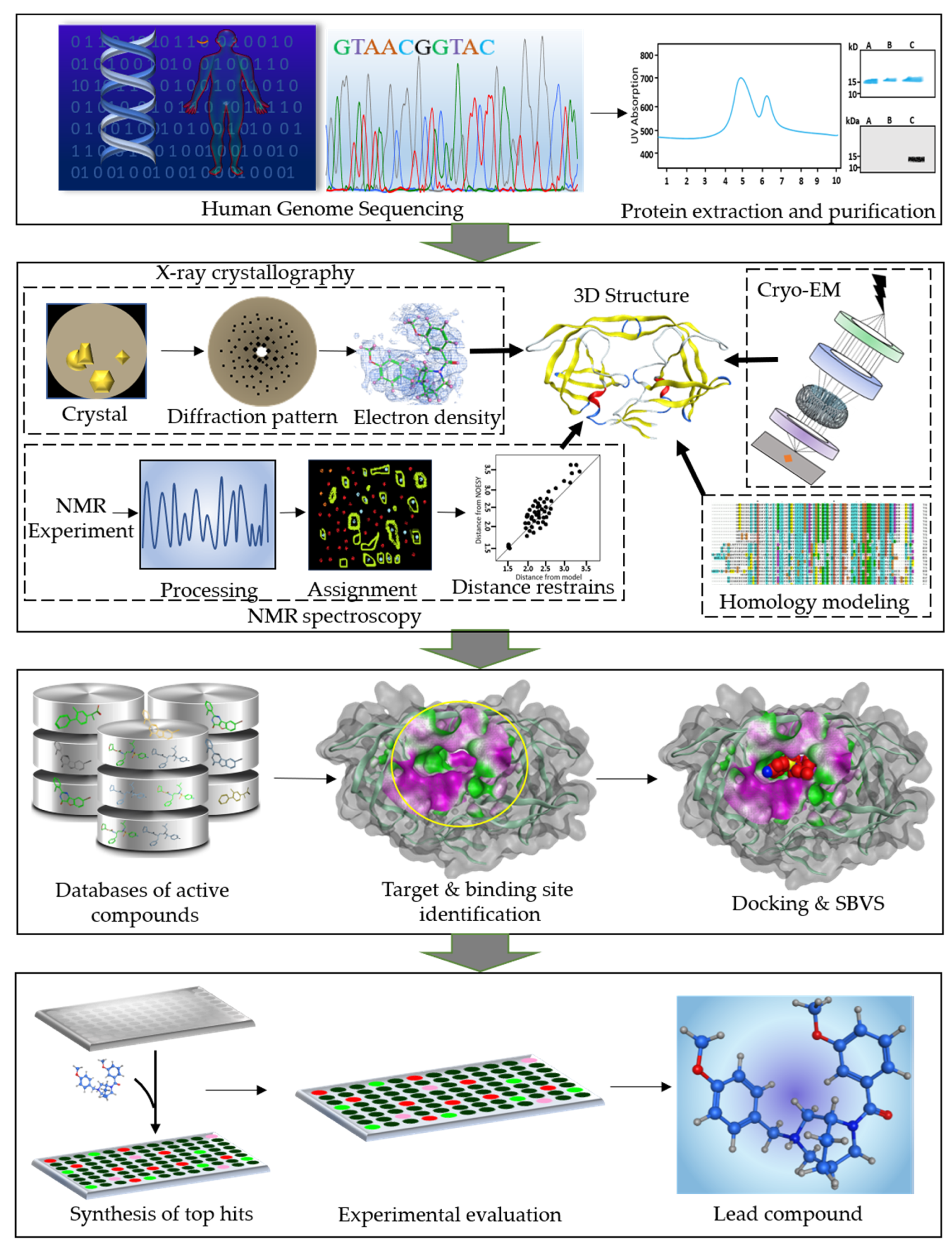
2. An Overview of SBDD Process
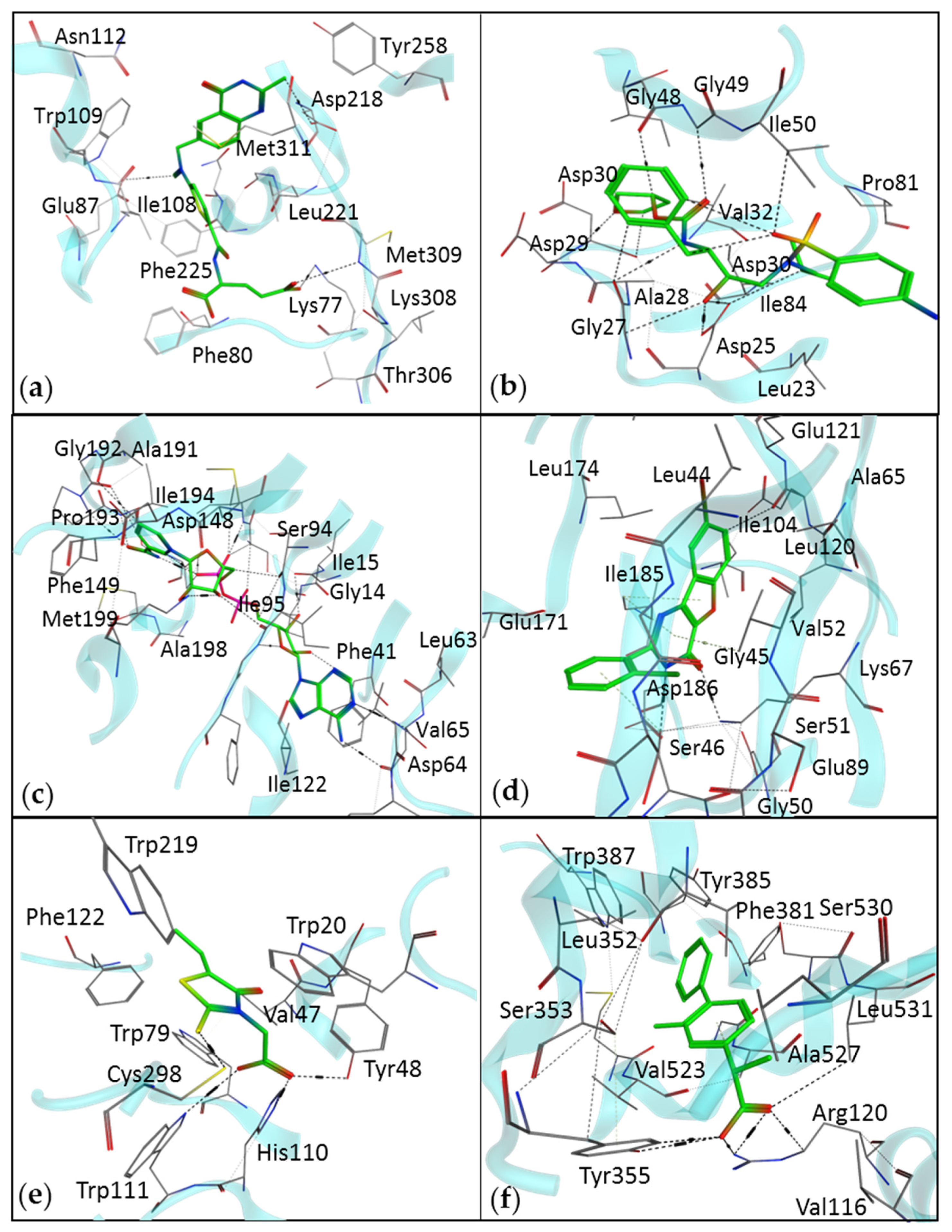
2.1. Target Protein and Binding Site Identification
2.2. Virtual Screening: A Lead Identification Approach
2.3. De Novo Drug Design
2.4. Molecular Docking
2.5. Scoring Functions
3. Big Data in Drug Discovery
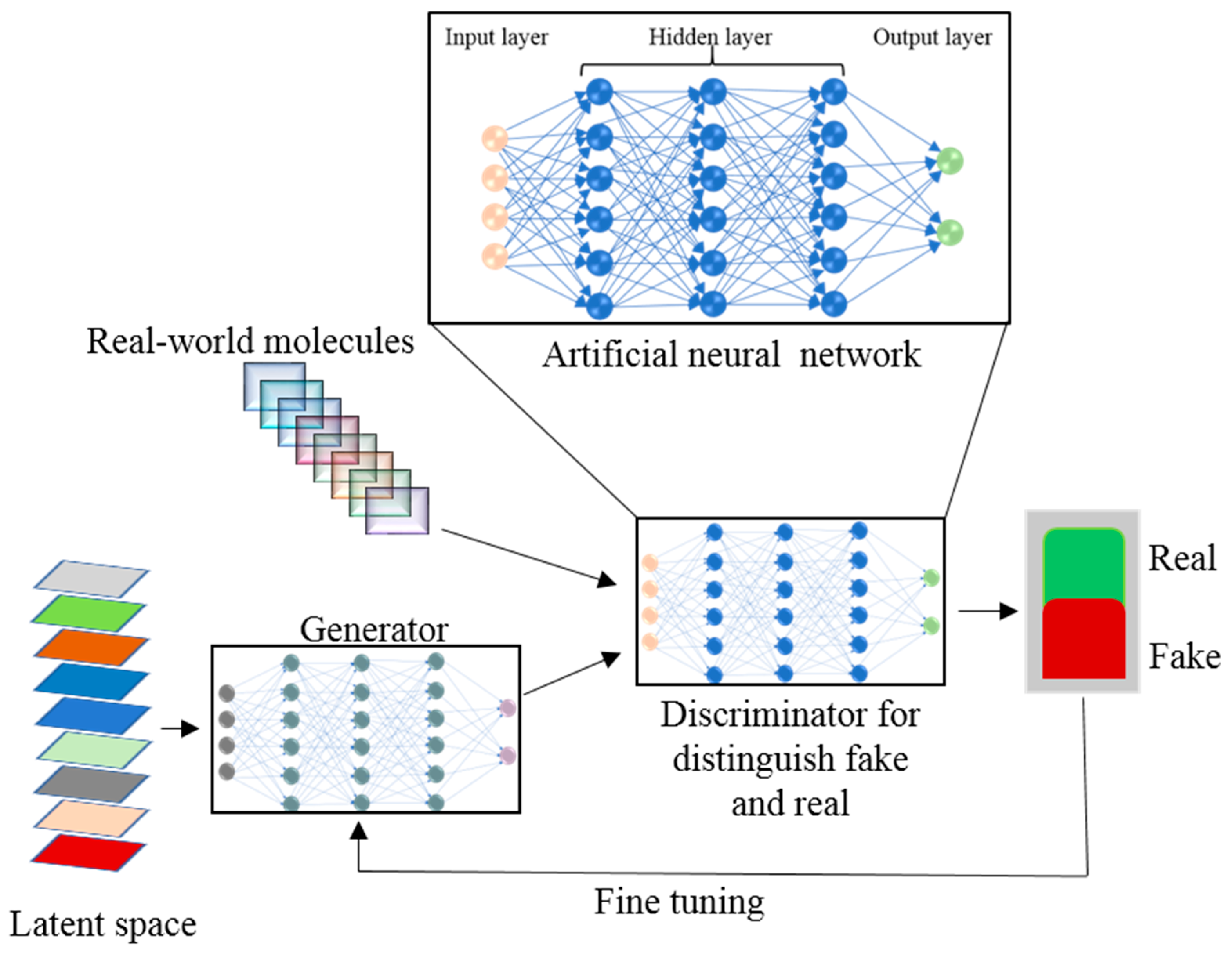
4. Artificial Intelligence and Machine Learning in Drug Discovery
5. The Role of Deep Learning in Drug Design
6. Challenges and Emerging Problems
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AI | Artificial intelligence |
| DL | Deep learning |
| HTS | High throughput screening |
| vdW | van der Waals |
| VS | Virtual screening |
| SBVS | Structure-based virtual screening |
| HIV | Human Immunodeficiency Virus |
| GA | Genetic algorithm |
| MC | Monte Carlo |
| SVM | Support vector machine |
| VAE | Variational autoencoder |
| RF | Random forest |
| ANN | Artificial neural network |
| DNN | Deep neural network |
| GAN | Generative adversarial network |
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| GSK | GlaxoSmithKline |
| RANC | Reinforced adversarial neural computer |
| RL | Reinforcement learning |
| MD | Molecular dynamics |
| GPCRs | G-protein-coupled receptors |
| STATs | Signal transducers and transcription activators |
| 3D | Three-dimensional |
| RNN | Recurrent neural network |
| ML | Machine learning |
| SBDD | Structure-based drug design |
| PDB | Protein data bank |
| NN | Neural Network |
| QSAR | Quantitative structure–activity relationship |
| QED | Quantitative estimate of drug-likeness |
| SMILES | Simplified molecular-input line-entry system |
References
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.J.; Zhang, H.; Anderson, G.; Alexander, G.C. Estimated costs of pivotal trials for novel therapeutic agents approved by the us food and drug administration, 2015–2016. JAMA Intern. Med. 2018, 178, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef]
- Searls, D.B. Data integration: Challenges for drug discovery. Nat. Rev. Drug Discov. 2005, 4, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Choi, S. Identification of druggable genome in staphylococcus aureus multidrug resistant strain. In Proceedings of the 2017 IEEE Life Sciences Conference (LSC), Sydney, NSW, Australia, 13–15 December 2017; pp. 270–273. [Google Scholar]
- Blaney, J. A very short history of structure-based design: How did we get here and where do we need to go? J. Comput. Aided Mol. Des. 2012, 26, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Moudgil, M.; Mandal, S.K. Rational drug design. Eur. J. Pharm. 2009, 625, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Urwyler, S. Allosteric modulation of family c g-protein-coupled receptors: From molecular insights to therapeutic perspectives. Pharm. Rev. 2011, 63, 59–126. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y. Ligand-receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 2012, 7, 969–988. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu Rev. Biophys Biomol. Struct. 1998, 27, 249–284. [Google Scholar] [CrossRef]
- Clark, D.E. What has computer-aided molecular design ever done for drug discovery? Expert Opin. Drug Discov. 2006, 1, 103–110. [Google Scholar] [CrossRef]
- Rutenber, E.E.; Stroud, R.M. Binding of the anticancer drug zd1694 to E. Coli thymidylate synthase: Assessing specificity and affinity. Structure 1996, 4, 1317–1324. [Google Scholar] [CrossRef]
- De Paulis, T. Drug evaluation: Prx-00023, a selective 5-ht1a receptor agonist for depression. Curr. Opin. Investig. Drugs 2007, 8, 78–86. [Google Scholar]
- Marrakchi, H.; Laneelle, G.; Quemard, A. Inha, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, fas-ii. Microbiology 2000, 146, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.X.; Li, L.L.; Zheng, R.L.; Xie, H.Z.; Cao, Z.X.; Feng, S.; Pan, Y.L.; Chen, X.; Wei, Y.Q.; Yang, S.Y. Discovery of novel pim-1 kinase inhibitors by a hierarchical multistage virtual screening approach based on svm model, pharmacophore, and molecular docking. J. Chem. Inf. Model. 2011, 51, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gu, Q.; Zheng, X.; Ye, J.; Liu, Z.; Li, J.; Hu, X.; Hagler, A.; Xu, J. Discovery of new selective human aldose reductase inhibitors through virtual screening multiple binding pocket conformations. J. Chem. Inf. Model. 2013, 53, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Dadashpour, S.; Tuylu Kucukkilinc, T.; Unsal Tan, O.; Ozadali, K.; Irannejad, H.; Emami, S. Design, synthesis and in vitro study of 5,6-diaryl-1,2,4-triazine-3-ylthioacetate derivatives as cox-2 and beta-amyloid aggregation inhibitors. Arch. Pharm. 2015, 348, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.; Kim, K.S.; Lee, D.M.; Kasam, V.; Baek, S.E.; Lee, K.H.; Zhang, Y.Y.; Ao, L.; Carmony, K.; Lee, N.R.; et al. Proteasome inhibitors with pyrazole scaffolds from structure-based virtual screening. J. Med. Chem. 2015, 58, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Masuda, Y.; Uehara, Y.; Sato, H.; Muroya, A.; Takahashi, O.; Yokotagawa, T.; Furuya, T.; Okawara, T.; Otsuka, M.; et al. Identification of a new series of stat3 inhibitors by virtual screening. ACS Med. Chem. Lett. 2010, 1, 371–375. [Google Scholar] [CrossRef]
- Grover, S.; Apushkin, M.A.; Fishman, G.A. Topical dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa. Am. J. Ophthalmol. 2006, 141, 850–858. [Google Scholar] [CrossRef]
- Grant, M.A. Protein structure prediction in structure-based ligand design and virtual screening. Comb. Chem. High Throughput Screen. 2009, 12, 940–960. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in casp8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using swiss-model workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef]
- Potapov, V.; Cohen, M.; Inbar, Y.; Schreiber, G. Protein structure modelling and evaluation based on a 4-distance description of side-chain interactions. BMC Bioinform. 2010, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Laurie, A.T.; Jackson, R.M. Q-sitefinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef]
- Wunberg, T.; Hendrix, M.; Hillisch, A.; Lobell, M.; Meier, H.; Schmeck, C.; Wild, H.; Hinzen, B. Improving the hit-to-lead process: Data-driven assessment of drug-like and lead-like screening hits. Drug Discov. Today 2006, 11, 175–180. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Phatak, S.S.; Stephan, C.C.; Cavasotto, C.N. High-throughput and in silico screenings in drug discovery. Expert. Opin. Drug Discov. 2009, 4, 947–959. [Google Scholar] [CrossRef]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.N.; Sastry, G.N. Virtual screening in drug discovery—A computational perspective. Curr. Protein Pept. Sci 2007, 8, 329–351. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Vistoli, G. Rescoring and linearly combining: A highly effective consensus strategy for virtual screening campaigns. Int. J. Mol. Sci 2019, 20, 2060. [Google Scholar] [CrossRef]
- Hartenfeller, M.; Schneider, G. De novo drug design. Methods Mol. Biol. 2011, 672, 299–323. [Google Scholar]
- Richardson, J.S.; Richardson, D.C. The de novo design of protein structures. Trends Biochem. Sci 1989, 14, 304–309. [Google Scholar] [CrossRef]
- Lameijer, E.W.; Tromp, R.A.; Spanjersberg, R.F.; Brussee, J.; Ijzerman, A.P. Designing active template molecules by combining computational de novo design and human chemist’s expertise. J. Med. Chem 2007, 50, 1925–1932. [Google Scholar] [CrossRef]
- Gillet, V.J. New directions in library design and analysis. Curr. Opin. Chem. Biol. 2008, 12, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Keseru, G.M.; Makara, G.M. Hit discovery and hit-to-lead approaches. Drug Discov. Today 2006, 11, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhu, W.; Chen, K.; Jiang, H. New technologies in computer-aided drug design: Toward target identification and new chemical entity discovery. Drug Discov. Today Technol. 2006, 3, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Prada-Gracia, D.; Huerta-Yepez, S.; Moreno-Vargas, L.M. Application of computational methods for anticancer drug discovery, design, and optimization. Bol. Med. Hosp. Infan.t Mex. 2016, 73, 411–423. [Google Scholar]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci 2010, 11, 3016–3034. [Google Scholar] [CrossRef]
- Lopez-Vallejo, F.; Caulfield, T.; Martinez-Mayorga, K.; Giulianotti, M.A.; Nefzi, A.; Houghten, R.A.; Medina-Franco, J.L. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Comb. Chem. High. Throughput Screen. 2011, 14, 475–487. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development (caddd): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.D.; Jewsbury, P.J.; Essex, J.W. A review of protein-small molecule docking methods. J. Comput. Aided Mol. Des. 2002, 16, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, C.M.; Kuntz, I.D.; Dixon, J.S. Flexible ligand docking using a genetic algorithm. J. Comput. Aided Mol. Des. 1995, 9, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.N.; Read, R.J. A multiple-start monte carlo docking method. Proteins 1992, 13, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput Mol. Sci 2015, 5, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharm. 2008, 153, 7–26. [Google Scholar] [CrossRef]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical scoring functions for structure-based virtual screening: Applications, critical aspects, and challenges. Front. Pharm. 2018, 9, 1089. [Google Scholar] [CrossRef]
- Muegge, I. Pmf scoring revisited. J. Med. Chem. 2006, 49, 5895–5902. [Google Scholar] [CrossRef]
- Li, H.; Peng, J.; Leung, Y.; Leung, K.S.; Wong, M.H.; Lu, G.; Ballester, P.J. The impact of protein structure and sequence similarity on the accuracy of machine-learning scoring functions for binding affinity prediction. Biomolecules 2018, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- David, H.; Gary, B.F. Computational intelligence methods for docking scores. Curr. Comput. Aided Drug Des. 2009, 5, 56–68. [Google Scholar]
- Cheng, T.; Li, X.; Li, Y.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on a diverse test set. J. Chem. Inf. Model. 2009, 49, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, P.; Gohlke, H.; Price, D.J.; Klebe, G.; Brooks, C.L., 3rd. Assessing scoring functions for protein-ligand interactions. J. Med. Chem. 2004, 47, 3032–3047. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Fang, X.; Wang, S. An extensive test of 14 scoring functions using the pdbbind refined set of 800 protein-ligand complexes. J. Chem. Inf. Comput. Sci 2004, 44, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci 2001, 41, 1422–1426. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein-ligand interactions. J. Chem. Inf. Model. 2010, 50, 262–273. [Google Scholar] [CrossRef]
- Raub, S.; Steffen, A.; Kamper, A.; Marian, C.M. Aiscore chemically diverse empirical scoring function employing quantum chemical binding energies of hydrogen-bonded complexes. J. Chem. Inf. Model. 2008, 48, 1492–1510. [Google Scholar] [CrossRef]
- Seifert, M.H. Targeted scoring functions for virtual screening. Drug Discov. Today 2009, 14, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Martínez, F.D.; López-López, E.; Eurídice Juárez-Mercado, K.; Medina-Franco, J.L. Chapter 2—computational drug design methods—current and future perspectives. In In silico drug design; Roy, K., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 19–44. [Google Scholar]
- Akoka, J.; Comyn-Wattiau, I.; Laoufi, N. Research on big data—A systematic mapping study. Comput. Stand. Interfaces 2017, 54, 105–115. [Google Scholar] [CrossRef]
- Secchi, P. On the role of statistics in the era of big data: A call for a debate. Stat. Probab. Lett. 2018, 136, 10–14. [Google Scholar] [CrossRef]
- Cox, D.R.; Kartsonaki, C.; Keogh, R.H. Big data: Some statistical issues. Stat. Probab. Lett. 2018, 136, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Bornmann, L. Measuring the societal impact of research. EMBO Rep. 2012, 13, 673. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, P.; Fors, U.; Wallin, S.-B.; Zander, U.; Nilsson, G.H. Evaluating research: A multidisciplinary approach to assessing research practice and quality. Res. Policy 2016, 45, 593–603. [Google Scholar] [CrossRef]
- Cabrera, M.T.; Brewer, E.M.; Grant, L.; Tarczy-Hornoch, K. Exudative retinal detachment documented by handheld spectral domain optical coherence tomography after retinal laser photocoagulation for retinopathy of prematurity. Retin. Cases Brief. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent progress in the development of HIV-1 protease inhibitors for the treatment of hiv/aids. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef]
- Barmania, F.; Pepper, M.S. C-c chemokine receptor type five (ccr5): An emerging target for the control of hiv infection. Appl. Transl. Genom 2013, 2, 3–16. [Google Scholar] [CrossRef]
- MacArthur, R.D.; Novak, R.M. Reviews of anti-infective agents: Maraviroc: The first of a new class of antiretroviral agents. Clin. Infect. Dis. 2008, 47, 236–241. [Google Scholar] [CrossRef]
- Kuritzkes, D.; Kar, S.; Kirkpatrick, P. Maraviroc. Nat. Rev. Drug Discov. 2008, 7, 15. [Google Scholar] [CrossRef]
- Lusher, S.J.; McGuire, R.; van Schaik, R.C.; Nicholson, C.D.; de Vlieg, J. Data-driven medicinal chemistry in the era of big data. Drug Discov. Today 2014, 19, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Ebejer, J.P.; Fulle, S.; Morris, G.M.; Finn, P.W. The emerging role of cloud computing in molecular modelling. J. Mol. Graph. Model. 2013, 44, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Kissin, I. What can big data on academic interest reveal about a drug? Reflections in three major us databases. Trends Pharm. Sci 2018, 39, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Pichika, M.R. Artificial intelligence in drug development: Present status and future prospects. Drug Discov. Today 2019, 24, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.M. Model-based machine learning. Philos Trans. A Math. Phys. Eng. Sci 2013, 371, 20120222. [Google Scholar] [CrossRef] [PubMed]
- Duch, W.; Swaminathan, K.; Meller, J. Artificial intelligence approaches for rational drug design and discovery. Curr. Pharm. Des. 2007, 13, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Probst, C.; Schneider, S.; Loskill, P. High-throughput Organ-on-a-chip systems: Current status and remaining challenges. Curr. Opin. Biomed. Eng. 2018, 6, 33–41. [Google Scholar] [CrossRef]
- IBM. Ibm Watson. Available online: https://www.ibm.com/watson (accessed on 1 May 2019).
- Smalley, E. Ai-powered drug discovery captures pharma interest. Nat. Biotechnol. 2017, 35, 604–605. [Google Scholar] [CrossRef]
- Exscientia. At the forefront of small molecule drug discovery. Available online: https://www.exscientia.co.uk/ (accessed on 1 May 2019).
- Fleming, N. How artificial intelligence is changing drug discovery. Nature 2018, 557, 55–57. [Google Scholar] [CrossRef]
- Exscientia. Celgene and exscientia enter 3-year ai drug discovery collaboration focused on accelerating drug discovery in oncology and autoimmunity. Available online: https://www.exscientia.co.uk/news (accessed on 1 May 2019).
- Exscientia. Exscientia achieves molecule discovery milestone as part of gsk collaboration. Available online: https://www.exscientia.co.uk/news (accessed on 1 May 2019).
- Guncar, G.; Kukar, M.; Notar, M.; Brvar, M.; Cernelc, P.; Notar, M.; Notar, M. An application of machine learning to haematological diagnosis. Sci Rep. 2018, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Byvatov, E.; Fechner, U.; Sadowski, J.; Schneider, G. Comparison of support vector machine and artificial neural network systems for drug/nondrug classification. J. Chem. Inf. Comput. Sci 2003, 43, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Zernov, V.V.; Balakin, K.V.; Ivaschenko, A.A.; Savchuk, N.P.; Pletnev, I.V. Drug discovery using support vector machines. The case studies of drug-likeness, agrochemical-likeness, and enzyme inhibition predictions. J. Chem. Inf. Comput. Sci 2003, 43, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Warmuth, M.K.; Liao, J.; Ratsch, G.; Mathieson, M.; Putta, S.; Lemmen, C. Active learning with support vector machines in the drug discovery process. J. Chem. Inf. Comput. Sci 2003, 43, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Gilson, M.K. Virtual screening of molecular databases using a support vector machine. J. Chem. Inf. Model. 2005, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Koohy, H. The rise and fall of machine learning methods in biomedical research. F1000Res 2017, 6, 2012. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Cai, C.; Lu, X. Unsupervised deep learning reveals prognostically relevant subtypes of glioblastoma. BMC Bioinform. 2017, 18, 381. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Lima, A.N.; Philot, E.A.; Trossini, G.H.; Scott, L.P.; Maltarollo, V.G.; Honorio, K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 225–239. [Google Scholar] [CrossRef]
- Ma, X.H.; Jia, J.; Zhu, F.; Xue, Y.; Li, Z.R.; Chen, Y.Z. Comparative analysis of machine learning methods in ligand-based virtual screening of large compound libraries. COMB Chem. High Throughput Screen. 2009, 12, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Han, L.Y.; Ma, X.H.; Lin, H.H.; Jia, J.; Zhu, F.; Xue, Y.; Li, Z.R.; Cao, Z.W.; Ji, Z.L.; Chen, Y.Z. A support vector machines approach for virtual screening of active compounds of single and multiple mechanisms from large libraries at an improved hit-rate and enrichment factor. J. Mol. Graph. Model. 2008, 26, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Ma, X.H.; Tan, C.Y.; Jiang, Y.Y.; Go, M.L.; Low, B.C.; Chen, Y.Z. Virtual screening of abl inhibitors from large compound libraries by support vector machines. J. Chem. Inf. Model. 2009, 49, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Van Gerven, M.; Bohte, S. Artificial Neural Networks as Models of Neural Information Processing; Frontiers Media SA: Lausanne, Switzerland, 2018. [Google Scholar]
- Dreyfus, S. The computational solution of optimal control problems with time lag. IEEE Trans. Autom. Control. 1973, 18, 383–385. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of deep learning in biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Howard, J. The business impact of deep learning. In Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Chicago, IL, USA, 11–14 August 2013; ACM: New York, NY, USA, 2013; p. 1135. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Atomwise. Artificial intelligence for drug discovery. Available online: https://www.atomwise.com/ (accessed on 24 April 2019).
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. Deeptox: Toxicity prediction using deep learning. Front. Env. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Suthaharan, S. Support vector machine. In Machine Learning Models and Algorithms for Big Data Classification: Thinking with Examples for Effective Learning; Springer US: Boston, MA, USA, 2016; pp. 207–235. [Google Scholar]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Kruger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The chembl bioactivity database: An update. Nucleic Acids Res. 2014, 42, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. Drugan: An advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Jiang, D.; Nambiar, D.K.; Liew, L.P.; Hay, M.P.; Bloomstein, J.; Lu, P.; Turner, B.; Le, Q.T.; Tibshirani, R.; et al. Chemical space mimicry for drug discovery. J. Chem. Inf. Model. 2017, 57, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Nieto-Draghi, C.; Fayet, G.; Creton, B.; Rozanska, X.; Rotureau, P.; de Hemptinne, J.C.; Ungerer, P.; Rousseau, B.; Adamo, C. A general guidebook for the theoretical prediction of physicochemical properties of chemicals for regulatory purposes. Chem. Rev. 2015, 115, 13093–13164. [Google Scholar] [CrossRef]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef]
- Pereira, J.C.; Caffarena, E.R.; dos Santos, C.N. Boosting docking-based virtual screening with deep learning. J. Chem. Inf. Model. 2016, 56, 2495–2506. [Google Scholar] [CrossRef]
- Hughes, T.B.; Miller, G.P.; Swamidass, S.J. Modeling epoxidation of drug-like molecules with a deep machine learning network. ACS Cent. Sci 2015, 1, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. Let’s not forget tautomers. J. Comput. Aided Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Mangiatordi, G.F.; Alberga, D.; Altomare, C.D.; Carotti, A.; Catto, M.; Cellamare, S.; Gadaleta, D.; Lattanzi, G.; Leonetti, F.; Pisani, L.; et al. Mind the gap! A journey towards computational toxicology. Mol. Inf. 2016, 35, 294–308. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Drug Target | Target Disease | Technique | Ref. |
|---|---|---|---|---|
| Raltitrexed | Thymidylate synthase | Human immunodeficiency virus (HIV) | SBDD | [8] |
| Amprenavir | Antiretroviral protease | HIV | Protein modeling and molecular dynamics (MD) | [18,19] |
| Isoniazid | InhA | Tuberculosis | Structure-based virtual screening (SBVS) and pharmacophore modeling | [22] |
| Pim-1 Kinase Inhibitors | Pim-1 Kinase | Cancer | Hierarchical multistage virtual screening (VS) | [23] |
| Epalrestat 2 | Aldose Reductase | Diabetic neuropathy | MD and SBVS | [24] |
| Flurbiprofen | Cyclooxygenase-2 | Rheumatoid arthritis, Osteoarthritis | Molecular docking | [25,26] |
| STX-0119 | STAT3 1 | Lymphoma | SBVS | [27] |
| Norfloxacin | Topoisomerase II, IV | Urinary tract infection | SBVS | |
| Dorzolamide | Carbonic anhydrase | Glaucoma, cystoid macular edema | Fragment-based screening | [28] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. https://doi.org/10.3390/ijms20112783
Batool M, Ahmad B, Choi S. A Structure-Based Drug Discovery Paradigm. International Journal of Molecular Sciences. 2019; 20(11):2783. https://doi.org/10.3390/ijms20112783
Chicago/Turabian StyleBatool, Maria, Bilal Ahmad, and Sangdun Choi. 2019. "A Structure-Based Drug Discovery Paradigm" International Journal of Molecular Sciences 20, no. 11: 2783. https://doi.org/10.3390/ijms20112783
APA StyleBatool, M., Ahmad, B., & Choi, S. (2019). A Structure-Based Drug Discovery Paradigm. International Journal of Molecular Sciences, 20(11), 2783. https://doi.org/10.3390/ijms20112783