Parkin Mutation Affects Clock Gene-Dependent Energy Metabolism

 , ,
, ,  , and
, and 
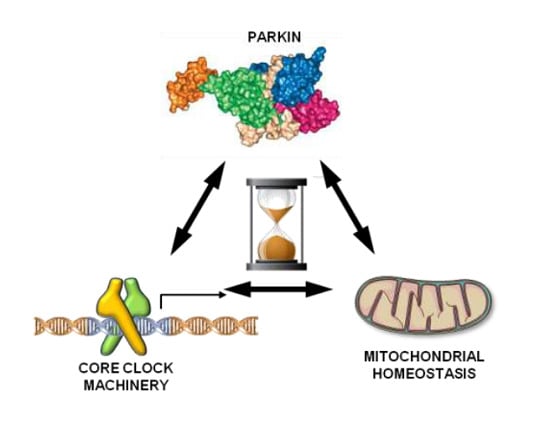
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
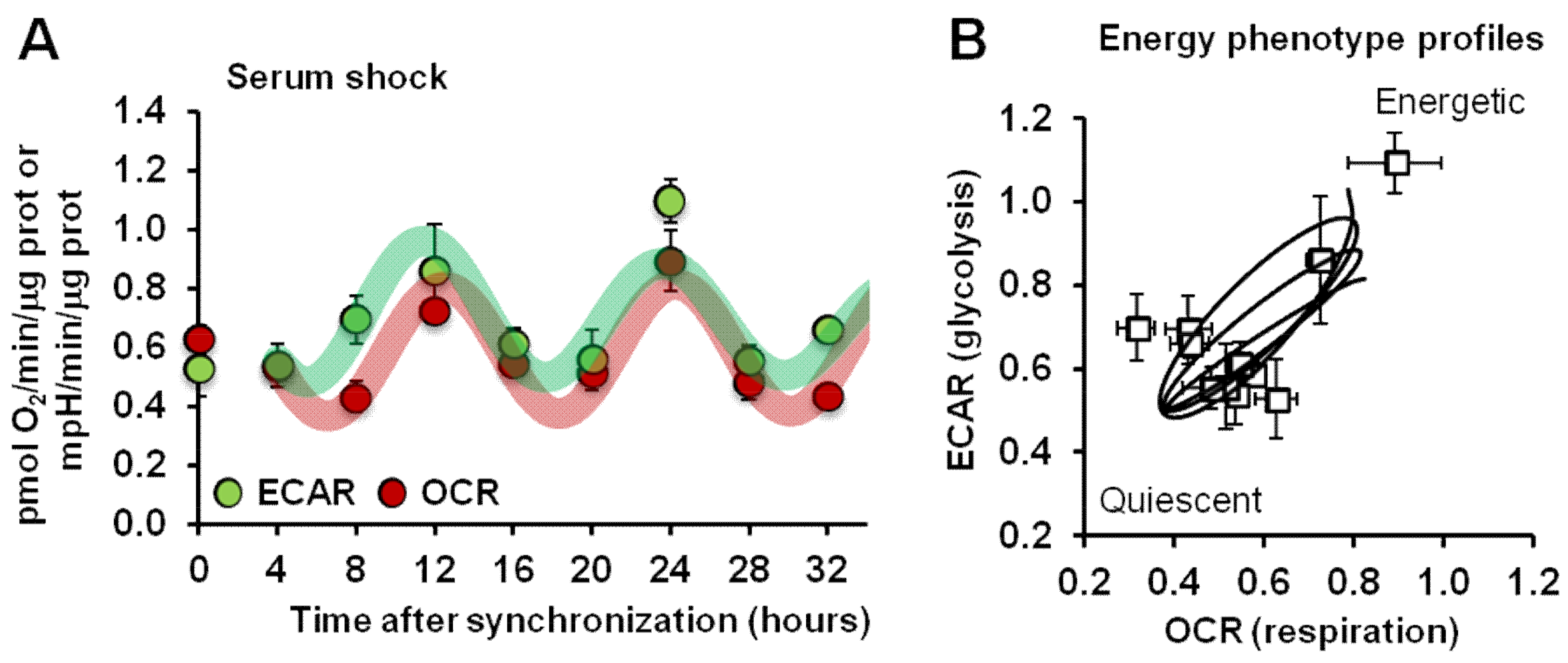
2.1. Synchronized Fibroblasts Exhibit Autonomous Oscillatory Mitochondrial Respiration and Glycolytic Activity
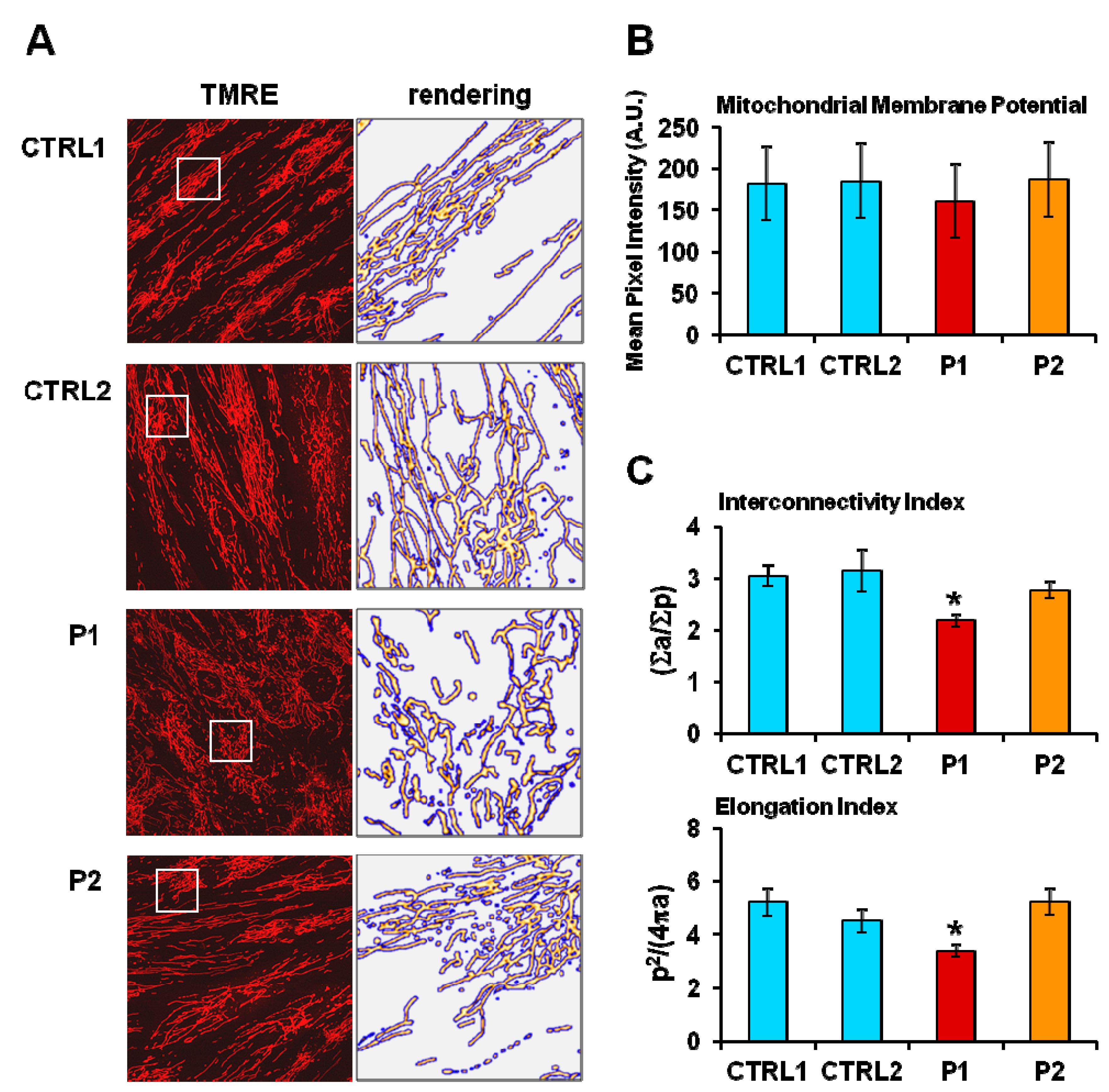
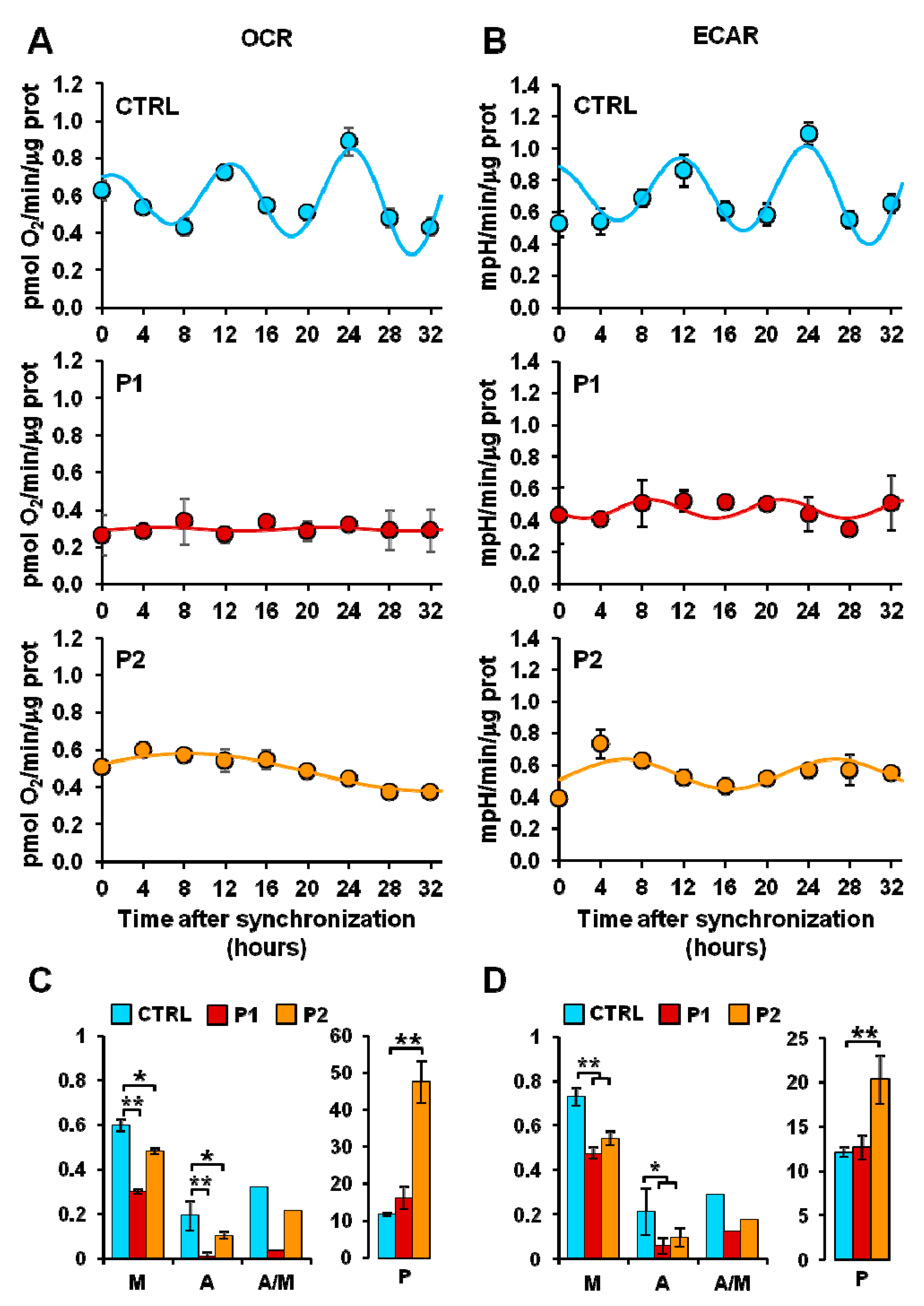
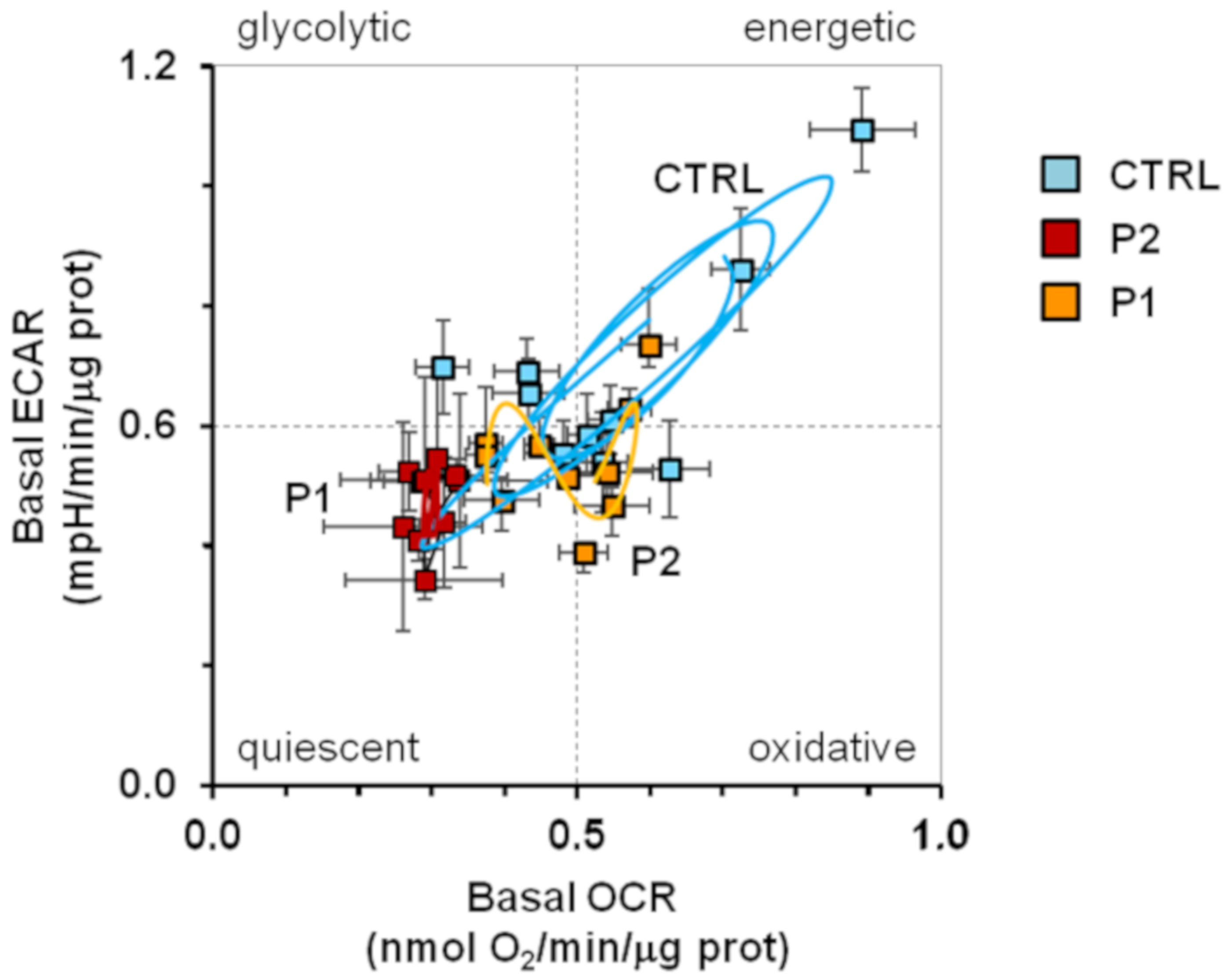
2.2. Energy Metabolism in Patient Fibroblasts with the Parkin Mutation
2.3. Circadian Clock-Dependent Energy Metabolism in Patients’ Fibroblasts with Mutated Parkin
2.4. Circadian Expression of Genes Involved in Mitochondrial Quality Control
2.5. Circadian Clock-Gene Expression in Patient Fibroblasts is Deregulated in Parkin-Null Fibrolasts
2.6. Core Clock Genes Expression in Normal and PD Patient Fibroblast-Derived Induced Pluripotent Stem Cells and Neurospheres
3. Discussion
4. Materials and Methods
4.1. Skin Fibroblast Harvesting and Culture Conditions
4.2. Seahorse Analysis
4.3. Laser Scanning Confocal Microscopy (LSCM) Live Cell Imaging of Mitochondrial Membrane Potential and Mitochondrial Morphology Analysis
4.4. Real-Time PCR
4.5. Fibroblast Reprogramming
4.6. Immunofluorescence Staining
4.7. In Vitro Spontaneous Differentiation
4.8. Karyotyping
4.9. Teratoma Formation
4.10. iPSC Neuralization
4.11. Protein Measurement
4.12. Cosinor Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BMAL1/ARNTL | aryl hydrocarbon receptor nuclear translocator like |
| CLOCK | circadian locomotor cycle kaput |
| CRY1,2 | cryptochrome circadian regulator 1 and 2 |
| NR1D1 | nuclear receptor subfamily 1 group D member 1 |
| iPSC | induced pluripotent stem cells |
| iNSC | induced neural stem cells |
| NHDF | normal human dermal fibroblasts |
| PER 1,2,3 | period circadian clock 1,2,3 |
| PGC-1α | peroxisome proliferator-activated receptor (PPAR)-γ coactivator 1 alpha |
| PD | Parkinson’s disease |
| SIRT1 | sirtuin 1 |
References
- Lesage, S. Genetics of Mendelian Forms of Parkinson’s Disease. In Movement Disorder Genetics; Schneider, S.A., Brás, J.M.T., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–18. ISBN 978-3-319-17223-1. [Google Scholar]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J.; Haass, C. How does parkin ligate ubiquitin to Parkinson’s disease? EMBO Rep. 2004, 5, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [PubMed]
- Giguère, N.; Pacelli, C.; Saumure, C.; Bourque, M.-J.; Matheoud, D.; Levesque, D.; Slack, R.S.; Park, D.S.; Trudeau, L.-É. Comparative analysis of Parkinson’s disease-associated genes in mice reveals altered survival and bioenergetics of Parkin-deficient dopamine neurons. J. Biol. Chem. 2018, 293, 9580–9593. [Google Scholar] [CrossRef] [PubMed]
- Botella, J.A.; Bayersdorfer, F.; Gmeiner, F.; Schneuwly, S. Modelling Parkinson’s disease in Drosophila. Neuromolecular Med. 2009, 11, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Müftüoglu, M.; Elibol, B.; Dalmizrak, O.; Ercan, A.; Kulaksiz, G.; Ogüs, H.; Dalkara, T.; Ozer, N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord. 2004, 19, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Thomas, K.J.; Koopman, W.J.H.; Klaffke, S.; Abou-Sleiman, P.; Olpin, S.; Wood, N.W.; Willems, P.H.G.M.; Smeitink, J.A.M.; Cookson, M.R.; et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 2008, 64, 555–565. [Google Scholar] [CrossRef]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1α dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA. 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [PubMed]
- Levine, B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar] [PubMed]
- Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway: a mitochondrial quality control system? J. Bioenerg. Biomembr. 2009, 41, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J.; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008, 3, 309–323. [Google Scholar] [PubMed]
- Sandebring, A.; Dehvari, N.; Perez-Manso, M.; Thomas, K.J.; Karpilovski, E.; Cookson, M.R.; Cowburn, R.F.; Cedazo-Mínguez, A. Parkin deficiency disrupts calcium homeostasis by modulating phospholipase C signalling. FEBS J. 2009, 276, 5041–5052. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 2013, 1832, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Tanzarella, P.; Ferretta, A.; Barile, S.N.; Ancona, M.; De Rasmo, D.; Signorile, A.; Papa, S.; Capitanio, N.; Pacelli, C.; Cocco, T. Increased Levels of cAMP by the Calcium-Dependent Activation of Soluble Adenylyl Cyclase in Parkin-Mutant Fibroblasts. Cells 2019, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Y.; Wang, F.; Hu, L.-F.; Liu, C.-F. A New Perspective for Parkinson’s Disease: Circadian Rhythm. Neurosci. Bull. 2017, 33, 62–72. [Google Scholar] [CrossRef] [PubMed]
- McClung, C.A.; Sidiropoulou, K.; Vitaterna, M.; Takahashi, J.S.; White, F.J.; Cooper, D.C.; Nestler, E.J. Regulation of dopaminergic transmission and cocaine reward by the Clock gene. Proc. Natl. Acad. Sci. USA 2005, 102, 9377–9381. [Google Scholar] [CrossRef] [PubMed]
- Yujnovsky, I.; Hirayama, J.; Doi, M.; Borrelli, E.; Sassone-Corsi, P. Signaling mediated by the dopamine D2 receptor potentiates circadian regulation by clock: BMAL1. Proc. Natl. Acad. Sci. USA 2006, 103, 6386–6391. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Loh, D.H.; Truong, D.; Wu, Y.; Colwell, C.S. Circadian dysfunction in a mouse model of Parkinson’s disease. Exp. Neurol. 2011, 232, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Lax, P.; Esquiva, G.; Esteve-Rudd, J.; Otalora, B.B.; Madrid, J.A.; Cuenca, N. Circadian Dysfunction in a Rotenone-Induced Parkinsonian Rodent Model. Chronobiol. Int. 2012, 29, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Monaca, C.; Laloux, C.; Jacquesson, J.-M.; Gelé, P.; Maréchal, X.; Bordet, R.; Destée, A.; Derambure, P. Vigilance states in a parkinsonian model, the MPTP mouse. Eur. J. Neurosci. 2004, 20, 2474–2478. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, D.; Liu, W.; Li, S.; Chen, J.; Shen, Y.; Wang, F.; Hu, L.-F.; Liu, C.-F. Disruption of the Circadian Clock Alters Antioxidative Defense via the SIRT1-BMAL1 Pathway in 6-OHDA-Induced Models of Parkinson’s Disease. Available online: https://www.hindawi.com/journals/omcl/2018/4854732/ (accessed on 10 September 2018).
- Breen, D.P.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.M.; Reddy, A.B.; Barker, R.A. Sleep and Circadian Rhythm Regulation in Early Parkinson Disease. JAMA Neurol 2014, 71, 589–595. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, S.; Sothern, R.B.; Xu, S.; Chan, P. Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease. Eur. J. Neurol. 2010, 17, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Zhao, C.; Ding, H.; Liang, K.; Xue, J.; Chan, P.; Cai, Y. Pyrosequencing analysis of methylation levels of clock genes in leukocytes from Parkinson’s disease patients. Neurosci. Lett. 2018, 668, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Scrima, R.; Cela, O.; Merla, G.; Augello, B.; Rubino, R.; Quarato, G.; Fugetto, S.; Menga, M.; Fuhr, L.; Relógio, A.; et al. Clock-genes and mitochondrial respiratory activity: Evidence of a reciprocal interplay. Biochim. Biophys. Acta 2016, 1857, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys. Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef]
- Balsalobre, A.; Damiola, F.; Schibler, U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 1998, 93, 929–937. [Google Scholar] [CrossRef]
- Balsalobre, A.; Marcacci, L.; Schibler, U. Multiple signaling pathways elicit circadian gene expression in cultured Rat-1 fibroblasts. Curr. Biol. 2000, 10, 1291–1294. [Google Scholar] [CrossRef]
- Manella, G.; Asher, G. The Circadian Nature of Mitochondrial Biology. Front Endocrinol. 2016, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Sardon Puig, L.; Valera-Alberni, M.; Cantó, C.; Pillon, N.J. Circadian Rhythms and Mitochondria: Connecting the Dots. Front Genet. 2018, 9, 452. [Google Scholar] [CrossRef] [PubMed]
- Reinke, H.; Asher, G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 2019, 20, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol Cells 2018, 41, 18–26. [Google Scholar]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Parkinsons Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef]
- Cornelissen, G. Cosinor-based rhythmometry. Theor. Biol. Med. Model 2014, 11, 16. [Google Scholar] [CrossRef]
- De Goede, P.; Wefers, J.; Brombacher, E.C.; Schrauwen, P.; Kalsbeek, A. Circadian rhythms in mitochondrial respiration. J. Mol. Endocrinol. 2018, 60, R115–R130. [Google Scholar] [CrossRef]
- Schmitt, K.; Grimm, A.; Dallmann, R.; Oettinghaus, B.; Restelli, L.M.; Witzig, M.; Ishihara, N.; Mihara, K.; Ripperger, J.A.; Albrecht, U.; et al. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 2018, 27, 657–666. [Google Scholar] [CrossRef]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian Control of the NAD+ Salvage Pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Bellet, M.M.; Orozco-Solis, R.; Sahar, S.; Eckel-Mahan, K.; Sassone-Corsi, P. The Time of Metabolism: NAD+, SIRT1, and the Circadian Clock. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Zhao, T.; Cui, K.; Hu, G.; Chen, Q.; Chen, W.; Wang, X.-W.; Soto-Gutierrez, A.; Zhao, K.; Deng, C.-X. Negative reciprocal regulation between Sirt1 and Per2 modulates the circadian clock and aging. Sci. Rep. 2016, 6, 28633. [Google Scholar] [CrossRef] [PubMed]
- Partch, C.L.; Green, C.B.; Takahashi, J.S. Molecular Architecture of the Mammalian Circadian Clock. Trends Cell Biol. 2014, 24, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Rosati, J.; Ferrari, D.; Altieri, F.; Tardivo, S.; Ricciolini, C.; Fusilli, C.; Zalfa, C.; Profico, D.C.; Pinos, F.; Bernardini, L.; et al. Establishment of stable iPS-derived human neural stem cell lines suitable for cell therapies. Cell Death Dis. 2018, 9, 937. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Golombek, D. Circadian Dysregulation in Parkinson’s Disease. Neurobiol. Sleep Circadian Rhythm. 2017, 2, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Buhr, E.D.; Takahashi, J.S. Molecular components of the mammalian circadian clock. In Circadian clocks; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–27. [Google Scholar]
- Fuhr, L.; El-Athman, R.; Scrima, R.; Cela, O.; Carbone, A.; Knoop, H.; Li, Y.; Hoffmann, K.; Laukkanen, M.O.; Corcione, F.; et al. The Circadian Clock Regulates Metabolic Phenotype Rewiring Via HKDC1 and Modulates Tumor Progression and Drug Response in Colorectal Cancer. EBioMedicine 2018, 33, 105–121. [Google Scholar] [CrossRef]
- Peek, C.B.; Affinati, A.H.; Ramsey, K.M.; Kuo, H.-Y.; Yu, W.; Sena, L.A.; Ilkayeva, O.; Marcheva, B.; Kobayashi, Y.; Omura, C.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013, 342, 1243417. [Google Scholar] [CrossRef]
- Neufeld-Cohen, A.; Robles, M.S.; Aviram, R.; Manella, G.; Adamovich, Y.; Ladeuix, B.; Nir, D.; Rousso-Noori, L.; Kuperman, Y.; Golik, M.; et al. Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. PNAS 2016, 113, E1673–E1682. [Google Scholar] [CrossRef]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. PNAS 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature 2007, 447, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Woldt, E.; Sebti, Y.; Solt, L.A.; Duhem, C.; Lancel, S.; Eeckhoute, J.; Hesselink, M.K.C.; Paquet, C.; Delhaye, S.; Shin, Y.; et al. Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat. Med. 2013, 19, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Belden, W.J.; Dunlap, J.C. SIRT1 is a circadian deacetylase for core clock components. Cell 2008, 134, 212–214. [Google Scholar] [CrossRef]
- Mattam, U.; Jagota, A. Daily rhythms of serotonin metabolism and the expression of clock genes in suprachiasmatic nucleus of rotenone-induced Parkinson’s disease male Wistar rat model and effect of melatonin administration. Biogerontology 2015, 16, 109–123. [Google Scholar] [CrossRef]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar] [CrossRef]
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 2007, 450, 1086–1090. [Google Scholar] [CrossRef]
- Griffin, P.; Dimitry, J.M.; Sheehan, P.W.; Lananna, B.V.; Guo, C.; Robinette, M.L.; Hayes, M.E.; Cedeño, M.R.; Nadarajah, C.J.; Ezerskiy, L.A.; et al. Circadian clock protein Rev-erbα regulates neuroinflammation. PNAS 2019, 116, 5102–5107. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef] [PubMed]
- Harms, E.; Kivimäe, S.; Young, M.W.; Saez, L. Posttranscriptional and Posttranslational Regulation of Clock Genes. J. Biol. Rhythms 2004, 19, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.; Sassone-Corsi, P. Metabolism control by the circadian clock and vice versa. Nat. Struct. Mol. Biol. 2009, 16, 462–467. [Google Scholar] [CrossRef]
- Sison, S.L.; Vermilyea, S.C.; Emborg, M.E.; Ebert, A.D. Using Patient-Derived Induced Pluripotent Stem Cells to Identify Parkinson’s Disease-Relevant Phenotypes. Curr. Neurol. Neurosci. Rep. 2018, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef]
- Xu, X.; Duan, S.; Yi, F.; Ocampo, A.; Liu, G.-H.; Izpisua Belmonte, J.C. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013, 18, 325–332. [Google Scholar] [CrossRef]
- Son, M.J.; Jeong, B.R.; Kwon, Y.; Cho, Y.S. Interference with the mitochondrial bioenergetics fuels reprogramming to pluripotency via facilitation of the glycolytic transition. Int. J. Biochem. Cell Biol. 2013, 45, 2512–2518. [Google Scholar] [CrossRef] [PubMed]
- Yagita, K.; Horie, K.; Koinuma, S.; Nakamura, W.; Yamanaka, I.; Urasaki, A.; Shigeyoshi, Y.; Kawakami, K.; Shimada, S.; Takeda, J.; et al. Development of the circadian oscillator during differentiation of mouse embryonic stem cells in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 3846–3851. [Google Scholar] [CrossRef]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Jamasbi, R.J.; Kondratov, R.V.; Geusz, M.E. Development of circadian oscillators in neurosphere cultures during adult neurogenesis. PLoS ONE 2015, 10, e0122937. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Thakkar, A. Mitochondria/metabolic reprogramming in the formation of neurons from peripheral cells: Cause or consequence and the implications to their utility. Neurochem. Int. 2018, 117, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Weger, M.; Diotel, N.; Dorsemans, A.-C.; Dickmeis, T.; Weger, B.D. Stem cells and the circadian clock. Dev. Biol. 2017, 431, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Dierickx, P.; Vermunt, M.W.; Muraro, M.J.; Creyghton, M.P.; Doevendans, P.A.; van Oudenaarden, A.; Geijsen, N.; Van Laake, L.W. Circadian networks in human embryonic stem cell-derived cardiomyocytes. EMBO Rep. 2017, 18, 1199–1212. [Google Scholar] [CrossRef] [PubMed]
- Vallone, D.; Lahiri, K.; Dickmeis, T.; Foulkes, N.S. Start the clock! Circadian rhythms and development. Dev. Dyn. 2007, 236, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Umemura, Y.; Koike, N.; Ohashi, M.; Tsuchiya, Y.; Meng, Q.J.; Minami, Y.; Hara, M.; Hisatomi, M.; Yagita, K. Involvement of posttranscriptional regulation of Clock in the emergence of circadian clock oscillation during mouse development. Proc. Natl. Acad. Sci. USA 2017, 114, E7479–E7488. [Google Scholar] [CrossRef] [PubMed]
- Paulose, J.K.; Rucker, E.B.; Cassone, V.M. Toward the beginning of time: circadian rhythms in metabolism precede rhythms in clock gene expression in mouse embryonic stem cells. PLoS ONE 2012, 7, e49555. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacelli, C.; Rotundo, G.; Lecce, L.; Menga, M.; Bidollari, E.; Scrima, R.; Cela, O.; Piccoli, C.; Cocco, T.; Vescovi, A.L.; et al. Parkin Mutation Affects Clock Gene-Dependent Energy Metabolism. Int. J. Mol. Sci. 2019, 20, 2772. https://doi.org/10.3390/ijms20112772
Pacelli C, Rotundo G, Lecce L, Menga M, Bidollari E, Scrima R, Cela O, Piccoli C, Cocco T, Vescovi AL, et al. Parkin Mutation Affects Clock Gene-Dependent Energy Metabolism. International Journal of Molecular Sciences. 2019; 20(11):2772. https://doi.org/10.3390/ijms20112772
Chicago/Turabian StylePacelli, Consiglia, Giovannina Rotundo, Lucia Lecce, Marta Menga, Eris Bidollari, Rosella Scrima, Olga Cela, Claudia Piccoli, Tiziana Cocco, Angelo Luigi Vescovi, and et al. 2019. "Parkin Mutation Affects Clock Gene-Dependent Energy Metabolism" International Journal of Molecular Sciences 20, no. 11: 2772. https://doi.org/10.3390/ijms20112772
APA StylePacelli, C., Rotundo, G., Lecce, L., Menga, M., Bidollari, E., Scrima, R., Cela, O., Piccoli, C., Cocco, T., Vescovi, A. L., Mazzoccoli, G., Rosati, J., & Capitanio, N. (2019). Parkin Mutation Affects Clock Gene-Dependent Energy Metabolism. International Journal of Molecular Sciences, 20(11), 2772. https://doi.org/10.3390/ijms20112772