Oestrogen Non-Genomic Signalling is Activated in Tamoxifen-Resistant Breast Cancer



Abstract
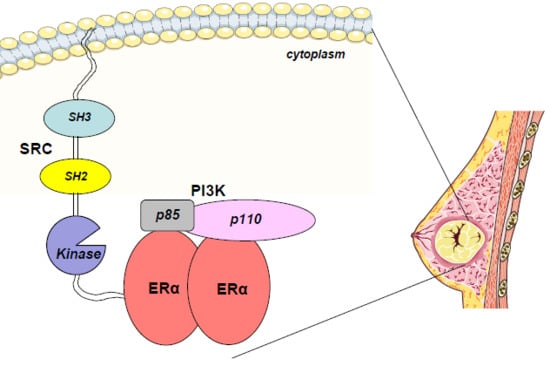
1. Introduction
2. Results
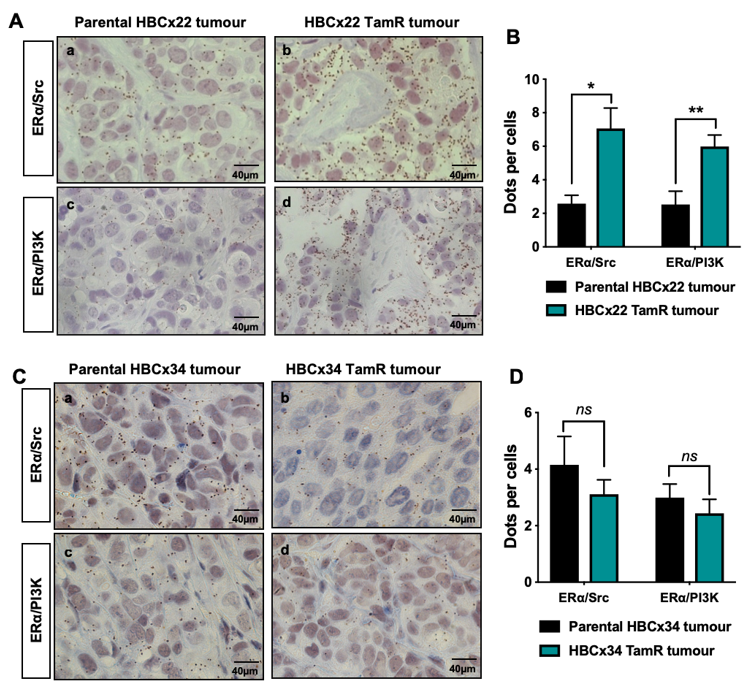
2.1. ERα–Src–PI3K Expression in Patient-Derived Breast Cancer with Acquired Resistance to Tamoxifen
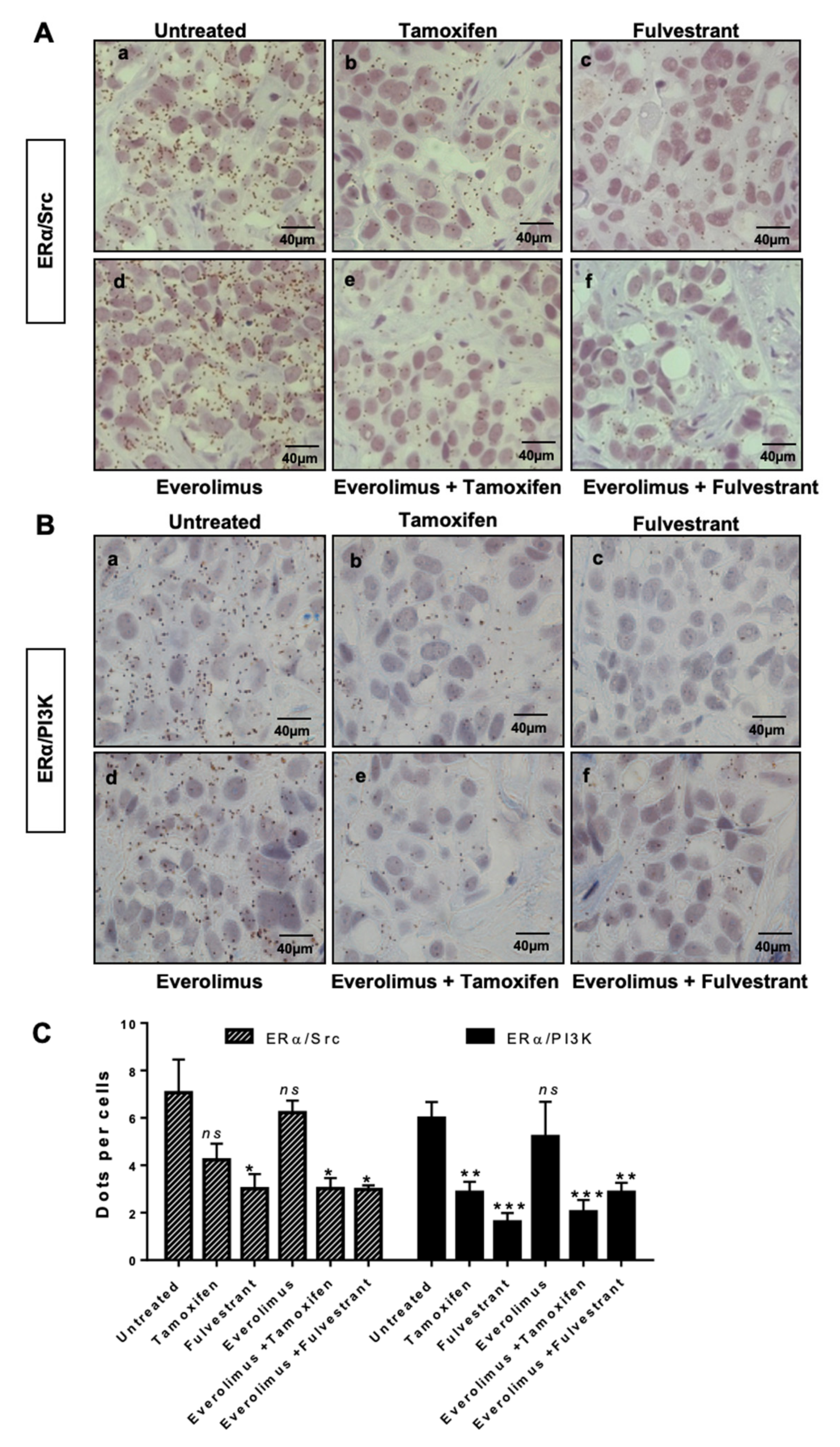
2.2. ERα/Src/PI3K Activation in HBCx22 TamR Treated with Everolimus and/or with Endocrine Therapies
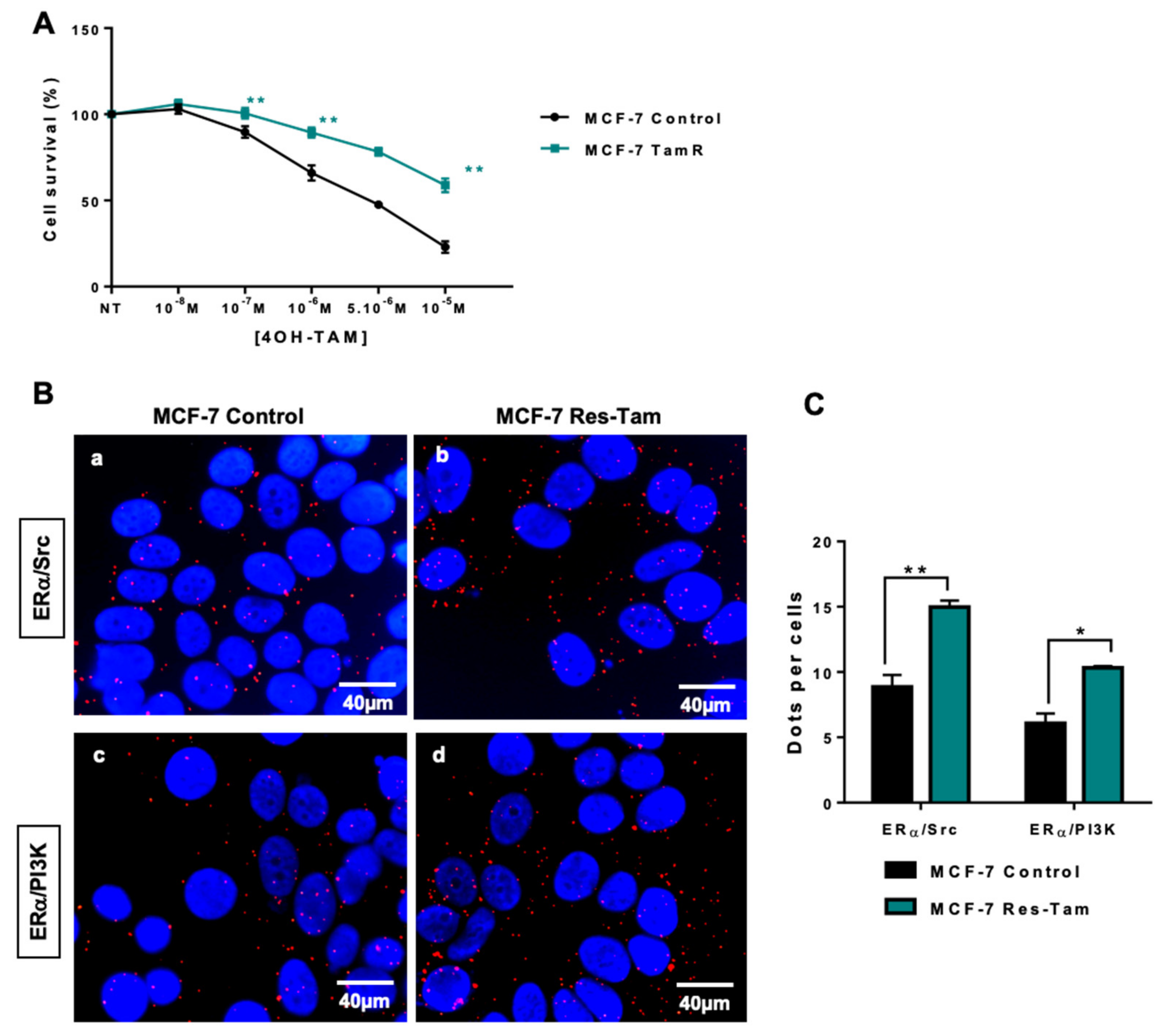
2.3. ERα–Src–PI3K Interaction in MCF-7 Cells Resistant to Endocrine Therapy
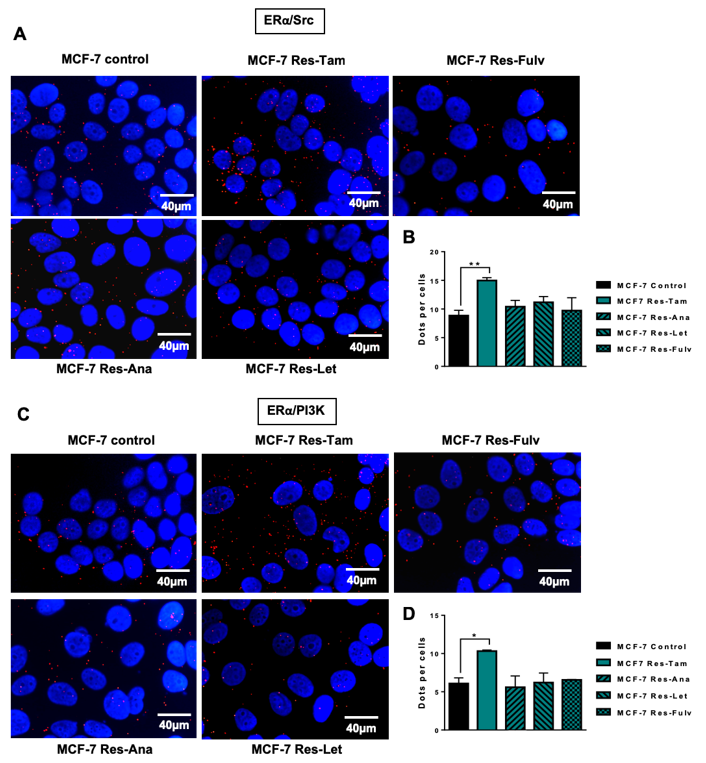
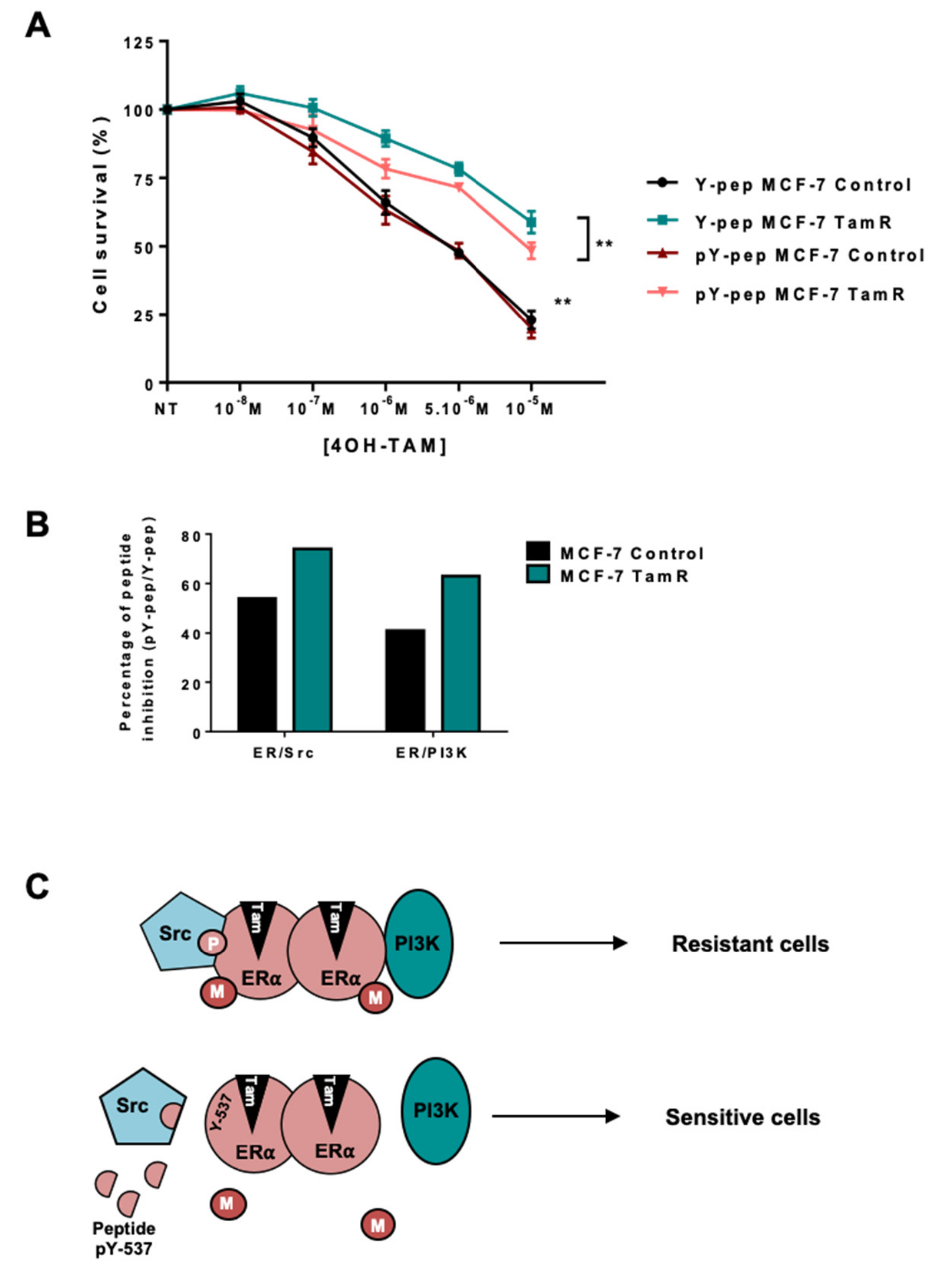
2.4. Targeting the ERα/Src/PI3K Complex
3. Discussion
4. Materials and Methods
4.1. Establishment of Xenografts Models Resistant to Tamoxifen (TamR)
4.2. In Vivo Treatment Studies
4.3. Cell Culture
4.4. Antibodies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Supplier | Origin | Dilution for PLA | Dilution for WB | Dilution for IF | Dilution for IHC |
|---|---|---|---|---|---|---|
| PI3K p85 ab-22653 | Abcam | mouse | 1/30 | |||
| c-Src (B12) sc-8056 | Santa Cruz | mouse | 1/150 | 1/1000 | 1/1000 | |
| ERα (HC20) sc-542 | Santa Cruz | rabbit | 1/75 | |||
| ERα (F10) sc-542 | Santa Cruz | mouse | 1/200 | 1/200 | ||
| GAPDH Sc-47724 | Santa Cruz | mouse | 1/2000 | |||
| PI3K p85 05-212 | Millipore | mouse | 1/1000 | 1/100 | ||
| ERα-36 | Custom made [40] | rabbit | 1/100 | |||
| p-Akt 4060 | Cell Signaling | rabbit | 1/1000 | |||
| Akt 4691 | Cell Signaling | rabbit | 1/1000 |
4.5. Proximity Ligation Assay
4.6. Western Blotting
4.7. Immunofluorescence
4.8. Cytotoxicity Assay
4.9. Image Acquisition and Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Boyd, N.F.; Chlebowski, R.T.; Cummings, S.; Cuzick, J.; Dowsett, M.; Easton, D.; Forbes, J.F.; Key, T.; Hankinson, S.E.; et al. Critical assessment of new risk factors for breast cancer: Considerations for development of an improved risk prediction model. Endocrine-Related Cancer 2007, 14, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocrine-Related Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef]
- Zhang, X.H.-F.; Giuliano, M.; Trivedi, M.V.; Schiff, R.; Osborne, C.K. Metastasis Dormancy in Estrogen Receptor-Positive Breast Cancer. Clin. Breast Cancer 2013, 19, 6389–6397. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, cloning, and expression of human estrogen receptor-α36, a novel variant of human estrogen receptor-α66. Biochem. Biophys. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A variant of estrogen receptor-, hER- 36: Transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Acad. Sci. 2006, 103, 9063–9068. [Google Scholar] [CrossRef]
- Shi, L.; Dong, B.; Li, Z.; Lu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Expression of ER-α36, a Novel Variant of Estrogen Receptor α, and Resistance to Tamoxifen Treatment in Breast Cancer. J. Clin. Oncol. 2009, 27, 3423–3429. [Google Scholar] [CrossRef]
- Teymourzadeh, A.; Mansouri, S.; Farahmand, L.; Hosseinzade, A.; Majidzadeh-A, K. ER-α36 Interactions With Cytosolic Molecular Network in Acquired Tamoxifen Resistance. Clin. Breast Cancer 2017, 17, 403–407. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.F.; et al. Tamoxifen enhances stemness and promotes metastasis of ERalpha36(+) breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. 2018, 28, 336–358. [Google Scholar] [CrossRef]
- Hsu, L.-H.; Chu, N.-M.; Lin, Y.-F.; Kao, S.-H. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef]
- Björnström, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar]
- Greger, J.G.; Fursov, N.; Cooch, N.; McLarney, S.; Freedman, L.P.; Edwards, D.P.; Cheskis, B.J. Phosphorylation of MNAR promotes estrogen activation of phosphatidylinositol 3-kinase. Mol. Cell Biol. 2007, 27, 1904–1913. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; De Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K.; Brazil, D.P.; et al. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nat. Cell Boil. 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Song, R.X.-D.; Zhang, Z.; Santen, R.J. Estrogen rapid action via protein complex formation involving ERα and Src. Trends Endocrinol. Metab. 2005, 16, 347–353. [Google Scholar] [CrossRef]
- Le Romancer, M.; Treilleux, I.; Bouchekioua-Bouzaghou, K.; Sentis, S.; Corbo, L. Methylation, a key step for nongenomic estrogen signaling in breast tumors. Steroids 2010, 75, 560–564. [Google Scholar] [CrossRef]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef]
- Söderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstråle, K.; Leuchowius, K.-J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.-G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef]
- Poulard, C.; Treilleux, I.; Lavergne, E.; Bouchekioua-Bouzaghou, K.; Goddard-Léon, S.; Chabaud, S.; Trédan, O.; Corbo, L.; Le Romancer, M. Activation of rapid oestrogen signalling in aggressive human breast cancers. EMBO Mol. Med. 2012, 4, 1200–1213. [Google Scholar] [CrossRef]
- Poulard, C.; Rambaud, J.; Le Romancer, M.; Corbo, L. Proximity Ligation Assay to Detect and Localize the Interactions of ERα with PI3-K and Src in Breast Cancer Cells and Tumor Samples. Methods Mol. Biol. 2014, 1204, 135–143. [Google Scholar]
- Dobrolecki, L.E.; Airhart, S.D.; Alferez, D.G.; Aparicio, S.; Behbod, F.; Bentires-Alj, M.; Brisken, C.; Bult, C.J.; Cai, S.; Clarke, R.B.; et al. Patient-derived Xenograft (PDX) Models In Basic and Translational Breast Cancer Research. Cancer Metastasis Rev. 2016, 35, 547–573. [Google Scholar] [CrossRef]
- Marangoni, E.; Vincent-Salomon, A.; Auger, N.; Degeorges, A.; Assayag, F.; De Cremoux, P.; De Plater, L.; Guyader, C.; De Pinieux, G.; Judde, J.-G.; et al. A New Model of Patient Tumor-Derived Breast Cancer Xenografts for Preclinical Assays. Clin. Cancer Res. 2007, 13, 3989–3998. [Google Scholar] [CrossRef]
- Marangoni, E.; Poupon, M.-F. Patient-derived tumour xenografts as models for breast cancer drug development. Curr. Opin. Oncol. 2014, 26, 556–561. [Google Scholar] [CrossRef]
- Cottu, P.; Bièche, I.; Assayag, F.; El Botty, R.; Chateau-Joubert, S.; Thuleau, A.; Bagarre, T.; Albaud, B.; Rapinat, A.; Gentien, D.; et al. Acquired Resistance to Endocrine Treatments Is Associated with Tumor-Specific Molecular Changes in Patient-Derived Luminal Breast Cancer Xenografts. Clin. Cancer Res. 2014, 20, 4314–4325. [Google Scholar] [CrossRef]
- Sanchez, C.G.; Ma, C.X.; Crowder, R.J.; Guintoli, T.; Phommaly, C.; Gao, F.; Lin, L.; Ellis, M.J. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011, 13, R21. [Google Scholar] [CrossRef]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-Talk between Estrogen Receptor and Growth Factor Pathways as a Molecular Target for Overcoming Endocrine Resistance. Clin. Cancer Res. 2004, 10, 331S–336. [Google Scholar] [CrossRef]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.-M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.-C.; Debled, M.; Spaeth, D.; et al. Randomized Phase II Trial of Everolimus in Combination With Tamoxifen in Patients With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer With Prior Exposure to Aromatase Inhibitors: A GINECO Study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., III; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Varricchio, L.; Migliaccio, A.; Castoria, G.; Yamaguchi, H.; De Falco, A.; Di Domenico, M.; Giovannelli, P.; Farrar, W.; Appella, E.; Auricchio, F. Inhibition of Estradiol Receptor/Src Association and Cell Growth by an Estradiol Receptor Tyrosine-Phosphorylated Peptide. Mol. Cancer Res. 2007, 5, 1213–1221. [Google Scholar] [CrossRef]
- Johnston, S.R. Enhancing Endocrine Therapy for Hormone Receptor-Positive Advanced Breast Cancer: Cotargeting Signaling Pathways. J. Natl. Cancer. Inst. 2015, 107, djv212. [Google Scholar] [CrossRef]
- Fan, P.; Wang, J.; Santen, R.J.; Yue, W. Long-term Treatment with Tamoxifen Facilitates Translocation of Estrogen Receptor α out of the Nucleus and Enhances its Interaction with EGFR in MCF-7 Breast Cancer Cells. Cancer Res. 2007, 67, 1352–1360. [Google Scholar] [CrossRef]
- Poulard, C.; Rambaud, J.; Hussein, N.; Corbo, L.; Le Romancer, M. JMJD6 Regulates ERα Methylation on Arginine. PLoS ONE 2014, 9, 87982. [Google Scholar] [CrossRef]
- Cabodi, S. p130Cas interacts with estrogen receptor and modulates non-genomic estrogen signaling in breast cancer cells. J. Cell Sci. 2004, 117, 1603–1611. [Google Scholar] [CrossRef]
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Trédan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The arginine methyltransferase PRMT1 regulates IGF-1 signaling in breast cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Cottu, P.; Marangoni, E.; Assayag, F.; De Cremoux, P.; Vincent-Salomon, A.; Guyader, C.; De Plater, L.; Elbaz, C.; Karboul, N.; Fontaine, J.J.; et al. Modeling of response to endocrine therapy in a panel of human luminal breast cancer xenografts. Breast Cancer Res. Treat 2012, 133, 595–606. [Google Scholar] [CrossRef]
- Itoh, T.; Karlsberg, K.; Kijima, I.; Yuan, Y.-C.; Smith, D.; Ye, J.; Chen, S. Letrozole-, anastrozole-, and tamoxifen-responsive genes in MCF-7aro cells: A microarray approach. Mol. Cancer Res. 2005, 3, 203–218. [Google Scholar]
- Vilquin, P.; Villedieu, M.; Grisard, E.; Ben Larbi, S.; Ghayad, S.E.; Heudel, P.-E.; Bachelot, T.; Corbo, L.; Treilleux, I.; Vendrell, J.A.; et al. Molecular characterization of anastrozole resistance in breast cancer: Pivotal role of the Akt/mTOR pathway in the emergence ofde novoor acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int. J. Cancer 2013, 133, 1589–1602. [Google Scholar] [CrossRef]
- Vilquin, P.; Donini, C.F.; Villedieu, M.; Grisard, E.; Corbo, L.; Bachelot, T.; A Vendrell, J.; A Cohen, P. MicroRNA-125b upregulation confers aromatase inhibitor resistance and is a novel marker of poor prognosis in breast cancer. Breast Cancer Res. 2015, 17, 13. [Google Scholar] [CrossRef]
- Omarjee, S.; Jacquemetton, J.; Poulard, C.; Rochel, N.; Dejaegere, A.; Chebaro, Y.; Treilleux, I.; Marangoni, E.; Corbo, L.; Le Romancer, M. The molecular mechanisms underlying the ERalpha-36-mediated signaling in breast cancer. Oncogene 2017, 36, 2503–2514. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poulard, C.; Jacquemetton, J.; Trédan, O.; Cohen, P.A.; Vendrell, J.; Ghayad, S.E.; Treilleux, I.; Marangoni, E.; Le Romancer, M. Oestrogen Non-Genomic Signalling is Activated in Tamoxifen-Resistant Breast Cancer. Int. J. Mol. Sci. 2019, 20, 2773. https://doi.org/10.3390/ijms20112773
Poulard C, Jacquemetton J, Trédan O, Cohen PA, Vendrell J, Ghayad SE, Treilleux I, Marangoni E, Le Romancer M. Oestrogen Non-Genomic Signalling is Activated in Tamoxifen-Resistant Breast Cancer. International Journal of Molecular Sciences. 2019; 20(11):2773. https://doi.org/10.3390/ijms20112773
Chicago/Turabian StylePoulard, Coralie, Julien Jacquemetton, Olivier Trédan, Pascale A. Cohen, Julie Vendrell, Sandra E. Ghayad, Isabelle Treilleux, Elisabetta Marangoni, and Muriel Le Romancer. 2019. "Oestrogen Non-Genomic Signalling is Activated in Tamoxifen-Resistant Breast Cancer" International Journal of Molecular Sciences 20, no. 11: 2773. https://doi.org/10.3390/ijms20112773
APA StylePoulard, C., Jacquemetton, J., Trédan, O., Cohen, P. A., Vendrell, J., Ghayad, S. E., Treilleux, I., Marangoni, E., & Le Romancer, M. (2019). Oestrogen Non-Genomic Signalling is Activated in Tamoxifen-Resistant Breast Cancer. International Journal of Molecular Sciences, 20(11), 2773. https://doi.org/10.3390/ijms20112773