Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
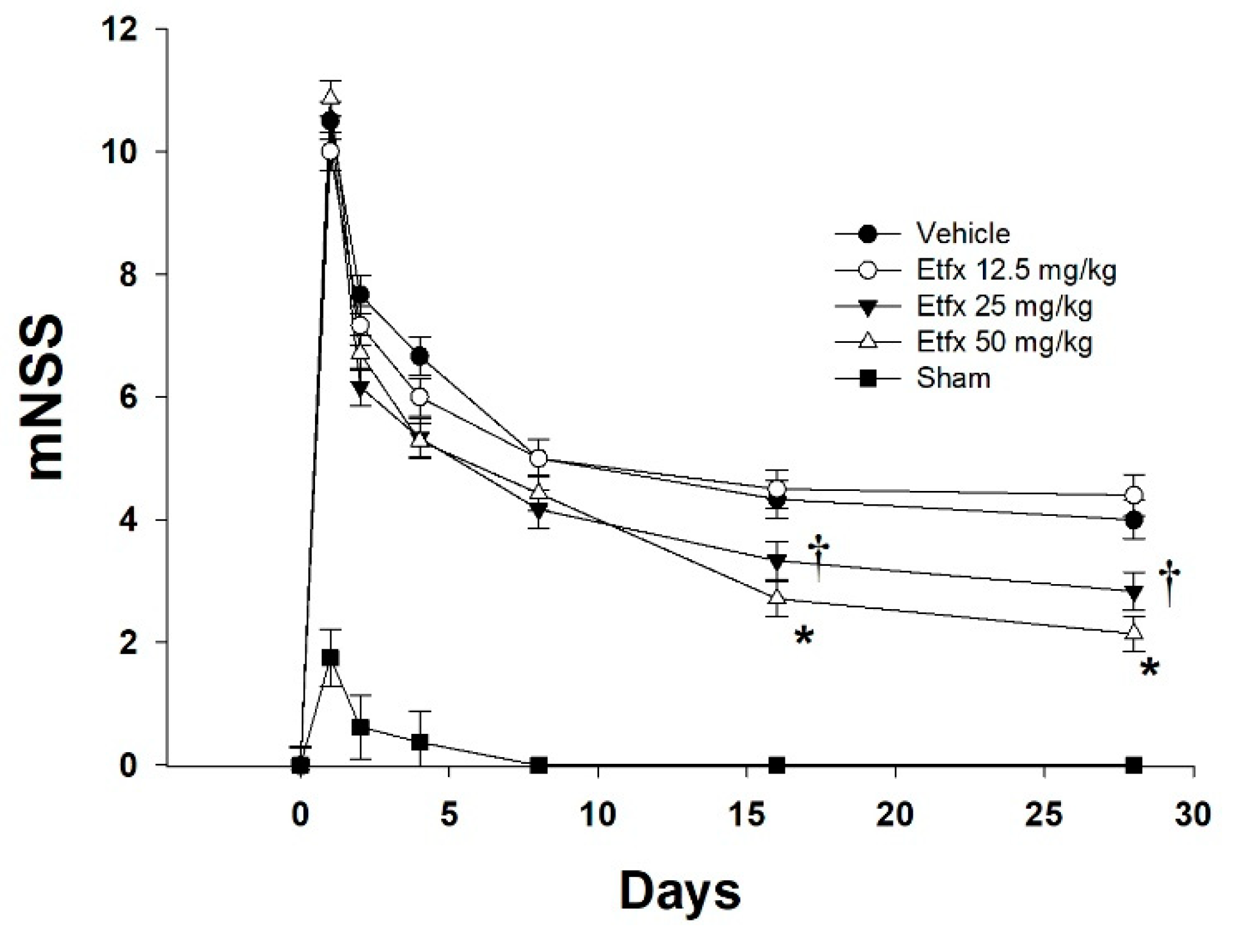
2.1. Motor and Behavioral Assessment
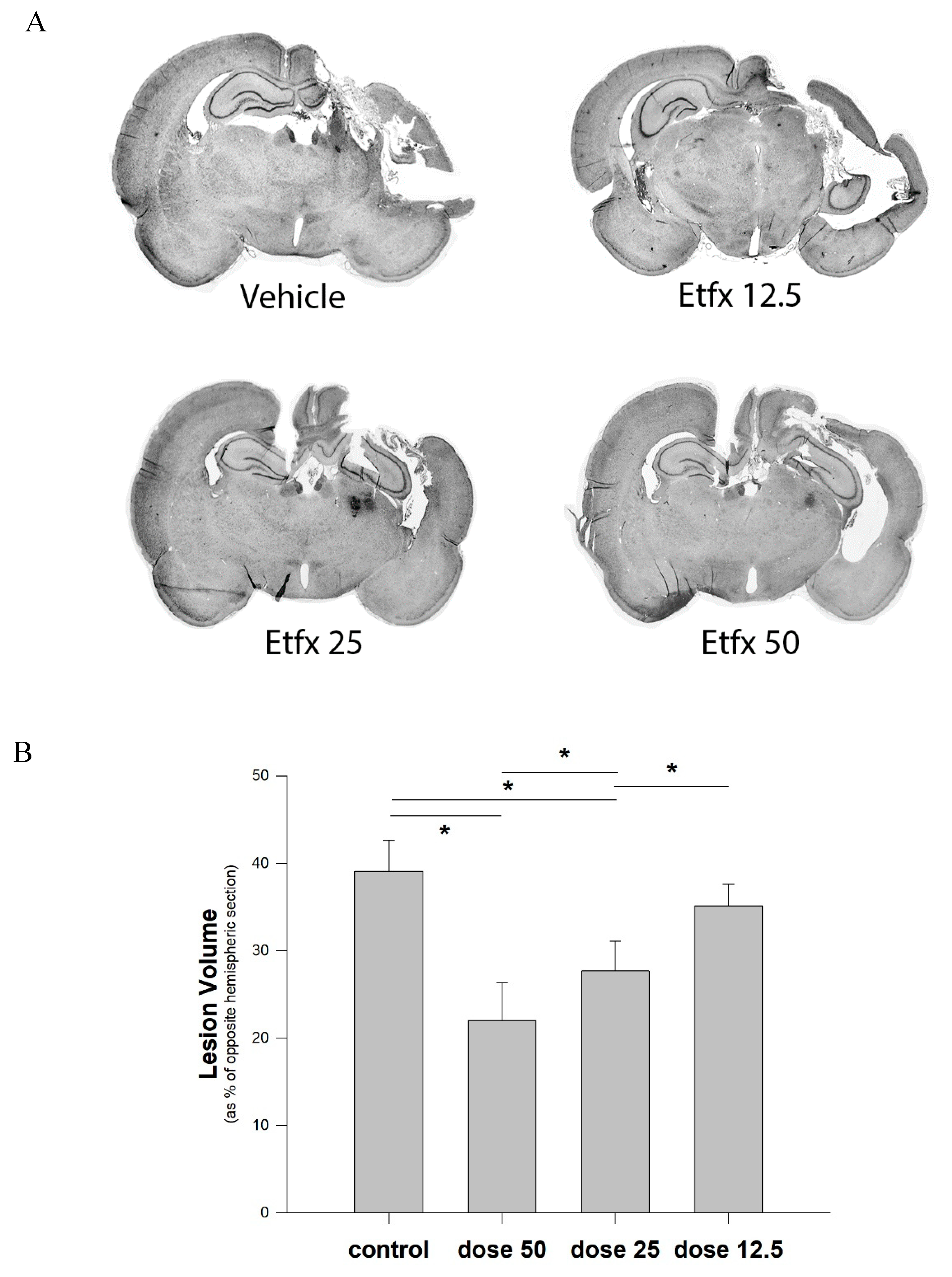
2.2. Lesion Volume
2.3. Histological Studies
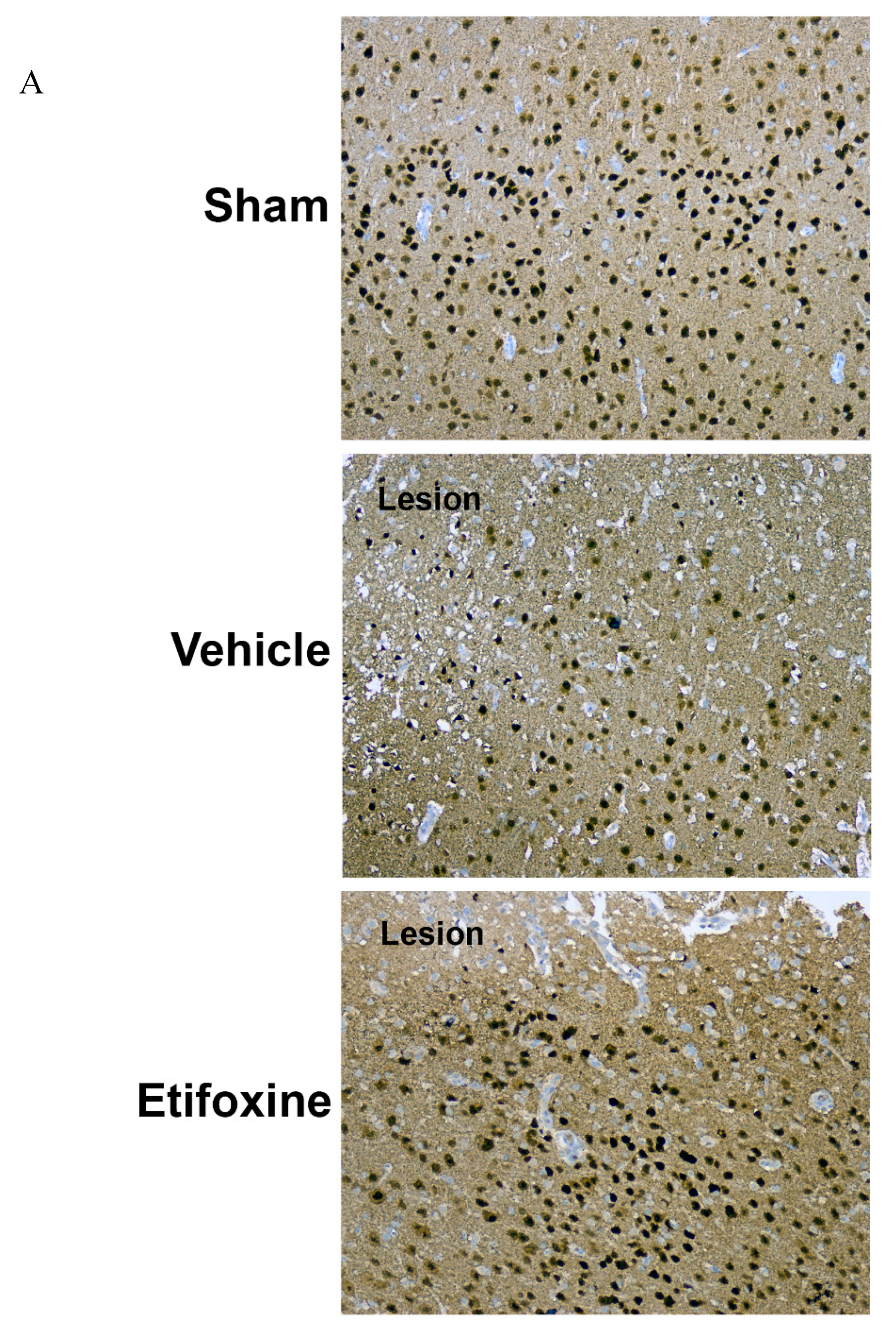
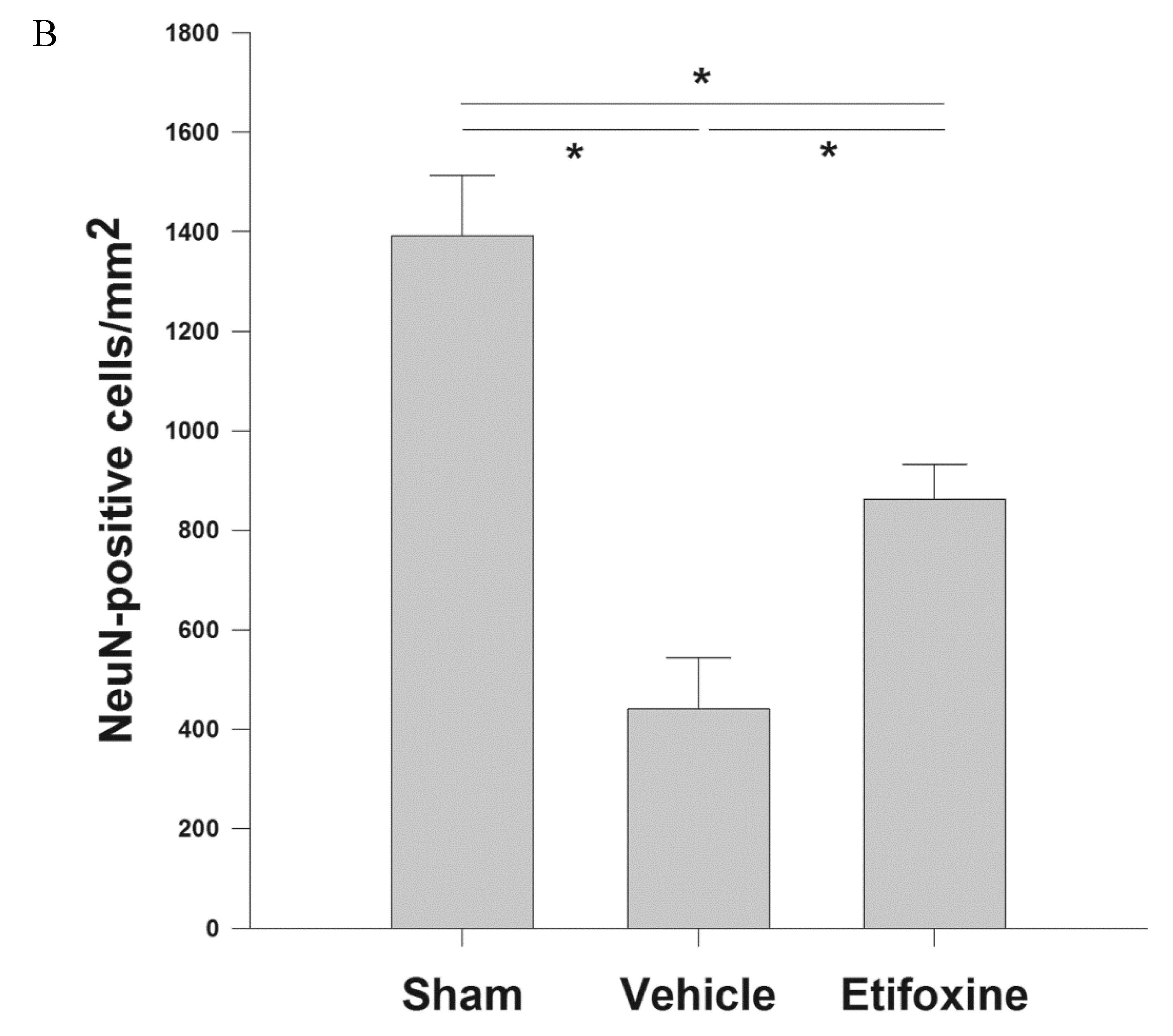
2.3.1. Neuronal Density
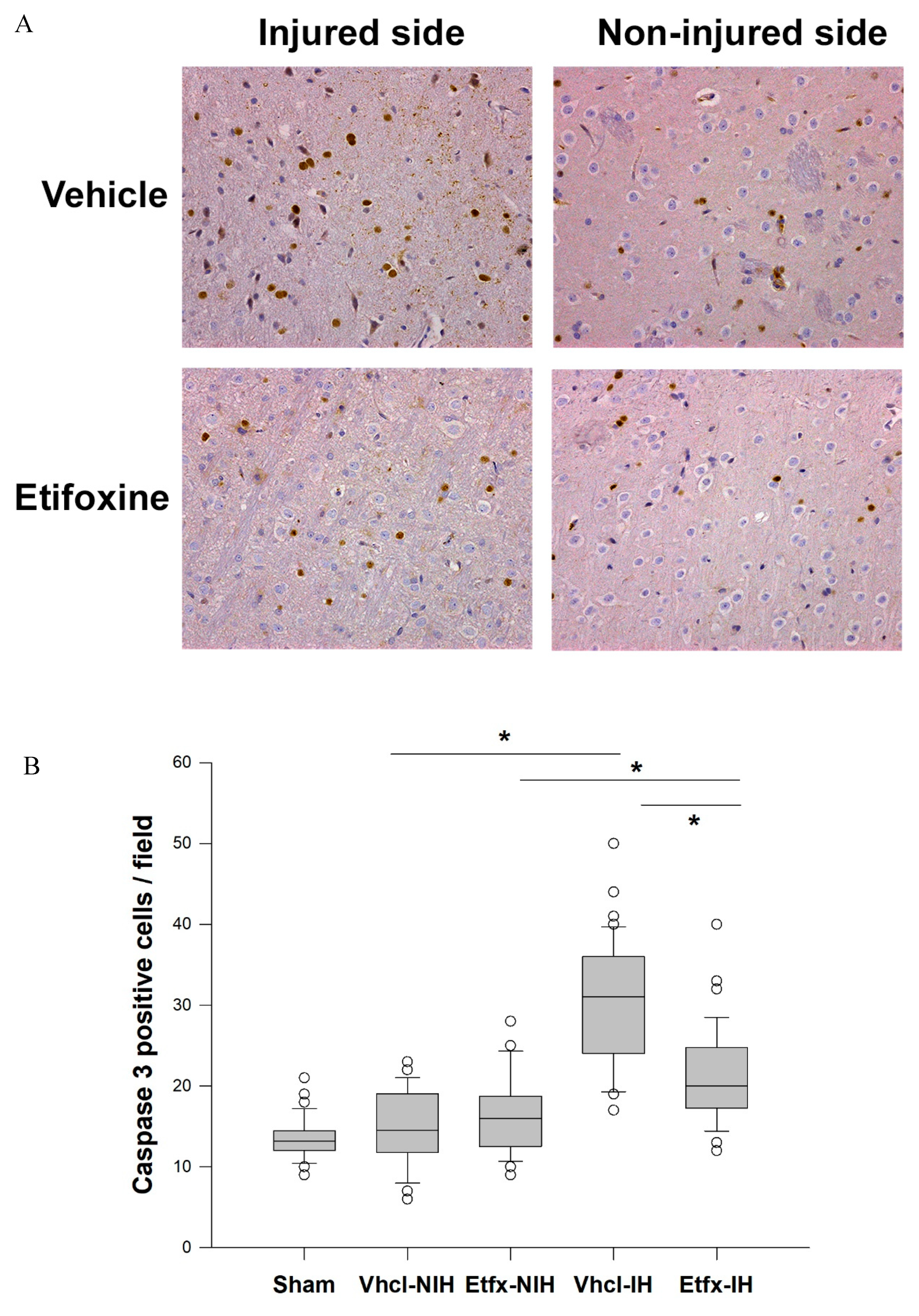
2.3.2. Caspase-3 Activity
3. Discussion
4. Material and Methods
4.1. Brain Injury Model
4.2. In Vivo Study
4.2.1. Animal Grouping and Treatment
4.2.2. Motor and Behavioral Assessment
4.2.3. Lesion Volume
4.3. Immunohistochemistry
4.3.1. Brain Removal and Fixation
4.3.2. NeuN Staining
4.3.3. Caspase-3
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Kinnally, K.W.; Zorov, D.B.; Antonenko, Y.N.; Snyder, S.H.; McEnery, M.W.; Tedeschi, H. Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc. Natl. Acad. Sci. USA 1993, 90, 1374–1378. [Google Scholar] [CrossRef]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef]
- Batra, S.; Iosif, C.S. Elevated concentrations of mitochondrial peripheral benzodiazepine receptors in ovarian tumors. Int. J. Oncol. 1998, 12, 1295–1298. [Google Scholar] [CrossRef]
- Carmel, I.; Fares, F.A.; Leschiner, S.; Scherubl, H.; Weisinger, G.; Gavish, M. Peripheral-type benzodiazepine receptors in the regulation of proliferation of MCF-7 human breast carcinoma cell line. Biochem. Pharm. 1999, 58, 273–278. [Google Scholar] [CrossRef]
- Hardwick, M.; Fertikh, D.; Culty, M.; Li, H.; Vidic, B.; Papadopoulos, V. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: Correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999, 59, 831–842. [Google Scholar]
- Katz, Y.; Eitan, A.; Gavish, M. Increase in peripheral benzodiazepine binding sites in colonic adenocarcinoma. Oncology 1990, 47, 139–142. [Google Scholar] [CrossRef]
- Sutter, A.P.; Maaser, K.; Grabowski, P.; Bradacs, G.; Vormbrock, K.; Hopfner, M.; Krahn, A.; Heine, B.; Stein, H.; Somasundaram, R.; et al. Peripheral benzodiazepine receptor ligands induce apoptosis and cell cycle arrest in human hepatocellular carcinoma cells and enhance chemosensitivity to paclitaxel, docetaxel, doxorubicin and the Bcl-2 inhibitor HA14-1. J. Hepatol. 2004, 41, 799–807. [Google Scholar] [CrossRef]
- Venturini, I.; Zeneroli, M.L.; Corsi, L.; Avallone, R.; Farina, F.; Alho, H.; Baraldi, C.; Ferrarese, C.; Pecora, N.; Frigo, M.; et al. Up-regulation of peripheral benzodiazepine receptor system in hepatocellular carcinoma. Life Sci. 1998, 63, 1269–1280. [Google Scholar] [CrossRef]
- Vlodavsky, E.; Soustiel, J.F. Immunohistochemical expression of peripheral benzodiazepine receptors in human astrocytomas and its correlation with grade of malignancy, proliferation, apoptosis and survival. J. Neurooncol. 2007, 81, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Banker, D.E.; Cooper, J.J.; Fennell, D.A.; Willman, C.L.; Appelbaum, F.R.; Cotter, F.E. PK11195, a peripheral benzodiazepine receptor ligand, chemosensitizes acute myeloid leukemia cells to relevant therapeutic agents by more than one mechanism. Leuk. Res. 2002, 26, 91–106. [Google Scholar] [CrossRef]
- Decaudin, D.; Castedo, M.; Nemati, F.; Beurdeley-Thomas, A.; De Pinieux, G.; Caron, A.; Pouillart, P.; Wijdenes, J.; Rouillard, D.; Kroemer, G.; et al. Peripheral benzodiazepine receptor ligands reverse apoptosis resistance of cancer cells in vitro and in vivo. Cancer Res. 2002, 62, 1388–1393. [Google Scholar] [PubMed]
- Gonzalez-Polo, R.A.; Carvalho, G.; Braun, T.; Decaudin, D.; Fabre, C.; Larochette, N.; Perfettini, J.L.; Djavaheri-Mergny, M.; Youlyouz-Marfak, I.; Codogno, P.; et al. PK11195 potently sensitizes to apoptosis induction independently from the peripheral benzodiazepin receptor. Oncogene 2005, 24, 7503–7513. [Google Scholar] [CrossRef]
- Hirsch, T.; Decaudin, D.; Susin, S.A.; Marchetti, P.; Larochette, N.; Resche-Rigon, M.; Kroemer, G. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp. Cell Res. 1998, 241, 426–434. [Google Scholar] [CrossRef]
- Maaser, K.; Hopfner, M.; Jansen, A.; Weisinger, G.; Gavish, M.; Kozikowski, A.P.; Weizman, A.; Carayon, P.; Riecken, E.O.; Zeitz, M.; et al. Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human colorectal cancer cells. Br. J. Cancer 2001, 85, 1771–1780. [Google Scholar] [CrossRef]
- Jorda, E.G.; Jimenez, A.; Verdaguer, E.; Canudas, A.M.; Folch, J.; Sureda, F.X.; Camins, A.; Pallas, M. Evidence in favour of a role for peripheral-type benzodiazepine receptor ligands in amplification of neuronal apoptosis. Apoptosis 2005, 10, 91–104. [Google Scholar] [CrossRef]
- Fennell, D.A.; Corbo, M.; Pallaska, A.; Cotter, F.E. Bcl-2 resistant mitochondrial toxicity mediated by the isoquinoline carboxamide PK11195 involves de novo generation of reactive oxygen species. Br. J. Cancer 2001, 84, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Bono, F.; Lamarche, I.; Prabonnaud, V.; Le Fur, G.; Herbert, J.M. Peripheral benzodiazepine receptor agonists exhibit potent antiapoptotic activities. Biochem. Biophys. Res. Commun. 1999, 265, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Mestre, M.; Carriot, T.; Belin, C.; Uzan, A.; Renault, C.; Dubroeucq, M.C.; Gueremy, C.; Doble, A.; Le Fur, G. Electrophysiological and pharmacological evidence that peripheral type benzodiazepine receptors are coupled to calcium channels in the heart. Life Sci. 1985, 36, 391–400. [Google Scholar] [CrossRef]
- Azarashvili, T.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Yurkov, I.; Papadopoulos, V.; Reiser, G. The peripheral-type benzodiazepine receptor is involved in control of Ca2+-induced permeability transition pore opening in rat brain mitochondria. Cell Calcium 2007, 42, 27–39. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Zaaroor, M.; Vlodavsky, E.; Veenman, L.; Weizman, A.; Gavish, M. Neuroprotective effect of Ro5-4864 following brain injury. Exp. Neurol. 2008, 214, 201–208. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Vlodavsky, E.; Milman, F.; Gavish, M.; Zaaroor, M. Improvement of cerebral metabolism mediated by Ro5-4864 is associated with relief of intracranial pressure and mitochondrial protective effect in experimental brain injury. Pharm. Res. 2011, 28, 2945–2953. [Google Scholar] [CrossRef]
- Nakamoto, Y.; Watabe, S.; Shiotani, T.; Yoshii, M. Peripheral-type benzodiazepine receptors in association with epileptic seizures in EL mice. Brain Res. 1996, 717, 91–98. [Google Scholar] [CrossRef]
- Shiotani, T.; Nakamoto, Y.; Watabe, S.; Yoshii, M.; Nabeshima, T. Anticonvulsant actions of nefiracetam on epileptic EL mice and their relation to peripheral-type benzodiazepine receptors. Brain Res. 2000, 859, 255–261. [Google Scholar] [CrossRef]
- Stein, D.J. Etifoxine versus alprazolam for the treatment of adjustment disorder with anxiety: A randomized controlled trial. Adv Ther. 2015, 32, 57–68. [Google Scholar] [CrossRef]
- Verleye, M.; Akwa, Y.; Liere, P.; Ladurelle, N.; Pianos, A.; Eychenne, B.; Schumacher, M.; Gillardin, J.M. The anxiolytic etifoxine activates the peripheral benzodiazepine receptor and increases the neurosteroid levels in rat brain. Pharm. Biochem. Behav. 2005, 82, 712–720. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Lecanu, L. Translocator protein (18 kDa) TSPO: An emerging therapeutic target in neurotrauma. Exp. Neurol. 2009, 219, 53–57. [Google Scholar] [CrossRef]
- Girard, P.; Pansart, Y.; Gillardin, J.M. Preventive and curative effects of etifoxine in a rat model of brain oedema. Clin. Exp. Pharm. Physiol. 2009, 36, 655–661. [Google Scholar] [CrossRef]
- Aouad, M.; Petit-Demouliere, N.; Goumon, Y.; Poisbeau, P. Etifoxine stimulates allopregnanolone synthesis in the spinal cord to produce analgesia in experimental mononeuropathy. Eur. J. Pain 2014, 18, 258–268. [Google Scholar] [CrossRef]
- Aouad, M.; Zell, V.; Juif, P.E.; Lacaud, A.; Goumon, Y.; Darbon, P.; Lelievre, V.; Poisbeau, P. Etifoxine analgesia in experimental monoarthritis: A combined action that protects spinal inhibition and limits central inflammatory processes. Pain 2014, 155, 403–412. [Google Scholar] [CrossRef]
- Girard, C.; Liu, S.; Cadepond, F.; Adams, D.; Lacroix, C.; Verleye, M.; Gillardin, J.M.; Baulieu, E.E.; Schumacher, M.; Schweizer-Groyer, G. Etifoxine improves peripheral nerve regeneration and functional recovery. Proc. Natl. Acad. Sci. USA 2008, 105, 20505–20510. [Google Scholar] [CrossRef]
- Zhou, X.; He, B.; Zhu, Z.; He, X.; Zheng, C.; Xu, J.; Jiang, L.; Gu, L.; Zhu, J.; Zhu, Q.; et al. Etifoxine provides benefits in nerve repair with acellular nerve grafts. Muscle Nerve 2014, 50, 235–243. [Google Scholar] [CrossRef]
- Ganau, M.; Lavinio, A.; Prisco, L. Delirium and agitation in traumatic brain injury patients: An update on pathological hypotheses and treatment options. Minerva Anestesiol. 2018, 84, 632–640. [Google Scholar]
- Szabo, I.; Zoratti, M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J. Biol. Chem. 1991, 266, 3376–3379. [Google Scholar]
- Le Fur, G.; Vaucher, N.; Perrier, M.L.; Flamier, A.; Benavides, J.; Renault, C.; Dubroeucq, M.C.; Gueremy, C.; Uzan, A. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci. 1983, 33, 449–457. [Google Scholar] [CrossRef]
- Campiani, G.; Cappelli, A.; Nacci, V.; Anzini, M.; Vomero, S.; Hamon, M.; Cagnotto, A.; Fracasso, C.; Uboldi, C.; Caccia, S.; et al. Novel and highly potent 5-HT3 receptor agonists based on a pyrroloquinoxaline structure. J. Med. Chem. 1997, 40, 3670–3678. [Google Scholar] [CrossRef] [PubMed]
- Campiani, G.; Nacci, V.; Fiorini, I.; De Filippis, M.P.; Garofalo, A.; Ciani, S.M.; Greco, G.; Novellino, E.; Williams, D.C.; Zisterer, D.M.; et al. Synthesis, biological activity, and SARs of pyrrolobenzoxazepine derivatives, a new class of specific “peripheral-type” benzodiazepine receptor ligands. J. Med. Chem. 1996, 39, 3435–3450. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Ma, D.; Brewer, J.; Sun, S.; Costa, E.; Romeo, E.; Guidotti, A. Chemistry, binding affinities, and behavioral properties of a new class of "antineophobic" mitochondrial DBI receptor complex (mDRC) ligands. J. Med. Chem. 1993, 36, 2908–2920. [Google Scholar] [CrossRef]
- Trapani, G.; Franco, M.; Ricciardi, L.; Latrofa, A.; Genchi, G.; Sanna, E.; Tuveri, F.; Cagetti, E.; Biggio, G.; Liso, G. Synthesis and binding affinity of 2-phenylimidazo[1,2-alpha]pyridine derivatives for both central and peripheral benzodiazepine receptors. A new series of high-affinity and selective ligands for the peripheral type. J. Med. Chem. 1997, 40, 3109–3118. [Google Scholar] [CrossRef]
- Okuyama, S.; Chaki, S.; Yoshikawa, R.; Ogawa, S.; Suzuki, Y.; Okubo, T.; Nakazato, A.; Nagamine, M.; Tomisawa, K. Neuropharmacological profile of peripheral benzodiazepine receptor agonists, DAA1097 and DAA1106. Life Sci. 1999, 64, 1455–1464. [Google Scholar] [CrossRef]
- Selleri, S.; Bruni, F.; Costagli, C.; Costanzo, A.; Guerrini, G.; Ciciani, G.; Costa, B.; Martini, C. 2-Arylpyrazolo[1,5-a]pyrimidin-3-yl acetamides. New potent and selective peripheral benzodiazepine receptor ligands. Bioorg. Med. Chem. 2001, 9, 2661–2671. [Google Scholar] [CrossRef]
- Taliani, S.; Simorini, F.; Sergianni, V.; La Motta, C.; Da Settimo, F.; Cosimelli, B.; Abignente, E.; Greco, G.; Novellino, E.; Rossi, L.; et al. New fluorescent 2-phenylindolglyoxylamide derivatives as probes targeting the peripheral-type benzodiazepine receptor: Design, synthesis, and biological evaluation. J. Med. Chem. 2007, 50, 404–407. [Google Scholar] [CrossRef]
- Daugherty, D.J.; Selvaraj, V.; Chechneva, O.V.; Liu, X.B.; Pleasure, D.E.; Deng, W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol. Med. 2013, 5, 891–903. [Google Scholar] [CrossRef]
- Aouad, M.; Charlet, A.; Rodeau, J.L.; Poisbeau, P. Reduction and prevention of vincristine-induced neuropathic pain symptoms by the non-benzodiazepine anxiolytic etifoxine are mediated by 3alpha-reduced neurosteroids. Pain 2009, 147, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Liu, S.; Adams, D.; Lacroix, C.; Sineus, M.; Boucher, C.; Papadopoulos, V.; Rupprecht, R.; Schumacher, M.; Groyer, G. Axonal regeneration and neuroinflammation: Roles for the translocator protein 18 kDa. J. Neuroendocr. 2012, 24, 71–81. [Google Scholar] [CrossRef]
- Zhou, X.; He, X.; He, B.; Zhu, Z.; Zheng, C.; Xu, J.; Jiang, L.; Gu, L.; Zhu, J.; Zhu, Q.; et al. Etifoxine promotes glialderived neurotrophic factorinduced neurite outgrowth in PC12 cells. Mol. Med. Rep. 2013, 8, 75–80. [Google Scholar] [CrossRef]
- Simon-O’Brien, E.; Gauthier, D.; Riban, V.; Verleye, M. Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J. Neuroinflamm. 2016, 13, 203. [Google Scholar] [CrossRef]
- Marmarou, A.; Foda, M.A.; van den Brink, W.; Campbell, J.; Kita, H.; Demetriadou, K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J. Neurosurg. 1994, 80, 291–300. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Wang, L.; Zhang, Z.; Lu, D.; Lu, M.; Chopp, M. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 2001, 32, 1005–1011. [Google Scholar] [CrossRef]
- Sutton, R.L.; Lescaudron, L.; Stein, D.G. Unilateral cortical contusion injury in the rat: Vascular disruption and temporal development of cortical necrosis. J. Neurotrauma 1993, 10, 135–149. [Google Scholar] [CrossRef]
- Duan, W.R.; Garner, D.S.; Williams, S.D.; Funckes-Shippy, C.L.; Spath, I.S.; Blomme, E.A. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J. Pathol. 2003, 199, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ganau, M.; Syrmos, N.; Paris, M.; Ganau, L.; Ligarotti, G.K.I.; Moghaddamjou, A.; Chibbaro, S.; Soddu, A.; Ambu, R.; Prisco, L. Current and Future Applications of Biomedical Engineering for Proteomic Profiling: Predictive Biomarkers in Neuro-Traumatology. Medicines 2018, 5, 19. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shehadeh, M.; Palzur, E.; Apel, L.; Soustiel, J.F. Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine. Int. J. Mol. Sci. 2019, 20, 2639. https://doi.org/10.3390/ijms20112639
Shehadeh M, Palzur E, Apel L, Soustiel JF. Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine. International Journal of Molecular Sciences. 2019; 20(11):2639. https://doi.org/10.3390/ijms20112639
Chicago/Turabian StyleShehadeh, Mona, Eilam Palzur, Liat Apel, and Jean Francois Soustiel. 2019. "Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine" International Journal of Molecular Sciences 20, no. 11: 2639. https://doi.org/10.3390/ijms20112639
APA StyleShehadeh, M., Palzur, E., Apel, L., & Soustiel, J. F. (2019). Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine. International Journal of Molecular Sciences, 20(11), 2639. https://doi.org/10.3390/ijms20112639