Abstract
Neuroinflammation is an inflammatory response in the brain and spinal cord, which can involve the activation of microglia and astrocytes. It is a common feature of many central nervous system disorders, including a range of neurodegenerative disorders. An overlap between activated microglia, pro-inflammatory cytokines and translocator protein (TSPO) ligand binding was shown in early animal studies of neurodegeneration. These findings have been translated in clinical studies, where increases in TSPO positron emission tomography (PET) signal occur in disease-relevant areas across a broad spectrum of neurodegenerative diseases. While this supports the use of TSPO PET as a biomarker to monitor response in clinical trials of novel neurodegenerative therapeutics, the clinical utility of current TSPO PET radioligands has been hampered by the lack of high affinity binding to a prevalent form of polymorphic TSPO (A147T) compared to wild type TSPO. This review details recent developments in exploration of ligand-sensitivity to A147T TSPO that have yielded ligands with improved clinical utility. In addition to developing a non-discriminating TSPO ligand, the final frontier of TSPO biomarker research requires developing an understanding of the cellular and functional interpretation of the TSPO PET signal. Recent insights resulting from single cell analysis of microglial phenotypes are reviewed.
1. Neuroinflammation in Neurodegenerative Disorders
1.1. Neuroinflammation Overview
Neuroinflammation involves a complex multi-stage physiological response triggered by cell damaging processes in the brain. These can include infection, toxins, autoimmunity, trauma, and responses to processes that change neuronal activity. Neuroinflammation is choreographed by the reactive morphology of resident central nervous system (CNS) innate immune glial cells, predominantly microglia and astrocytes, accompanied by a dynamic biochemical cascade of inflammatory factors that modify the CNS microenvironment. Varying degrees of neuroinflammation exist and are dependent on multiple factors such as the duration of the inflammatory response, its course, and the circumstances underlying the primary insult [1]. The neuroinflammatory response is aimed at mitigating triggering factors by evoking CNS immunity to defend against potential harm and to maintain and restore homeostasis. Despite this, neuroinflammation has the potential to be both beneficial and damaging [2,3].
The dynamic neuroinflammatory response occurs via activation of microglial pattern recognition receptors (PRRs, including toll-like receptors (TLRs), pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs)) [4,5,6] in addition to glial production of a milieu of inflammatory factors including cytokines, chemokines, secondary messengers, and reactive oxygen species [7,8]. Microglia and astrocytes function as both the target and source of these inflammatory cytokines and chemokines [9,10]. If the neuroinflammatory response is not transient or tightly controlled within the CNS, then an uncontrolled, chronic neuroinflammatory response ensues, constituted by the prolonged overactivation of glial cells. This chronic state is deleterious due to the excessive and dysregulated production of pro-inflammatory factors, resulting in prohibited neuronal repair, synaptic impairment, oxidative damage and mitochondrial dysfunction which can lead to or exacerbate neurodegeneration [11,12,13,14]. This highly damaging, chronic response may also result in involvement of adaptive immunity, with the recruitment and infiltration of peripheral immune cells, via disruption of the blood-brain barrier (BBB), which can further initiate neurodegenerative mechanisms [15,16].
Microglia are at the center of the CNS immune response, given they are the chief and resident immunocompetent cells. They conduct a diversity of functions, surveying their surroundings with cellular processes [17,18,19] and adapting quickly to perturbations such as disease, infection, and aging. The activation and adaptation of microglia promotes an inflammatory response that further engages the immune system, involving changes to their morphology and migration to the site of injury to initiate tissue repair within the CNS [19]. While microglia are central to the neuroimmune response, the cellular microenvironment involved in the inflammatory response within the CNS is not limited to microglia, and also includes astrocytes, oligodendrocytes, and peripherally-derived immune cells, the latter constituted by the adaptive immune response.
Increased glial activation and thus neuroinflammation are components of various pathologies and disease states, including but not limited to cancer, traumatic brain injury, stroke, psychiatric disorders, and neurodegenerative diseases, which are the focus of this review.
1.2. Neuroinflammation in Neurodegenerative Diseases
Despite different etiologies, neuroinflammation is considered a pathological hallmark across the spectrum of neurodegenerative diseases [20,21,22], including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD). A primary risk factor for these neurodegenerative diseases is advancing in age [23,24] and there is evidence that the brain undergoes ‘inflamm-aging’, progressively acquiring a pro-inflammatory environment across the lifespan, which may contribute to this age-related risk of sporadic neurodegeneration. Specifically, microglia and endothelial cells continue to change phenotypes with age and experience in humans [25,26].
Whether neuroinflammation is a primary or secondary event and whether it has an overall beneficial or detrimental effect is hotly debated [3,27,28]. It has been hypothesized that immune activation is an early cause, as opposed to a late consequence of neurodegeneration [10]. In support of this, inflammation has been reported in the early stages of AD prior to the onset of dementia [29,30], similarly in PD, early inflammation in the brainstem has been shown prior to extending cortically with disease progression [31,32] and recently an early role for microglial activation that precedes clinical symptom onset has been shown in FTD [33]. An alternative hypothesis suggests the initial pathological insult, dependent on the phenotype of disease (e.g., amyloid plaques, α-synuclein, tau etc.) may induce an ongoing cytotoxic response, resulting in secondary chronic neuroinflammation, with altered neuronal function in predilection sites also dependent on the phenotype of the disease [28,34,35].
Despite the unknowns regarding timing and precise mechanisms underlying the neuroinflammatory response in neurodegeneration, there is mounting evidence across the spectrum of neurodegenerative diseases indicating that chronic neuroinflammation plays an instrumental role in the pathogenesis and progression of neurodegeneration. Findings from animal models of disease, particularly models of AD and PD, have been extensively reviewed and show early activation of inflammatory processes preceding neurodegeneration and in some instances concurrent with the production and accumulation of intracellular deposits [36,37,38,39]. Post-mortem human brain tissue from patients with neurodegenerative diseases show upregulation of pro-inflammatory cytokines, TLR subtypes, activated microglia, and astrocytes [10,40,41,42,43]. In addition to increased glial activation, post-mortem studies have shown dysregulated inflammatory factors, including, but not limited to, pro-inflammatory cytokines, chemokines, and complement in the blood, serum, and cerebrospinal fluid of disease affected individuals across the spectrum of neurodegenerative diseases [44,45,46,47,48,49]. Perhaps the most compelling evidence to date however, comes from genome-wide association studies (GWAS) reporting the expression of function and disease-causative mutations or associated polymorphisms in genes that are implicated in neuroinflammation and a significant overlap with neurodegenerative diseases and human leukocyte antigen (HLA)-loci (immune system) [23,50,51,52,53,54,55,56,57]. It is also becoming increasingly more understood that this chronic neuroinflammatory component is not restricted to a compromised CNS microenvironment and neuronal function, yet it also involves complex interactions with immunological mechanisms throughout both the central and peripheral nervous systems [8,11,58].
Collectively, investigation across animal studies, post-mortem studies and GWAS, indicate a shift towards a more pro-inflammatory environment in neurodegeneration and further support the importance of neuroinflammation in the pathophysiology of neurodegeneration and its potential as a useful biomarker, and therefore treatment target, across the spectrum of neurodegenerative diseases.
1.3. Monitoring Neuroinflammation as A Biomarker in Neurodegenerative Diseases
While it is likely that a panel of tools will be necessary to monitor and assess disease status in neurodegeneration, the in vivo monitoring of neuroinflammation in neurodegenerative diseases may provide a number of uses. As mentioned, neuroinflammation is a common finding across multiple neurodegenerative diseases, decreasing the need for development of separate tools for each disease or for multiple imaging of patients with unclear diagnosis. Being able to monitor the trajectory of neuroinflammation as a disease progresses will help understand the degree to which neuroinflammation is a causative or reactive process. Identification of inflammation as early as possible in the primary stages of the neurodegenerative process could enable early delivery of therapeutics, potentially in the prodromal phase of disease [39]. As agents that modulate neuroinflammation are being assessed in clinical trials in diseases such as ALS [59], monitoring neuroinflammation could assist in these trials by allowing tracking of disease progression and providing an indication of therapeutic response [27,60]. Furthermore, clinical trials could be streamlined to assist therapeutic efficacy, by providing insights into the underlying pathophysiology of neuroinflammatory and regulatory pathways [27,39,61].
For this to occur, however, an adequate biomarker of neuroinflammation is needed. One protein that has shown promise as a biomarker for neuroinflammation is the translocator protein (TSPO).
2. TSPO as a Biomarker For Neuroinflammation
The translocator protein (TSPO), originally named the peripheral benzodiazepine receptor, is an 18 kDa outer mitochondrial membrane protein. It is implicated in a number of functions relevant to neurodegeneration, such as redox homeostasis [62,63,64] and neurosteroidogenesis [65], however the direct role of TSPO in neurosteroidogenesis is the subject of current debate [66,67,68,69]. Clinical interest in TSPO stems from a wide body of evidence that it is upregulated in neuroinflammation, and hence may be a suitable neuroinflammation biomarker. Increased TSPO expression, indexed with immunohistochemistry and TSPO ligand positron emission tomography (PET) imaging, has been observed in a wide variety of animal models of neuroinflammatory conditions, including AD, stroke, brain injury, experimental autoimmune encephalitis, and epilepsy. This increased PET signal is in contrast with the low background expression of TSPO seen in quiescent CNS tissue, and it overlaps both with areas of brain pathology and with areas of increased immunohistochemical staining of TSPO [70,71,72,73,74,75,76,77,78,79,80,81,82]. Furthermore, the TSPO PET signal decreased in successful preclinical trials of novel therapeutics in HD and AD models, suggesting that it could be used to monitor treatment progress in clinical trials [83,84].
These promising preclinical studies prompted clinical studies using TSPO ligands to detect neuroinflammation. Early clinical studies, however, produced mixed results. While several studies using the first-generation radioligand [11C]PK 11195 reported higher TSPO PET brain signal in amyotrophic lateral sclerosis, AD, PD, and in brains of people at risk of Huntington’s disease when compared to controls [31,32,85,86,87,88,89,90], other studies using second-generation ligands reported no difference in TSPO PET signal in AD and multiple sclerosis [45,91,92,93,94].
One source of failure in these clinical trials was attributed to large inter-individual variability in PET signal. Second-generation TSPO ligands such as PBR-28, DPA-713, and DAA1106 showed three different binding patterns in tissue from human donors. These ligands bound with high affinity to the tissue from ~50–65% of donors (high affinity binders; HABs) and with low-affinity, or not at all, to tissue from ~5–25% of donors (low affinity binders; LABs) (Figure 1) [95,96,97]. A third group (~30%; mixed affinity binders; MABs) displayed binding of these ligands in a two-site manner for some ligands, or with a Ki close to half-way between HABs and LABs for other ligands [95,96,97].
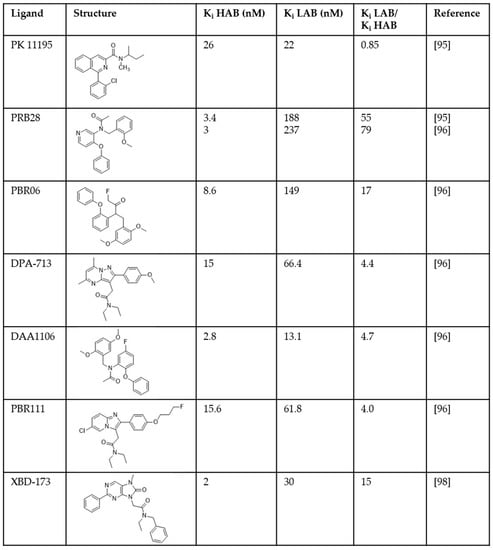
Figure 1.
Distribution of affinities of translocator protein (TSPO) ligands measured in homogenates prepared from post-mortem human brain tissue. HAB = high affinity binders; LAB = low affinity binders [95,96,98].
This binding affinity distribution can be predicted by the presence of a single nucleotide polymorphism (SNP) in the TSPO gene. This rs6971 SNP causes a non-conservative substitution of alanine for threonine at the 147th amino acid (A147T) of TSPO. According to the Hapmap database (http://hapmap.ncbi.nlm.nih.gov), it is present in 30% of Caucasians, 25% of Africans, 4% of Japanese, and 2% of Han Chinese. Patients who are HABs to second-generation ligands are homozygous for wild type TSPO, MABs are heterozygous and LABS are homozygous for the A147T TSPO [99,100].
Identification of the impact of A147T TSPO to second-generation ligand binding initiated a change in approach to clinical studies, with studies using second-generation ligands either being stratified for genotype or excluding LABs. This approach showed more consistent success using second-generation radioligands, with higher TSPO PET brain signal found in multiple sclerosis, ALS, mild cognitive impairment, and AD compared to controls [30,101,102,103,104,105,106,107,108,109,110]. These results suggest that TSPO is a good target for development of neuroinflammation imaging agents, however the SNP sensitivity of current TSPO PET radioligands complicate interpretation of results and necessitate genotyping of patients.
3. Third-Generation Ligands to Overcome The Challenge of A147T TSPO
3.1. ER176 and GE-180
Although stratifying for genotype has opened the door to TSPO PET radioligands being used to monitor therapeutic impact in neurodegeneration clinical trials [111], the ideal situation would see the development of a TSPO ligand with good PET imaging properties, which binds equally highly to TSPO WT and A147T. Two recent ligands, coined ‘third-generation TSPO ligands’, have showed improved detection of TSPO signal in LABs. The first developed out of a strategy to improve the first-generation TSPO ligand PK 11195. In both membrane preparations from human brain tissue and clinical PET scans, [11C]PK 11195 does not show binding sensitivity to A147T TSPO (Figure 1; [95]). It is limited as a PET imaging agent, though, by its high non-specific binding, high plasma protein binding and low brain permeability, which make it difficult to accurately quantify [11C]-PK 11195 signal in the brain [112]. Zanotti-Fregonara et al [113] examined PK 11195 analogs in a search for one that retained the lack of sensitivity to A147T TSPO but displayed improved imaging characteristics. They identified [11C]ER176 (Figure 2), a quinazoline analog of PK 11195, with equally high binding at WT and A147T TSPO in membranes prepared from human brain tissue, and improved lipophilicity (logD decreased from 3.97 to 3.55). In comparison to PK 11195, [11C]ER176 shows higher plasma free fractions, improved PET signal in monkey brain, and 80% of this signal can be blocked by administration of unlabeled PK 11195, suggesting specificity [113]. Furthermore, this ligand has the added benefit of increased accuracy of quantification as it does not produce radiometabolites that can enter the brain, unlike other second-generation ligands like DPA-713, PBR28, and PK 11195 [114]. However, despite the lack of sensitivity to A147T in human brain-derived membrane preparations, LABs unexpectedly had an [11C]ER176 binding potential (BPND) that was a third lower than HABs during a small first-in-man study [115]. This aside, the BPND of LABs for [11C]ER176, was approximately the same as that for HABs with the widely used [11C]PBR28, suggesting that although brain PET signal still needs to be corrected for genotype post-hoc, the BPND with ER176 should be sufficiently high to detect TSPO across all rs6971 genotypes, removing the need to exclude LABs [114,115]. As current clinical studies with ER176 have been performed on healthy controls, further evidence of the clinical utility of this ligand will be dependent on its performance in clinical studies using populations displaying neuroinflammation.
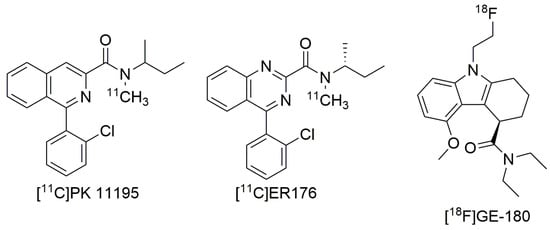
Figure 2.
Structures of [11C]PK 11195, [11C]ER176, and [18F]GE-180.
PET radioligands based on carbon-11 have a short half-life, restricting their use to facilities that have an on-site cyclotron [116]. Radioligands based on fluorine-18 have a half-life approximately five times longer, increasing their utility [117]. These ligands often have the added benefit of being more metabolically stable, reducing the effect of radiometabolites [118]. A third-generation TSPO ligand that incorporates fluorine-18 is the tricyclic indole [18F]GE-180 (flutriciclamide). Preclinical models of middle cerebral artery occlusion and AD showed high [18F]GE-180 signal in areas of neuroinflammation, low non-specific binding in unaffected brain tissue, and low contribution to the brain signal from radiometabolites [118,119,120]. In pre-clinical studies, this ligand showed better imaging characteristics than [11C]PK 11195 and [18F]DPA-714 [119,121], although it did not reflect the extent of microglial activation as accurately as [11C]DPA-713 [122]. A small clinical PET study measuring [18F]GE-180 signals in relapsing-remitting multiple sclerosis patients identified high signal to-noise ratio in focal lesions, with no significant difference in signal intensity across HABs, MABs and LABs [123]. This lack of sensitivity to the rs6971 genotype was surprising, as GE-180 drops off in affinity approximately 5-fold at A147T compared to WT TSPO in membrane radioligand binding studies [124]. Follow up studies on multiple sclerosis patients indicated that [18F]GE-180 had a low volume of distribution, suggesting low brain penetration [125,126], and it has been suggested that this low uptake generates an image quality that is not high enough to detect differences in binding to LABs and HABs [126]. Alternatively, the high variability in neuroinflammation among the multiple sclerosis patients may have reduced the power to detect significant differences in signal between genotypes [127]. These hypotheses can be examined by correlating [18F]GE-180 brain PET signal with immunohistochemical TSPO distribution in brain sections [126,127]. It will also be important to evaluate the potential of [18F]GE-180 as a neuroinflammation imaging agent in studies with larger sample sizes to increase their power to detect genotype differences. Another important future study is to compare the quantitative potential of second-generation ligands alongside [18F]GE-180 in clinical trials with patients experiencing neurodegenerative diseases that do not show as pronounced BBB breakdown as in multiple sclerosis, as it has been suggested that the high lesion-specific signal in these patients may be due to BBB breakdown allowing entrance of the radioligand into brain parenchyma in damaged areas, but not in intact areas [126].
3.2. Future Directions
While these novel third-generation ligands may be an improvement on second-generation ligands, as discussed above, they still show sensitivity to A147T TSPO. To advance on these, it will be vital to gain a greater understanding of how the binding requirements of WT and A147T TSPO differ. One approach to examining what drives the loss of affinity of TSPO ligands to A147T TSPO is examining the structural differences between WT and A147T TSPO. A crystal structure of the bacterial Rhodobacter sphaeroides TSPO (RsTSPO), which has 34% homology to human TSPO, shows marked differences between wild type TSPO and the A147T homolog, A139T. TSPO is comprised of five-transmembrane alpha-helices, and the gap between the second and fifth transmembrane segments is smaller in the mutant compared to the wild type RsTSPO [128]. In contrast, a 3D nuclear magnetic resonance characterization of mouse TSPO in complex with PK 11195 found the structures of A147T and WT TSPO closely resembled each other [129,130]. Given this contrast, future work resolving the human TSPO crystal structure in complex with discriminating second-generation ligands will provide valuable information about how the binding requirements of WT and A147T TSPO differ.
Another approach to generating insight about TSPO binding discrimination is high throughput screening of ligand binding at WT and A147T TSPO. This has been performed using a lower-throughput approach with membranes derived from tissue donated by HABs and LABs. This approach has shown endogenous TSPO ligands such as the protoporphyrin X and diazepam binding inhibitor do not discriminate between TSPO WT and SNP, in contrast to synthetic ligands [131], possibly providing a clue about binding requirements. A higher throughput approach using cell lines transfected with WT and A147T TSPO that show similar binding affinities to that generated with donated human tissue, has resulted in a derivative of GE-180 with minimal reduction in binding to A147T TSPO. This ligand, GE-387, crosses into the rat brain in preclinical PET studies, although future studies are required to examine its clinical utility [124]. Structure-activity relationships describing binding affinity differences at A147T vs. WT TSPO generated using this approach are slowly appearing in the literature [124,132]. This approach may yield new ligands that do not discriminate between WT and A147T. Such ligands may also emerge by targeting drug discovery to binding sites other than the PK 11195 binding site. These sites may include the cholesterol recognition amino acid consensus motif (for example, see [133]), or to sites at the interface with TSPO’s association partners, such as the voltage-dependent anion channel and the adenine nucleotide transporter [134].
Furthermore, it should be considered that optimizing for affinity determined using TSPO membrane preparations alone may not translate to equal binding potential in PET signals from HABs and LABs. As discussed, despite ER176’s almost identical affinity for TSPO WT and A147T in membrane preparations, LABs still showed lower BPND than HABs in vivo [115]. One reason for this may be that allosteric TSPO modulators such as cholesterol have a greater effect on second-generation ligand dissociation rate in LABs than HABs or MABs, and if the influence of kinetics and physiologically-relevant modulators are ignored in initial radioligand binding screens on membrane preparations then a pure affinity-based screen may miss this [135]. Secondly, TSPO may be in a different activity state in vivo because of associations with other proteins that are disrupted in the process of membrane preparation for binding assays [114]. Lastly, the production of brain-permeant radiometabolites most effects accurate quantification of PET signal from LABs, and these metabolites may not be generated in radioligand binding studies on membrane preparations [114].
This suggests that a wider drug discovery approach may accelerate development of translatable TSPO PET radioligands that are insensitive to the rs6971 genotype. In combination with the high throughput affinity screening approach to identify novel leads described above, it may be fruitful to include membrane binding studies measuring kinetics of candidate ligands and conducting binding studies with more physiological buffers containing allosteric modulators like cholesterol. Ex vivo autoradiography assessment of lead ligands should be conducted so affinity of these ligands can be determined for TSPO when it is in complex with other proteins. Finally, the brain-permeability of radiometabolite production should be investigated for identified lead molecules to ensure radiometabolites do not complicate analysis of brain signals.
4. Cellular and Functional Interpretation of TSPO PET Signals
In addition to developing a non-discriminating TSPO ligand, the final frontier of research into neuroinflammation-related TSPO PET imaging requires development of an understanding of the cellular and functional interpretation of the TSPO PET signal. At present, it is not clear whether the presence of TSPO PET signal indicates a predominantly pro-inflammatory or anti-inflammatory state, nor whether the signal indicates the presence of destructive or reparative cell states.
4.1. Microglial Phenotypes
A major brain cell type that displays TSPO upregulation in neuroinflammation is microglia [122,136,137,138,139,140,141]. Given microglia adapt to a variety of challenges across their lifespan, it is not surprising that they are a heterogeneous population consisting of multiple phenotypes [142]. Some of these microglial phenotypes enact useful functions, such as maintaining homeostasis, regulating synaptogenesis, and refining synapses [143]. On the other hand, some have a harmful pro-inflammatory function, and some phenotypes perhaps even carry out both useful and harmful actions. Due to this, an understanding of what functional state the TSPO signal in the brain represents will involve discovering which microglial phenotypes upregulate TSPO in neuroinflammation.
Until recently, microglial phenotypes were described using similar nomenclature to that which originally defined subpopulations of peripheral macrophages (Figure 3), with classification based on the presence of a limited number of cell markers and cytokines. M0 microglia were described as resting, ramified microglia that maintained homeostasis. Two types of activated microglia were suggested to arise from M0 microglia. A destructive pro-inflammatory phenotype (M1) was suggested to arise on exposure to pathogens, threats or pro-inflammatory stimuli. These M1 microglia were characterized by release of pro-inflammatory cytokines such as IL-1β and TNF-α. A protective anti-inflammatory phenotype (M2) was proposed to emerge on exposure to anti-inflammatory cytokines such as interleukin-4 (IL-4), IL-10, and IL-13 [144].
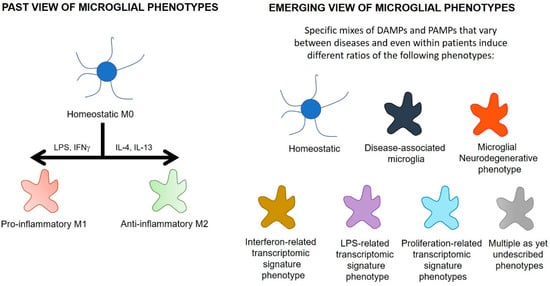
Figure 3.
The changing paradigm of microglial phenotypes. DAMPs = danger-associated molecular patterns; PAMPs = pathogen-associated molecular patterns; LPS = lipopolysaccharide; IFNγ = interferon-γ, IL-4 = interleukin-4, IL-13 = interleukin-13.
The phenotypic expression of TSPO has predominantly been examined within this M0/M1/M2 schema. In murine cells, there appears to be a selective upregulation of TSPO in pro-inflammatory M1 microglia. Cultured primary mouse microglia exposed to the pro-inflammatory lipopolysaccharide (LPS), or IL-1β with interferon-γ (IFNγ), upregulated expression of TSPO transcripts, while treatment with the anti-inflammatory IL-4 did not change TSPO transcript levels [145]. Injection of IL-4 into the lateral ventricle of mice did not alter parenchymal TSPO levels, while knockout of peroxisomal multifunctional protein-2 to induce neuroinflammation led to an increase in TSPO expression [145]. Similarly, TSPO expression increased approximately ten-fold in rodent-derived macrophages after a variety of pro-inflammatory stimuli, including LPS and IFNγ [146].
In contrast to these consistent findings in mice and rats, there is little consensus about changes in TSPO expression in human microglia and related macrophages, which is in accordance with observations that microglia from human samples often behave differently to rodent microglia [147]. Intraperitoneal delivery of LPS into human volunteers led to an upregulation of TSPO PET signal in the brain, although, as discussed below, this may not solely reflect microglial expression [148]. On the other hand, human microglia grown from a surgical resection did not show a change in TSPO expression even after intense pro-inflammatory induction involving a five-day exposure to granulocyte-macrophage colony-stimulating factor, followed by 1 h IFNγ and 48 h LPS [146]. TSPO expression even decreased after pro-inflammatory stimulation of human macrophages and did not change after anti-inflammatory stimulation [146,149]. Human microglia are known to change physiological properties on removal from the body and subsequent culturing [150,151], so different preparation methods may partly underlie these conflicting findings.
This conflict may also be a result of the over-simplified theoretical basis of the M0/M1/M2 continuum. This continuum describes microglial phenotypes according to the expression of a few surface markers or cytokines, in response to defined stimuli such as LPS or IL-4. During neurodegenerative diseases, however, microglia are exposed to a complex range of stimuli such as protein oligomers, excitotoxic neurotransmitter levels, reactive oxygen species, and a mix of pro- and anti-inflammatory cytokines. Recent studies using genomic and proteomic techniques have indicated that the cocktail of these stimuli generate a more complex and heterogenous phenotypic distribution of microglia than that previously described in the M0/M1/M2 continuum. These more recent studies have identified over eight different microglial phenotypes (Figure 3) and these are likely to be a small portion of the total number of microglial phenotypes in neurodegenerative diseases [142,152,153,154]. Each phenotype is characterized on the basis of a distinct signature of dysregulated genes. Some of these phenotypes include disease-associated microglia, microglia neurodegenerative phenotype, LPS-related transcriptomic signature microglia, interferon-related transcriptomic signature microglia, and proliferation-related transcriptomic signature microglia (see Table 1 for more detail). Of these newly described phenotypes, disease-associated microglia, microglia neurodegenerative phenotype, and LPS-signature microglia feature an upregulation of TSPO [132,142,144] (Table 1). These studies also suggest some phenotypes of microglia enriched in neurodegenerative conditions do not display TSPO upregulation [142], so the TSPO PET signal might not represent all microglial phenotypes altered in neurodegenerative diseases. As uncharacterized microglial phenotypes start to be described and as research continues to examine the change in TSPO expression on different phenotypes with disease progression, an understanding of the function of the microglia that upregulate TSPO will grow. This holds promise in understanding the functional implications of increased TSPO PET signals.

Table 1.
Features of new microglial phenotypes from ‘omics studies of neurodegenerative diseases.
4.2. Astrocytes
As described in Section 1, while microglia are key contributors to neuroinflammation, astrocytes also play a role in neuroinflammation. Given this, immunohistochemistry has been used to examine whether the overexpression of TSPO in neuroinflammation is only seen in microglia, or also manifests on astrocytes. Most studies that have examined this question have found no overlap with TSPO and markers for astrocytes such as glial-fibrillary acidic protein (GFAP) [137,155,156,157], although a small number of studies find GFAP co-localises with TSPO [140,141,158,159,160]. Two of these studies were longitudinal and identified that two weeks after induction of demyelination in a cuprizone-induced demyelination model, TSPO-positive cells were predominantly microglia, whereas six-weeks after demyelination induction TSPO-positive cells were predominantly astrocytes [160]. Thus, it is likely that astrocytes may contribute to the TSPO PET signal in demyelinating pathologies. Future studies that examine the longitudinal overlap between astrocytic markers and TSPO expression in neurodegeneration models will inform the extent to which astrocytes contribute to the TSPO PET signal in other neurodegenerative diseases.
4.3. Neurons
Upregulation of TSPO has been reported in the periphery on dorsal root ganglia after sciatic nerve injury [161], and on neural precursor cells and immature neurons in vitro [162]. The majority of reports, however, suggest TSPO is not upregulated on CNS neurons in neuroinflammation [70,162,163,164,165], although one study found an overlap between TSPO immunohistochemical signal and staining for NeuN, a neuronal marker, with a polyclonal TSPO antibody and not a monoclonal TSPO antibody [70]. This highlights the need for careful choice of specific antibodies for investigating the cellular expression of TSPO.
5. Conclusions
Neuroinflammation is a common finding in many neurodegenerative disorders. Tracking neuroinflammation through PET imaging may provide an avenue for early delivery of therapeutics, and tracking disease progression and response to novel therapeutics in clinical trials. PET radioligands that target the TSPO have showed promise in the detection of neuroinflammation, however, their clinical utility has been hampered by the presence of a single nucleotide polymorphism producing A147T TSPO at which every disclosed second-generation TSPO PET ligand loses affinity. The recent development of two third-generation TSPO ligands has yielded less-discriminating imaging options, but development of truly non-discriminating ligands will require increased knowledge of differences in A147T and WT TSPO structure, and the development of structure-affinity relationships through high-throughput screening. Furthermore, functional interpretation of the TSPO PET signal may be possible in the future with increasing knowledge of TSPO expression on complex microglial phenotypes and of the temporal differences in the role of astrocytes and microglia in neuroinflammation.
Funding
The authors research is supported by the National Health and Medical Research Council of Australia (NHMRC) (APP1132524; APP1095127; APP1140708; APP1081916) and the University of Sydney. GMH is an NHMRC Senior Principal Research Fellow (APP1079679); LMI is an NHMRC Principal Research Fellow (APP1136241); OP is an NHMRC Senior Research Fellow (APP1103258); CTL is an NHMRC Dementia Fellow (1107657); MK is an NHMRC Principal Research Fellow (APP1154692).
Conflicts of Interest
Eryn Werry, Fiona Bright, Olivier Piguet, Lars Ittner, Glenda Halliday, Clement Loy, Jillian Krill and Michael Kassiou have no competing interests to declare. John R Hodges is an editorial board member of Cognitive Neuropsychiatry, Aphasiology, Cognitive Neuropsychology, Nature Reviews Neurology, Journal of Alzheimer’s Disease, Acta Neuropsychologica, ALS-FTD Journal, Neurology and Clinical Neuroscience, Dementia Neurospsychologia; received research support from the NHMRC, the Australian Research Council and institutional support from the University of Sydney. Matthew C Kiernan serves as Editor-in-Chief of the Journal of Neurology, Neurosurgery and Psychiatry (BMJ Publishers).
Abbreviations
| A147T TSPO | Ala147Thr translocator protein |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| BBB | blood-brain barrier |
| BPND | binding potential |
| CNS | central nervous system |
| DAMPs | danger-associated molecular patterns |
| FTD | frontotemporal dementia |
| GFAP | glial fibrillary acidic protein |
| GWAS | genome-wide association studies |
| HAB | high affinity binder |
| HLA | human leukocyte antigen |
| IFNγ | interferon-γ |
| IL | interleukin |
| Ki | affinity |
| LAB | low affinity binder |
| LPS | lipopolysaccharide |
| MAB | mixed affinity binder |
| PAMPs | pathogen-associated molecular patterns |
| PET | positron emission tomography |
| MAB | mixed affinity binder |
| PAMPs | pathogen-associated molecular patterns |
| PD | Parkinson’s disease |
| PET | positron emission tomography |
| PRRs | pattern recognition receptors |
| RsTSPO | Rhodobacter sphaeroides translocator protein |
| SNP | single nucleotide polymorphism |
| TLRs | toll-like receptors |
| TSPO | translocator protein |
| WT TSPO | wild type translocator protein |
References
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzeriat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1. [Google Scholar]
- O’Callaghan, J.P.; Sriram, K.; Miller, D.B. Defining “neuroinflammation”. Ann. N. Y. Acad. Sci. 2008, 1139, 318–330. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Meeter, L.H.; Kaat, L.D.; Rohrer, J.D.; van Swieten, J.C. Imaging and fluid biomarkers in frontotemporal dementia. Nat. Rev. Neurol. 2017, 13, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and mast cells: Two tracks on the road to neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef][Green Version]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Gratwicke, J.; Jahanshahi, M.; Foltynie, T. Parkinson’s disease dementia: A neural networks perspective. Brain J. Neurol. 2015, 138, 1454–1476. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Hernandez, D.G.; Nalls, M.A.; Rohrer, J.D.; Ramasamy, A.; Kwok, J.B.; Dobson-Stone, C.; Brooks, W.S.; Schofield, P.R.; Halliday, G.M.; et al. Frontotemporal dementia and its subtypes: A genome-wide association study. Lancet Neurol. 2014, 13, 686–699. [Google Scholar] [CrossRef]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Soreq, L.; Consortium, U.K.B.E.; North American Brain Expression, C.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Cerami, C.; Iaccarino, L.; Perani, D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging. Int. J. Mol. Sci. 2017, 18, 993. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, G.; Brooks, D.J.; Edison, P. The role of neuroinflammation in dementias. Curr. Neurol. Neurosci. Rep. 2015, 15, 17. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Bevan-Jones, W.R.; Cope, T.E.; Jones, P.S.; Passamonti, L.; Hong, Y.T.; Fryer, T.; Arnold, R.; Coles, J.P.; Aigbirhio, F.I.; O’Brien, J.T.; et al. In vivo evidence for pre-symptomatic neuroinflammation in a MAPT mutation carrier. Ann. Clin. Transl. Neurol. 2019, 6, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S. Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2007, 66, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Ono, K.; Suzuki, H.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. 2005, 109, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Saido, T.C. Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 74. [Google Scholar] [CrossRef]
- Cebrian, C.; Loike, J.D.; Sulzer, D. Neuroinflammation in Parkinson’s disease animal models: A cell stress response or a step in neurodegeneration? Curr. Top. Behav. Neurosci. 2015, 22, 237–270. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Paterson, R.W.; Warren, J.D.; Zetterberg, H.; O’Brien, J.T.; Fox, N.C.; Halliday, G.M.; Schott, J.M. Biomarkers in dementia: Clinical utility and new directions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1426–1434. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth Factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.M.; Ke, Y.D.; Vucic, S.; Ittner, L.M.; Seeley, W.; Hodges, J.R.; Piguet, O.; Halliday, G.; Kiernan, M.C. Physiological changes in neurodegeneration—Mechanistic insights and clinical utility. Nat. Rev. Neurol. 2018, 14, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.; da Cruz, E.S.O.A.B.; Henriques, A.G. Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr. Alzheimer Res. 2017, 14, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, B.; Vas, A.; Toth, M.; Takano, A.; Varrone, A.; Cselenyi, Z.; Schain, M.; Mattsson, P.; Halldin, C. Age and disease related changes in the translocator protein (TSPO) system in the human brain: Positron emission tomography measurements with [11C]vinpocetine. Neuroimage 2011, 56, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Chung, J.H.; Choi, T.K.; Suh, S.Y.; Oh, B.H.; Hong, C.H. Peripheral cytokines and chemokines in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Venturelli, E.; Fenoglio, C.; Guidi, I.; Villa, C.; Bergamaschini, L.; Cortini, F.; Scalabrini, D.; Baron, P.; Vergani, C.; et al. Intrathecal levels of IL-6, IL-11 and LIF in Alzheimer’s disease and frontotemporal lobar degeneration. J. Neurol. 2008, 255, 539–544. [Google Scholar] [CrossRef]
- Sjogren, M.; Folkesson, S.; Blennow, K.; Tarkowski, E. Increased intrathecal inflammatory activity in frontotemporal dementia: Pathophysiological implications. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1107–1111. [Google Scholar] [CrossRef]
- Mosley, R.L.; Hutter-Saunders, J.A.; Stone, D.K.; Gendelman, H.E. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009381. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Kannarkat, G.T.; Cook, D.A.; Lee, J.K.; Chang, J.; Chung, J.; Sandy, E.; Paul, K.C.; Ritz, B.; Bronstein, J.; Factor, S.A.; et al. Common Genetic Variant Association with Altered HLA Expression, Synergy with Pyrethroid Exposure, and Risk for Parkinson’s Disease: An Observational and Case-Control Study. NPJ Parkinsons Dis. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Neuroinflammation and ALS: Transcriptomic Insights into Molecular Disease Mechanisms and Therapeutic Targets. Mediat. Inflamm. 2017, 2017, 7070469. [Google Scholar] [CrossRef] [PubMed]
- Broce, I.; Karch, C.M.; Wen, N.; Fan, C.C.; Wang, Y.; Tan, C.H.; Kouri, N.; Ross, O.A.; Hoglinger, G.U.; Muller, U.; et al. Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 2018, 15, e1002487. [Google Scholar] [CrossRef]
- Gagliano, S.A.; Pouget, J.G.; Hardy, J.; Knight, J.; Barnes, M.R.; Ryten, M.; Weale, M.E. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann. Clin. Transl. Neurol. 2016, 3, 924–933. [Google Scholar] [CrossRef]
- Fogh, I.; Ratti, A.; Gellera, C.; Lin, K.; Tiloca, C.; Moskvina, V.; Corrado, L.; Soraru, G.; Cereda, C.; Corti, S.; et al. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 2220–2231. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef]
- Mora, J.S.; Barbeito, L.; Hermine, O. Masitinib as an Add-On Therapy to Riluzole Is Beneficial in the Treatment of Amyotrophic Lateral Sclerosis (ALS) with Acceptable Tolerability: Results from a Randomized Controlled Phase 3 Trial. European Network to Cure ALS (ENCALS). 2017. Available online: http://videolectures.net/encals2017_barbeito_mora_hermine_therapy/ (accessed on 19 May 2019).
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Forum on Neuroscience and Nervous System Disorders. Biomarkers of Neuroinflammation: Proceedings of a Workshop; National Academies Press: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Politis, M.; Su, P.; Piccini, P. Imaging of microglia in patients with neurodegenerative disorders. Front. Pharmacol. 2012, 3, 96. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Loth, M.K.; Guariglia, S.R. TSPO Finds NOX2 in Microglia for Redox Homeostasis. Trends Pharmacol. Sci. 2016, 37, 334–343. [Google Scholar] [CrossRef]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; East, D.A.; Singh, A.; Alvarez, M.S.; Frison, M.; Matic, I.; Ferraina, C.; Sampson, N.; Turkheimer, F.; Campanella, M. A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis. 2017, 8, e2896. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Campioli, E.; Midzak, A.; Culty, M.; Papadopoulos, V. Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc. Natl. Acad. Sci. USA 2015, 112, 7261–7266. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, V.; Tu, L.N.; Stocco, D.M. Crucial Role Reported for TSPO in Viability and Steroidogenesis is a Misconception. Commentary: Conditional Steroidogenic Cell-Targeted Deletion of TSPO Unveils a Crucial Role in Viability and Hormone-Dependent Steroid Formation. Front. Endocrinol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018, 30, e12500. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Da Pozzo, E.; Martini, C. Translocator protein and steroidogenesis. Biochem. J. 2018, 475, 901–904. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef]
- Abourbeh, G.; Theze, B.; Maroy, R.; Dubois, A.; Brulon, V.; Fontyn, Y.; Dolle, F.; Tavitian, B.; Boisgard, R. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J. Neurosci. 2012, 32, 5728–5736. [Google Scholar] [CrossRef]
- Amhaoul, H.; Hamaide, J.; Bertoglio, D.; Reichel, S.N.; Verhaeghe, J.; Geerts, E.; Van Dam, D.; De Deyn, P.P.; Kumar-Singh, S.; Katsifis, A.; et al. Brain inflammation in a chronic epilepsy model: Evolving pattern of the translocator protein during epileptogenesis. Neurobiol. Dis. 2015, 82, 526–539. [Google Scholar] [CrossRef]
- Brendel, M.; Probst, F.; Jaworska, A.; Overhoff, F.; Korzhova, V.; Albert, N.L.; Beck, R.; Lindner, S.; Gildehaus, F.J.; Baumann, K.; et al. Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple-Tracer PET Study. J. Nucl. Med. 2016, 57, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, D.J.; Selvaraj, V.; Chechneva, O.V.; Liu, X.B.; Pleasure, D.E.; Deng, W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol. Med. 2013, 5, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Dedeurwaerdere, S.; Callaghan, P.D.; Pham, T.; Rahardjo, G.L.; Amhaoul, H.; Berghofer, P.; Quinlivan, M.; Mattner, F.; Loc’h, C.; Katsifis, A.; et al. PET imaging of brain inflammation during early epileptogenesis in a rat model of temporal lobe epilepsy. EJNMMI Res. 2012, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Israel, I.; Ohsiek, A.; Al-Momani, E.; Albert-Weissenberger, C.; Stetter, C.; Mencl, S.; Buck, A.K.; Kleinschnitz, C.; Samnick, S.; Siren, A.L. Combined [18F]DPA-714 micro-positron emission tomography and autoradiography imaging of microglia activation after closed head injury in mice. J. Neuroinflamm. 2016, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Boisgard, R.; Theze, B.; Van Camp, N.; Kuhnast, B.; Damont, A.; Kassiou, M.; Dolle, F.; Tavitian, B. Evaluation of the PBR/TSPO radioligand [18F]DPA-714 in a rat model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Mattner, F.; Katsifis, A.; Staykova, M.; Ballantyne, P.; Willenborg, D.O. Evaluation of a radiolabelled peripheral benzodiazepine receptor ligand in the central nervous system inflammation of experimental autoimmune encephalomyelitis: A possible probe for imaging multiple sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, N.; Tang, S.P.; Ashworth, S.; Coello, C.; Plisson, C.; Passchier, J.; Selvaraj, V.; Tyacke, R.J.; Nutt, D.J.; Sastre, M. In vivo imaging of microglial activation by positron emission tomography with [11C]PBR28 in the 5XFAD model of Alzheimer’s disease. Glia 2016, 64, 993–1006. [Google Scholar] [CrossRef]
- Thomas, C.; Vercouillie, J.; Domene, A.; Tauber, C.; Kassiou, M.; Guilloteau, D.; Destrieux, C.; Serriere, S.; Chalon, S. Detection of Neuroinflammation in a Rat Model of Subarachnoid Hemorrhage Using [18F]DPA-714 PET Imaging. Mol. Imaging 2016, 15. [Google Scholar] [CrossRef]
- Toth, M.; Little, P.; Arnberg, F.; Haggkvist, J.; Mulder, J.; Halldin, C.; Gulyas, B.; Holmin, S. Acute neuroinflammation in a clinically relevant focal cortical ischemic stroke model in rat: Longitudinal positron emission tomography and immunofluorescent tracking. Brain Struct. Funct. 2016, 221, 1279–1290. [Google Scholar] [CrossRef]
- Tremoleda, J.L.; Thau-Zuchman, O.; Davies, M.; Foster, J.; Khan, I.; Vadivelu, K.C.; Yip, P.K.; Sosabowski, J.; Trigg, W.; Michael-Titus, A.T. In vivo PET imaging of the neuroinflammatory response in rat spinal cord injury using the TSPO tracer [18F]GE-180 and effect of docosahexaenoic acid. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1710–1722. [Google Scholar] [CrossRef]
- Simmons, D.A.; James, M.L.; Belichenko, N.P.; Semaan, S.; Condon, C.; Kuan, J.; Shuhendler, A.J.; Miao, Z.; Chin, F.T.; Longo, F.M. TSPO-PET imaging using [18F]PBR06 is a potential translatable biomarker for treatment response in Huntington’s disease: Preclinical evidence with the p75NTR ligand LM11A-31. Hum. Mol. Genet. 2018, 27, 2893–2912. [Google Scholar] [CrossRef] [PubMed]
- James, M.L.; Belichenko, N.P.; Shuhendler, A.J.; Hoehne, A.; Andrews, L.E.; Condon, C.; Nguyen, T.V.; Reiser, V.; Jones, P.; Trigg, W.; et al. [18F]GE-180 PET Detects Reduced Microglia Activation After LM11A-31 Therapy in a Mouse Model of Alzheimer’s Disease. Theranostics 2017, 7, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, B.; Toth, M.; Schain, M.; Airaksinen, A.; Vas, A.; Kostulas, K.; Lindstrom, P.; Hillert, J.; Halldin, C. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: A PET study with the TSPO molecular imaging biomarker [11C]vinpocetine. J. Neurol. Sci. 2012, 320, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, F.; Kosaka, J.; Ota, M.; Higuchi, M.; Ito, H.; Fujimura, Y.; Nozaki, S.; Takahashi, S.; Mizukami, K.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [11C]DAA1106. Psychiatry Res. 2012, 203, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef]
- Takano, A.; Piehl, F.; Hillert, J.; Varrone, A.; Nag, S.; Gulyas, B.; Stenkrona, P.; Villemagne, V.L.; Rowe, C.C.; Macdonell, R.; et al. In vivo TSPO imaging in patients with multiple sclerosis: A brain PET study with [18F]FEDAA1106. EJNMMI Res. 2013, 3, 30. [Google Scholar] [CrossRef]
- Golla, S.S.; Boellaard, R.; Oikonen, V.; Hoffmann, A.; van Berckel, B.N.; Windhorst, A.D.; Virta, J.; Haaparanta-Solin, M.; Luoto, P.; Savisto, N.; et al. Quantification of [18F]DPA-714 binding in the human brain: Initial studies in healthy controls and Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2015, 35, 766–772. [Google Scholar] [CrossRef]
- Varrone, A.; Mattsson, P.; Forsberg, A.; Takano, A.; Nag, S.; Gulyas, B.; Borg, J.; Boellaard, R.; Al-Tawil, N.; Eriksdotter, M.; et al. In vivo imaging of the 18-kDa translocator protein (TSPO) with [18F]FEDAA1106 and PET does not show increased binding in Alzheimer’s disease patients. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, F.; Van Camp, N.; Dolle, F.; Kuhnast, B.; Hinnen, F.; Damont, A.; Boutin, H.; James, M.; Kassiou, M.; Tavitian, B. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J. Nucl. Med. 2009, 50, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Howell, O.W.; Tang, S.P.; Wells, L.A.; Bennacef, I.; Bergstrom, M.; Gunn, R.N.; Rabiner, E.A.; Wilkins, M.R.; Reynolds, R.; et al. Two binding sites for [3H]PBR28 in human brain: Implications for TSPO PET imaging of neuroinflammation. J. Cereb. Blood Flow Metab. 2010, 30, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Gunn, R.N.; Rabiner, E.A.; Bennacef, I.; Fujita, M.; Kreisl, W.C.; Innis, R.B.; Pike, V.W.; Reynolds, R.; Matthews, P.M.; et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J. Nucl. Med. 2011, 52, 24–32. [Google Scholar] [CrossRef]
- Mizrahi, R.; Rusjan, P.M.; Kennedy, J.; Pollock, B.; Mulsant, B.; Suridjan, I.; De Luca, V.; Wilson, A.A.; Houle, S. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [18F]-FEPPA. J. Cereb. Blood Flow Metab. 2012, 32, 968–972. [Google Scholar] [CrossRef]
- Owen, D.R.; Lewis, A.J.; Reynolds, R.; Rupprecht, R.; Eser, D.; Wilkins, M.R.; Bennacef, I.; Nutt, D.J.; Parker, C.A. Variation in binding affinity of the novel anxiolytic XBD173 for the 18 kDa translocator protein in human brain. Synapse 2011, 65, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Colasanti, A.; Owen, D.R.; Onega, M.; Kamalakaran, A.; Bennacef, I.; Matthews, P.M.; Rabiner, E.A.; Turkheimer, F.E.; Gunn, R.N. Quantification of the specific translocator protein signal of 18F-PBR111 in healthy humans: A genetic polymorphism effect on in vivo binding. J. Nucl. Med. 2013, 54, 1915–1923. [Google Scholar] [CrossRef]
- Colasanti, A.; Guo, Q.; Muhlert, N.; Giannetti, P.; Onega, M.; Newbould, R.D.; Ciccarelli, O.; Rison, S.; Thomas, C.; Nicholas, R.; et al. In Vivo Assessment of Brain White Matter Inflammation in Multiple Sclerosis with 18F-PBR111 PET. J. Nucl. Med. 2014, 55, 1112–1118. [Google Scholar] [CrossRef]
- Zurcher, N.R.; Loggia, M.L.; Lawson, R.; Chonde, D.B.; Izquierdo-Garcia, D.; Yasek, J.E.; Akeju, O.; Catana, C.; Rosen, B.R.; Cudkowicz, M.E.; et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: Assessed with [11C]-PBR28. Neuroimage Clin. 2015, 7, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Suridjan, I.; Pollock, B.G.; Verhoeff, N.P.; Voineskos, A.N.; Chow, T.; Rusjan, P.M.; Lobaugh, N.J.; Houle, S.; Mulsant, B.H.; Mizrahi, R. In-vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: A positron emission tomography study with a novel radioligand, [18F]-FEPPA. Mol. Psychiatry 2015, 20, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.S.; Wei, M.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. 11C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Datta, G.; Colasanti, A.; Kalk, N.; Owen, D.; Scott, G.; Rabiner, E.A.; Gunn, R.N.; Lingford-Hughes, A.; Malik, O.; Ciccarelli, O.; et al. 11C-PBR28 and 18F-PBR111 Detect White Matter Inflammatory Heterogeneity in Multiple Sclerosis. J. Nucl. Med. 2017, 58, 1477–1482. [Google Scholar] [CrossRef]
- Varrone, A.; Oikonen, V.; Forsberg, A.; Joutsa, J.; Takano, A.; Solin, O.; Haaparanta-Solin, M.; Nag, S.; Nakao, R.; Al-Tawil, N.; et al. Positron emission tomography imaging of the 18-kDa translocator protein (TSPO) with [18F]FEMPA in Alzheimer’s disease patients and control subjects. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 438–446. [Google Scholar] [CrossRef]
- Lyoo, C.H.; Ikawa, M.; Liow, J.S.; Zoghbi, S.S.; Morse, C.L.; Pike, V.W.; Fujita, M.; Innis, R.B.; Kreisl, W.C. Cerebellum Can Serve As a Pseudo-Reference Region in Alzheimer Disease to Detect Neuroinflammation Measured with PET Radioligand Binding to Translocator Protein. J. Nucl. Med. 2015, 56, 701–706. [Google Scholar] [CrossRef]
- Yoder, K.K.; Nho, K.; Risacher, S.L.; Kim, S.; Shen, L.; Saykin, A.J. Influence of TSPO genotype on 11C-PBR28 standardized uptake values. J. Nucl. Med. 2013, 54, 1320–1322. [Google Scholar] [CrossRef]
- Sucksdorff, M.; Rissanen, E.; Tuisku, J.; Nuutinen, S.; Paavilainen, T.; Rokka, J.; Rinne, J.; Airas, L. Evaluation of the Effect of Fingolimod Treatment on Microglial Activation Using Serial PET Imaging in Multiple Sclerosis. J. Nucl. Med. 2017, 58, 1646–1651. [Google Scholar] [CrossRef]
- Chauveau, F.; Boutin, H.; Van Camp, N.; Dolle, F.; Tavitian, B. Nuclear imaging of neuroinflammation: A comprehensive review of [11C]PK11195 challengers. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2304–2319. [Google Scholar] [CrossRef]
- Zanotti-Fregonara, P.; Zhang, Y.; Jenko, K.J.; Gladding, R.L.; Zoghbi, S.S.; Fujita, M.; Sbardella, G.; Castellano, S.; Taliani, S.; Martini, C.; et al. Synthesis and evaluation of translocator 18 kDa protein (TSPO) positron emission tomography (PET) radioligands with low binding sensitivity to human single nucleotide polymorphism rs6971. ACS Chem. Neurosci. 2014, 5, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Kobayashi, M.; Ikawa, M.; Gunn, R.N.; Rabiner, E.A.; Owen, D.R.; Zoghbi, S.S.; Haskali, M.B.; Telu, S.; Pike, V.W.; et al. Comparison of four 11C-labeled PET ligands to quantify translocator protein 18 kDa (TSPO) in human brain: (R)-PK11195, PBR28, DPA-713, and ER176-based on recent publications that measured specific-to-non-displaceable ratios. EJNMMI Res. 2017, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Lohith, T.G.; Shrestha, S.; Telu, S.; Zoghbi, S.S.; Castellano, S.; Taliani, S.; Da Settimo, F.; Fujita, M.; Pike, V.W.; et al. 11C-ER176, a Radioligand for 18-kDa Translocator Protein, Has Adequate Sensitivity to Robustly Image All Three Affinity Genotypes in Human Brain. J. Nucl. Med. 2017, 58, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Stoll, H.P.; Hutchins, G.D.; Winkle, W.L.; Nguyen, A.T.; Appledorn, C.R.; Janzen, I.; Seifert, H.; Rube, C.; Schieffer, H.; March, K.L. Advantages of short-lived positron-emitting radioisotopes for intracoronary radiation therapy with liquid-filled balloons to prevent restenosis. J. Nucl. Med. 2001, 42, 1375–1383. [Google Scholar] [PubMed]
- Liu, B.; Le, K.X.; Park, M.A.; Wang, S.; Belanger, A.P.; Dubey, S.; Frost, J.L.; Holton, P.; Reiser, V.; Jones, P.A.; et al. In Vivo Detection of Age- and Disease-Related Increases in Neuroinflammation by 18F-GE180 TSPO MicroPET Imaging in Wild-Type and Alzheimer’s Transgenic Mice. J. Neurosci. 2015, 35, 15716–15730. [Google Scholar] [CrossRef]
- Boutin, H.; Murray, K.; Pradillo, J.; Maroy, R.; Smigova, A.; Gerhard, A.; Jones, P.A.; Trigg, W. 18F-GE-180: A novel TSPO radiotracer compared to 11C-R-PK11195 in a preclinical model of stroke. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Picon, F.R.; Snellman, A.; Eskola, O.; Helin, S.; Solin, O.; Haaparanta-Solin, M.; Rinne, J.O. Neuroinflammation Appears Early on PET Imaging and Then Plateaus in a Mouse Model of Alzheimer Disease. J. Nucl. Med. 2018, 59, 509–515. [Google Scholar] [CrossRef]
- Sridharan, S.; Lepelletier, F.X.; Trigg, W.; Banister, S.; Reekie, T.; Kassiou, M.; Gerhard, A.; Hinz, R.; Boutin, H. Comparative Evaluation of Three TSPO PET Radiotracers in a LPS-Induced Model of Mild Neuroinflammation in Rats. Mol. Imaging Biol. 2017, 19, 77–89. [Google Scholar] [CrossRef]
- Chaney, A.; Cropper, H.C.; Johnson, E.M.; Lechtenberg, K.J.; Peterson, T.C.; Stevens, M.Y.; Buckwalter, M.S.; James, M.L. 11C-DPA-713 Versus 18F-GE-180: A Preclinical Comparison of Translocator Protein 18 kDa PET Tracers to Visualize Acute and Chronic Neuroinflammation in a Mouse Model of Ischemic Stroke. J. Nucl. Med. 2019, 60, 122–128. [Google Scholar] [CrossRef]
- Unterrainer, M.; Mahler, C.; Vomacka, L.; Lindner, S.; Havla, J.; Brendel, M.; Boning, G.; Ertl-Wagner, B.; Kumpfel, T.; Milenkovic, V.M.; et al. TSPO PET with [18F]GE-180 sensitively detects focal neuroinflammation in patients with relapsing-remitting multiple sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Fisher, E.; McMurray, L.; Milicevic Sephton, S.; Hird, M.; Kuzhuppilly-Ramakrishnan, N.; Williamson, D.J.; Zhou, X.; Werry, E.; Kassiou, M.; et al. Radiosynthesis of (R,S)-[18F]GE387: A Potential PET Radiotracer for Imaging Translocator Protein 18 kDa (TSPO) with Low Binding Sensitivity to the Human Gene Polymorphism rs6971. ChemMedChem 2019, 14, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Raffel, J.; Nandoskar, A.; Record, C.; Brooks, D.J.; Owen, D.; Sharp, D.; Muraro, P.A.; Gunn, R.; Nicholas, R. Confirmation of Specific Binding of the 18-kDa Translocator Protein (TSPO) Radioligand [18F]GE-180: A Blocking Study Using XBD173 in Multiple Sclerosis Normal Appearing White and Grey Matter. Mol. Imaging Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zanotti-Fregonara, P.; Veronese, M.; Pascual, B.; Rostomily, R.C.; Turkheimer, F.; Masdeu, J.C. The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Albert, N.L.; Unterrainer, M.; Brendel, M.; Kaiser, L.; Zweckstetter, M.; Cumming, P.; Bartenstein, P. In response to: The validity of 18F-GE180 as a TSPO imaging agent. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, J.; Zheng, Y.; Garavito, R.M.; Ferguson-Miller, S. Protein structure. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, M.; Jaremko, L.; Giller, K.; Becker, S.; Zweckstetter, M. Backbone and side-chain resonance assignment of the A147T polymorph of mouse TSPO in complex with a high-affinity radioligand. Biomol. NMR Assign 2016, 10, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, M.; Jaremko, L.; Giller, K.; Becker, S.; Zweckstetter, M. Structural Integrity of the A147T Polymorph of Mammalian TSPO. Chembiochem 2015, 16, 1483–1489. [Google Scholar] [CrossRef]
- Berroteran-Infante, N.; Tadic, M.; Hacker, M.; Wadsak, W.; Mitterhauser, M. Binding Affinity of Some Endogenous and Synthetic TSPO Ligands Regarding the rs6971 Polymorphism. Int. J. Mol. Sci. 2019, 20, 563. [Google Scholar] [CrossRef]
- Sokias, R.; Werry, E.L.; Chua, S.W.; Reekie, T.A.; Munoz, L.; Wong, E.C.N.; Ittner, L.M.; Kassiou, M. Determination and reduction of translocator protein (TSPO) ligand rs6971 discrimination. Medchemcomm 2017, 8, 202–210. [Google Scholar] [CrossRef]
- Midzak, A.S.; Akula, N.; Rone, M.B.; Papadopoulos, V. Computational modeling and biological validation of novel non-steroidal ligands for the cholesterol recognition/interaction amino acid consensus (CRAC) motif of the mitochondrial translocator protein (TSPO). Pharmacol. Res. 2015, 99, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Rone, M.B.; Midzak, A.S.; Issop, L.; Rammouz, G.; Jagannathan, S.; Fan, J.; Ye, X.; Blonder, J.; Veenstra, T.; Papadopoulos, V. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol. Endocrinol. 2012, 26, 1868–1882. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Stathis, M.; Coughlin, J.M.; Pomper, M.; Slusher, B.S. The Low-Affinity Binding of Second Generation Radiotracers Targeting TSPO is Associated with a Unique Allosteric Binding Site. J. Neuroimmune Pharmacol. 2018, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Rigaud, D.; Fossey, C.; Cailly, T.; Fabis, F.; Pham, T.; Gregoire, M.C.; Kovari, E.; Moulin-Sallanon, M.; et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Schober, D.A.; Smalstig, E.B.; Mincy, R.E.; Gehlert, D.R.; Clemens, J.A. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J. Neurosci. 1995, 15, 5263–5274. [Google Scholar] [CrossRef] [PubMed]
- Vowinckel, E.; Reutens, D.; Becher, B.; Verge, G.; Evans, A.; Owens, T.; Antel, J.P. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurosci. Res. 1997, 50, 345–353. [Google Scholar] [CrossRef]
- Arlicot, N.; Katsifis, A.; Garreau, L.; Mattner, F.; Vergote, J.; Duval, S.; Kousignian, I.; Bodard, S.; Guilloteau, D.; Chalon, S. Evaluation of CLINDE as potent translocator protein (18 kDa) SPECT radiotracer reflecting the degree of neuroinflammation in a rat model of microglial activation. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Baidoo, K.; Verina, T.; Guilarte, T.R. Peripheral benzodiazepine receptor imaging in CNS demyelination: Functional implications of anatomical and cellular localization. Brain 2004, 127, 1379–1392. [Google Scholar] [CrossRef]
- Chen, M.K.; Guilarte, T.R. Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol. Sci. 2006, 91, 532–539. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Beckers, L.; Ory, D.; Geric, I.; Declercq, L.; Koole, M.; Kassiou, M.; Bormans, G.; Baes, M. Increased Expression of Translocator Protein (TSPO) Marks Pro-inflammatory Microglia but Does Not Predict Neurodegeneration. Mol. Imaging Biol. 2018, 20, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H.; et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Park, T.I.; Schweder, P.; Scotter, J.; Correia, J.; Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; et al. Isolation of highly enriched primary human microglia for functional studies. Sci. Rep. 2016, 6, 19371. [Google Scholar] [CrossRef] [PubMed]
- Sandiego, C.M.; Gallezot, J.D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.F.; Matuskey, D.; Lee, J.Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef]
- Narayan, N.; Mandhair, H.; Smyth, E.; Dakin, S.G.; Kiriakidis, S.; Wells, L.; Owen, D.; Sabokbar, A.; Taylor, P. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1’ human macrophages. PLoS ONE 2017, 12, e0185767. [Google Scholar] [CrossRef]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef]
- Chamberlain, L.M.; Holt-Casper, D.; Gonzalez-Juarrero, M.; Grainger, D.W. Extended culture of macrophages from different sources and maturation results in a common M2 phenotype. J. Biomed. Mater. Res. A 2015, 103, 2864–2874. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.L.; Gundlach, A.L.; Craven, J.A. Temporal changes in glial fibrillary acidic protein messenger RNA and [3H]PK11195 binding in relation to imidazoline-I2-receptor and alpha 2-adrenoceptor binding in the hippocampus following transient global forebrain ischaemia in the rat. Neuroscience 1998, 82, 805–817. [Google Scholar] [CrossRef]
- Myers, R.; Manjil, L.G.; Cullen, B.M.; Price, G.W.; Frackowiak, R.S.; Cremer, J.E. Macrophage and astrocyte populations in relation to [3H]PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J. Cereb. Blood Flow Metab. 1991, 11, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Domene, A.; Cavanagh, C.; Page, G.; Bodard, S.; Klein, C.; Delarasse, C.; Chalon, S.; Krantic, S. Expression of Phenotypic Astrocyte Marker Is Increased in a Transgenic Mouse Model of Alzheimer’s Disease versus Age-Matched Controls: A Presymptomatic Stage Study. Int. J. Alzheimers Dis. 2016, 2016, 5696241. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Kuhlmann, A.C.; O’Callaghan, J.P.; Miceli, R.C. Enhanced expression of peripheral benzodiazepine receptors in trimethyltin-exposed rat brain: A biomarker of neurotoxicity. Neurotoxicology 1995, 16, 441–450. [Google Scholar] [PubMed]
- Lavisse, S.; Guillermier, M.; Herard, A.S.; Petit, F.; Delahaye, M.; Van Camp, N.; Ben Haim, L.; Lebon, V.; Remy, P.; Dolle, F.; et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J. Neurosci. 2012, 32, 10809–10818. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol. Ther. 2019, 194, 44–58. [Google Scholar] [CrossRef]
- Karchewski, L.A.; Bloechlinger, S.; Woolf, C.J. Axonal injury-dependent induction of the peripheral benzodiazepine receptor in small-diameter adult rat primary sensory neurons. Eur. J. Neurosci. 2004, 20, 671–683. [Google Scholar] [CrossRef]
- Varga, B.; Marko, K.; Hadinger, N.; Jelitai, M.; Demeter, K.; Tihanyi, K.; Vas, A.; Madarasz, E. Translocator protein (TSPO 18kDa) is expressed by neural stem and neuronal precursor cells. Neurosci. Lett. 2009, 462, 257–262. [Google Scholar] [CrossRef]
- Bonsack, F.t.; Alleyne, C.H., Jr.; Sukumari-Ramesh, S. Augmented expression of TSPO after intracerebral hemorrhage: A role in inflammation? J. Neuroinflamm. 2016, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Mages, K.; Grassmann, F.; Jagle, H.; Rupprecht, R.; Weber, B.H.F.; Hauck, S.M.; Grosche, A. The agonistic TSPO ligand XBD173 attenuates the glial response thereby protecting inner retinal neurons in a murine model of retinal ischemia. J. Neuroinflamm. 2019, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Notter, T.; Coughlin, J.M.; Gschwind, T.; Weber-Stadlbauer, U.; Wang, Y.; Kassiou, M.; Vernon, A.C.; Benke, D.; Pomper, M.G.; Sawa, A.; et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol. Psychiatry 2018, 23, 323–334. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).