Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis


Abstract
1. Introduction
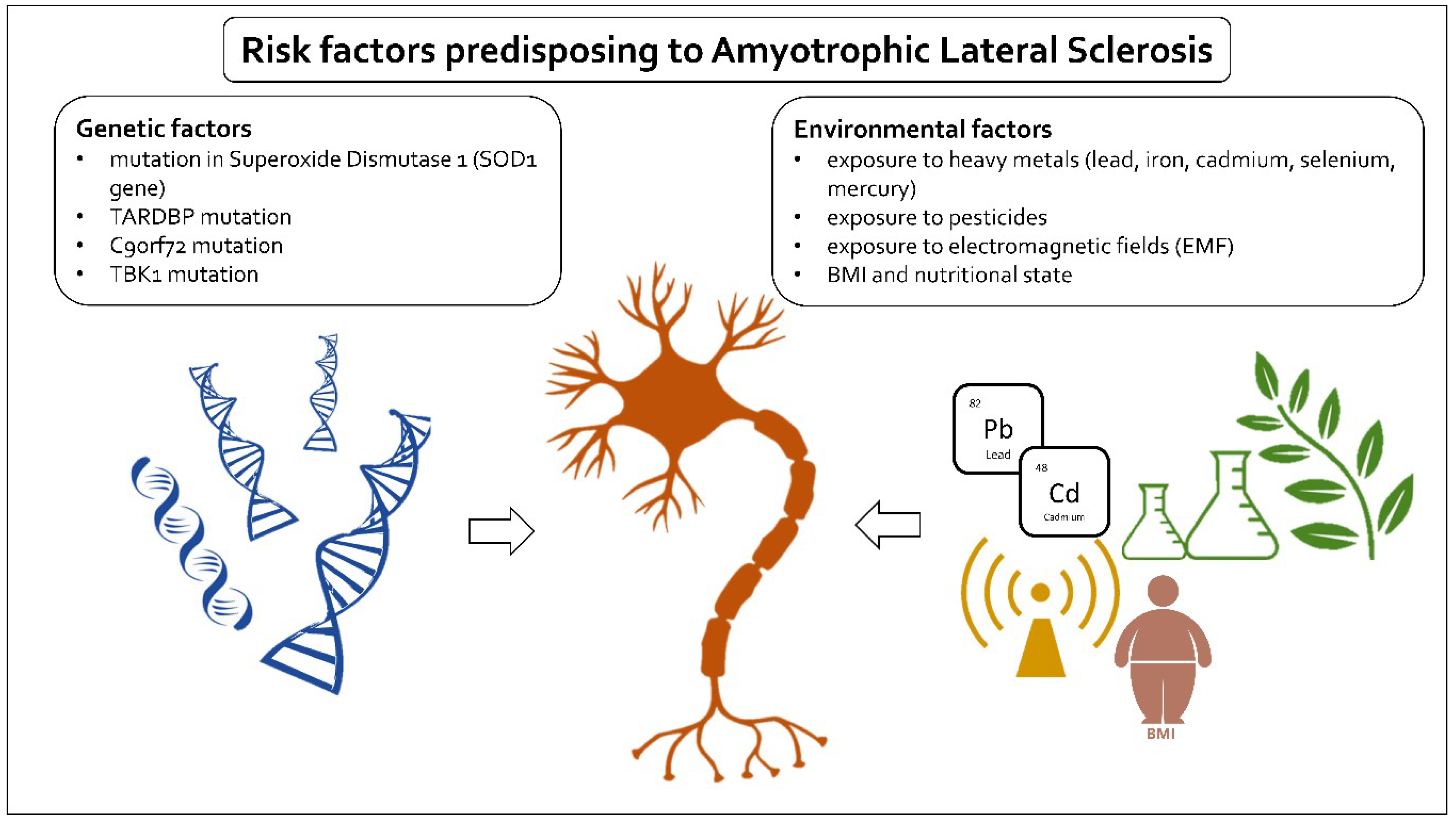
2. Risk Factors
3. Genetic Factors
3.1. Mutations in Superoxide Dismutase 1 (SOD1 Gene)
3.2. TARDBP Mutation
3.3. C9orf72 Mutation
3.4. TBK1 Mutation
4. Environmental Factors
4.1. Lead and Other Heavy Metals
4.2. BMI and Nutritional State
4.3. Pesticides
4.4. Electromagnetic Fields (EMF)
4.5. Oxidative Stress as a Common Aspect in Number of Environmental Risk Factors
5. ALS Treatments
5.1. Existing Disease-Modifying Treatments
5.2. Researched Treatments
5.2.1. Anti-Apoptotic
5.2.2. Anti-Inflammatory
5.2.3. Anti-Excitotoxicitory
5.2.4. Anti-Oxidant
5.2.5. Anti-Aggregation
5.2.6. Neurotrophic Growth Factors and Neuroprotection
5.3. Cell-Based Therapies
5.3.1. Glial-Restricted Precursors
5.3.2. Neural Progenitor Stem Cells
5.4. Gene Therapy Strategy
6. Factors in “ALS Reversal” Cases
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Tiwari, A.; Xu, Z.; Hayward, L.J. Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2005, 280, 29771–29779. [Google Scholar] [CrossRef]
- Siddique, T.; Dellefave, L. Amyotrophic lateral sclerosis. In Neurogenetics: Scientific and Clinical Advances; David, L., Jennifer, F., Eds.; Taylor and Francis: New York, NY, USA; London, UK, 2006; pp. 693–720. [Google Scholar]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chio, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E.; EURALS. Incidence of amyotrophic lateral sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2009, 81, 385–390. [Google Scholar] [CrossRef]
- Wang, M.D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 2017, 61, 101–130. [Google Scholar] [CrossRef]
- Hanspala, M.A.; Dobsona, C.M.; Yerburyb, J.J.; Kumita, J.R. The relevance of contact-independent cell-to-cell transfer of TDP-43 andSOD1 in amyotrophic lateral sclerosis. Bba - Mol. Basis Dis. 2017, 1863, 2762–2771. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Krishnan, A.V.; Kiernan, M.C. Fatigue and activity dependent changes in axonal excitability in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1202–1208. [Google Scholar] [CrossRef]
- Mitchell, J.D.; Borasio, G.D. Amyotrophic lateral sclerosis. Lancet 2007, 369, 2031–2041. [Google Scholar] [CrossRef]
- Armon, C. An evidence-based medicine approach to the evaluation of the role of exogenous risk factors in sporadic amyotrophic lateral sclerosis. Neuroepidemiology 2003, 22, 217–228. [Google Scholar] [CrossRef]
- Byrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. BMJ J. 2011, 82, 623–627. [Google Scholar] [CrossRef]
- Saddique, N.; Saddique, T. Genetics of Amyotrophic Lateral Sclerosis. Phys. Med. Rehabil. Clin. N. Am. 2008, 19, 429–439. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosen, D.R.; Saddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nat. Int. J. Sci. 1993, 362, 59–62. [Google Scholar]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci 2018, 19, 1345. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Zhang, Y.J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.; et al. Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nat. Int. J. Sci. 2010, 465, 223–226. [Google Scholar]
- Shaw, C.E.; Capturing, V.C.P. Another molecular piece in the ALS jigsaw puzzle. Neuron 2010, 68, 812–814. [Google Scholar] [CrossRef][Green Version]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, P.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Doucette, P.A.; Whitson, L.J.; Cao, X.; Schirf, V.; Demeler, B.; Valentine, J.S.; Hansen, J.C.; Hart, P.J. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J. Biol. Chem. 2004, 279, 54558–54566. [Google Scholar] [CrossRef]
- McCord, J.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.A.; Fisher, E.M.C.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain A J. Neurol. 2013, 136, 2342–2358. [Google Scholar] [CrossRef]
- Getzoff, E.D.; Tainer, J.A.; Stempien, M.M.; Bell, G.I.; Hallewell, R.A. Evolution of CuZn superoxide dismutase and the Greek key beta-barrel structural motif. Proteins 1989, 5, 322–336. [Google Scholar] [CrossRef]
- Zelko, I.N.; Thomas, J.M.; Rodney, J.F. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Hart, P.J. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. 2003, 100, 3617–3622. [Google Scholar] [CrossRef]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis—A valid pharmacological target? Br. J. Pharmacol. 2014, 171, 2191–2205. [Google Scholar] [CrossRef]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Invest. 2008, 118, 659–670. [Google Scholar] [CrossRef]
- Sasaki, S.; Horie, Y.; Iwata, M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007, 114, 633–639. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatr. 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Oztürk, A.; Hicks, G.G. FUS-regulatedRNAmetabolism and DNA damage repair: Implications for amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Rare Dis. 2014, 2, e29515. [Google Scholar] [CrossRef] [PubMed]
- Amlie-Wolf, A.; Ryvkin, P.; Tong, R.; Dragomir, I.; Suh, E.; Xu, Y.; Van Deerlin, V.M.; Gregory, B.D.; Kwong, L.K.; Trojanowski, J.Q.; et al. Transcriptomic changes due to cytoplasmic TDP-43 expression reveal dysregulation of Histone transcripts and nuclear chromatin. PLoS ONE 2015, 10, e141836. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008, 13, 867–878. [Google Scholar] [CrossRef]
- Cascella, R.; Capitini, C.; Fani, G.; Dobson, C.M.; Cecchi, C.; Chiti, F. Quantification of the relative contributions of loss-of-function and gain-of-function mechanisms in TAR DNA-binding protein 43 (TDP-43) proteinopathies. J. Biol. Chem. 2016, 291, 19437–19448. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9orf72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Bigio, E.H. Motor neuron disease: The C9orf72 hexanucleotide repeat expansion in FTD and ALS. Nat. Rev. Neurol. 2012, 8, 249–250. [Google Scholar] [CrossRef]
- Laaksovirta, H.; Peuralinna, T.; Schymick, J.C.; Scholz, S.W.; Lai, S.L.; Myllykangas, L.; Sulkava, R.; Jansson, L.; Hernandez, D.G.; Gibbs, J.R.; et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: A genome-wide association study. Lancet Neurol. 2010, 9, 978–985. [Google Scholar] [CrossRef]
- Hubers, A.; Marroquin, N.; Schmoll, B.; Vielhaber, S.; Just, M.; Mayer, B.; Högel, J.; Dorst, J.; Mertens, T.; Just, W.; et al. Polymerase chain reaction and Southern blot-based analysis of the C9orf72 hexa nucleotide repeat in different motor neuron diseases. Neurobiol. Aging 2014, 35, 1214e1–1214e6. [Google Scholar] [CrossRef]
- Bäumer, D.; Talbot, K.; Turner, M.R. Advances in motor neurone disease. J. R. Soc. Med. 2014, 107, 14–21. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Hewitt, C.; Highley, J.R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C.; et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansion in C9ORF72. Brain 2012, 135, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Seltman, R.E.; Matthews, B.R. Frontotemporal lobar degeneration: Epidemiology, pathology, diagnosis and management. Cns Drugs. 2012, 26, 841–870. [Google Scholar] [CrossRef]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Tu, D.; Zhu, Z.; Zhou, A.Y.; Yun, C.H.; Lee, K.E.; Toms, A.V.; Li, Y.; Dunn, G.P.; Chan, E.; Thai, T.; et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 2013, 3, 747–758. [Google Scholar] [CrossRef]
- Larabi, A.; Devos, J.M.; Ng, S.L.; Nanao, M.H.; Round, A.; Maniatis, T.; Panne, D. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 2013, 3, 734e746. [Google Scholar] [CrossRef]
- Weidberg, H.; Elazar, Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 2011, 4, pe39. [Google Scholar] [CrossRef]
- Li, F.; Xu, D.; Wang, Y.; Zhou, Z.; Liu, J.; Hu, S.; Gong, Y.; Yuan, J.; Pan, L. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1mediated phosphorylation. Autophagy 2018, 14, 66–79. [Google Scholar] [CrossRef]
- Pottier, C.; Bieniek, K.F.; Finch, N.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef]
- Ou, Y.H.; Torres, M.; Ram, R.; Formstecher, E.; Roland, C.; Cheng, T.; Brekken, R.; Wurz, R.; Tasker, A.; Polverino, T.; et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol. Cell. 2011, 41, 458–470. [Google Scholar] [CrossRef]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Gleason, C.E.; Ordureau, A.; Gourlay, R.; Arthur, J.S.; Cohen, P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase1 and production of interferon beta. J. Biol. Chem. 2011, 286, 35663–35674. [Google Scholar] [CrossRef]
- Edens, B.M.; Miller, N.; Ma, Y.C. Impaired autophagy and defective mitochondrial function: Converging paths on the road to motor neuron degeneration. Front Cell Neurosci. 2016, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Müller, K.; Ludolph, A.C.; Weishaupt, J.H.; Andersen, P.M. Association of mutations inTBK1 with sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. Jama Neurol. 2016, 74, 110e113. [Google Scholar]
- Cui, R.; Tuo, M.; Li1, P.; Zhou, C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: A meta-analysis. Neurol. Sci. 2018, 39, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.; Goutman, S.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef]
- Wang, M.D.; Gomes, J.; Cashman, N.R.; Little, J.; Krewski, D. A meta-analysis of observational studies of the association between chronic occupational exposure to lead and amyotrophic lateral sclerosis. J. Occup. Environ. Med. 2014, 56, 1235–1242. [Google Scholar] [CrossRef]
- Trojsi, F.; Monsurrò, M.R.; Tedeschi, G. Exposure to environmental toxicants and pathogenesis of amyotrophic lateral sclerosis: State of the art and research perspectives. Int. J. Mol. Sci. 2013, 14, 15286–15311. [Google Scholar] [CrossRef] [PubMed]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Papmphlett, R. Genetic susceptibility to environmental toxicants in ALS. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F.; Umbach, D.M.; Lehman, T.A.; Park, L.P.; Munsat, T.L.; Shefner, J.M.; Sandler, D.P.; Hu, H.; Taylor, J.A. Amyotrophic lateral sclerosis, lead, and genetic susceptibility: Polymorphisms in the delta-aminolevulinic acid dehydratase and vitamin D receptor genes. Environ. Health Perspect. 2003, 111, 1335–1339. [Google Scholar] [CrossRef]
- Fang, F.; Kwee, L.C.; Allen, K.D.; Umbach, D.M.; Weimin, Y.; Watson, M.; Keller, J.; Oddone, E.Z.; Sandler, D.P.; Schmidt, S.; et al. Association between blood lead and the risk of amyotrophic lateral sclerosis. Am. J. Epidemiol. 2010, 171, 1126–1133. [Google Scholar] [CrossRef]
- Kasarskis, E.J.; Berryman, S.; Vanderleest, J.G.; Schneider, A.R.; McClain, C.J. Nutritional status of patients with amyotrophic lateral sclerosis: Relation to the proximity of death. Am. J. Clin. Nutr. 1996, 63, 130–137. [Google Scholar] [CrossRef]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011, 44, 20–24. [Google Scholar] [CrossRef]
- Reich-Slotky, R.; Andrews, J.; Cheng, B.; Buchsbaum, R.; Levy, D.; Kaufmann, P.; Thompson, J.L. Body mass index (BMI) as predictor of ALSFRS-R score decline in ALS patients. Amyotroph Lateral Scler Front. Degener. 2013, 14, 212–216. [Google Scholar] [CrossRef]
- O’Reilly, É.J.; Wang, H.; Weisskopf, M.G.; Fitzgerald, K.C.; Falcone, G.; McCullough, M.L.; Thun, M.; Park, Y.; Kolonel, L.N.; Ascherio, A. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front. Degener. 2013, 14, 205–211. [Google Scholar] [CrossRef]
- Körner, S.; Hendricks, M.; Kollewe, K.; Zapf, A.; Dengler, R.; Silani, V.; Petri, S. Weight loss, dysphagia and supplement intake in patients with amyotrophic lateral sclerosis (ALS): Impact on quality of life and therapeutic options. BMC Neurol. 2013, 13, 84. [Google Scholar] [CrossRef]
- Paris, G.; Martinaud, O.; Petit, A.; Cuvelier, A.; Hannequin, D.; Roppeneck, P.; Verin, E. Oropharyngeal dysphagia in amyotrophic lateral sclerosis alters quality of life. J. Oral Rehabil. 2013, 40, 199–204. [Google Scholar] [CrossRef]
- Lindauer, E.; Dupuis, L.; Müller, H.P.; Neumann, H.; Ludolph, A.C.; Kassubek, J. Adipose Tissue Distribution Predicts Survival in Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e67783. [Google Scholar] [CrossRef]
- Patel, B.P.; Safdar, A.; Raha, S.; Tarnopolsky, M.A.; Hamadeh, M.J. Caloric Restriction Shortens Lifespan through an Increase in Lipid Peroxidation, Inflammation and Apoptosis in the G93A Mouse, an Animal Model of ALS. PLoS ONE 2010, 5, e9386. [Google Scholar] [CrossRef]
- Sutedja, N.A.; Veldink, J.H.; Fischer, K.; Kromhout, H.; Heederik, D.; Huisman, M.H.; Wokke, J.H.; van den Berg, L.H. Exposure to chemicals and metals and risk of amyotrophic lateral sclerosis: A systematic review. Amyotroph Lateral Scler. 2009, 10, 302–309. [Google Scholar] [CrossRef]
- Bozzoni, V.; Pansarasa, O.; Diamanti, L.; Nosari, G.; Cereda, C.; Ceroni, M. Amyotrophic lateral sclerosis and environmental factors. Funct Neurol. 2016, 31, 7–19. [Google Scholar] [CrossRef]
- Slowik, A.; Tomik, B.; Wolkow, P.P.; Partyka, D.; Turaj, W.; Malecki, M.T.; Pera, J.; Dziedzic, T.; Szczudlik, A.; Figlewicz, D.A. Paraoxonase gene polymorphisms and sporadic ALS. Neurology 2006, 67, 766–770. [Google Scholar] [CrossRef]
- Ticozzi, N.; LeClerc, A.L.; Keagle, P.J.; Glass, J.D.; Wills, A.M.; van Blitterswijk, M.; Bosco, D.A.; Rodriguez-Leyva, I.; Gellera, C.; Ratti, A.; et al. Paraoxonase gene mutations in amyotrophic lateral sclerosis. Ann. Neurol. 2010, 68, 102–107. [Google Scholar] [CrossRef]
- Gagliardi, S.; Abel, K.; Bianchi, M.; Milani, P.; Bernuzzi, S.; Corato, M.; Ceroni, M.; Cashman, J.R.; Cereda, C. Regulation of FMO and PON detoxication systems in ALS human tissues. Neurotox Res. 2013, 23, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Luo, D.; Yang, M.; Wang, Y.; Xiong, L.; Gao, L.; Liu, Y.; Liu, H. A Meta-Analysis on the Relationship of the PON Genes and Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2017, 30, 303–310. [Google Scholar] [CrossRef]
- McGuire, V.; Longstreth, W.T., Jr.; Nelson, L.M.; Koepsell, T.D.; Checkoway, H.; Morgan, M.S.; van Belle, G. Occupational exposures and amyotrophic lateral sclerosis. A populationbased case-control study. Am. J. Epidemiol. 1997, 145, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.D. Amyotrophic lateral sclerosis: Toxins and environment. Amyotroph. Lateral Scler. Other Motor. Neuron Disord. 2000, 1, 235–250. [Google Scholar] [CrossRef]
- Deapen, D.M.; Henderson, B.E. A case-control study of amyotrophic lateral sclerosis. Am. J. Epidemiol. 1986, 123, 790–799. [Google Scholar] [CrossRef]
- Johansen, C. Exposure to electromagnetic fields and risk of central nervous system disease in utility workers. Epidemiology 2000, 11, 539–543. [Google Scholar] [CrossRef]
- Li, C.Y.; Sung, F.C. Association between occupational exposure to power frequency electromagnetic fields and amyotrophic lateral sclerosis: A review. Am. J. Ind. Med. 2003, 43, 212–220. [Google Scholar] [CrossRef]
- Hakansson, N.; Gustavsson, P.; Johansen, C.; Floderus, B. Neurodegenerative diseases in welders and other workers exposed to high levels of magnetic fields. Epidemiology 2003, 14, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.G.; McCullough, M.L.; Morozova, N.; Calle, E.E.; Thun, M.J.; Ascherio, A. Prospective Study of Occupation and Amyotrophic Lateral Sclerosis Mortality. Am. J. Epidemiol. 2005, 162, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Park, R.M.; Schulte, P.A.; Bowman, J.D.; Walker, J.T.; Bondy, S.C.; Yost, M.G.; Touchstone, J.A.; Dosemeci, M. Potential occupational risks for neurodegenerative diseases. Am. J. Ind. Med. 2005, 48, 63–77. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, G.; Chen, C.; Yu, Y.; Xu, Z. Association between extremely low-frequency electromagnetic fields occupations and amyotrophic lateral sclerosis: A meta-analysis. PLoS ONE 2012, 7, e48354. [Google Scholar] [CrossRef]
- Oskarsson, B.; Horton, D.K.; Mitsumoto, H. Potential Environmental Factors in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 877–888. [Google Scholar] [CrossRef]
- Simpson, E.P.; Yen, A.A.; Appel, S.H. Oxidative stress: A common denominator in the pathogenesis of amyotrophic lateral sclerosis. Curr. Opin. Rheumatol. 2003, 15, 730–736. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef]
- Cacciapuoti, F. Oxidative Stress as “Mother” of Many Human Diseases at Strong Clinical Impact. J. Cardiovasc Med. Cardiol. 2016, 3, 001–006. [Google Scholar] [CrossRef]
- Matschke, V.; Theiss, C.; Matschke, J. Oxidative stress: The lowest common denominator of multiple diseases. Neural. Regen Res. 2019, 14, 238–241. [Google Scholar] [CrossRef]
- Ercal, N.; Gurer-Orhan, H.; Aykin-Burns, N. Toxic Metals and Oxidative Stress Part I: Mechanisms Involved in Me-tal induced Oxidative Damage. Curr. Top Med. Chem. 2001, 1, 529–539. [Google Scholar] [CrossRef]
- Mostafalou, S.; Abdollahi, M. The link of organophosphorus pesticides with neurodegenerative and neurodevelopmental diseases based on evidence and mechanisms. Toxicology 2018, 409, 44–52. [Google Scholar] [CrossRef]
- Simkó, M.; Mats-Olof, M. Extremely low frequency electromagnetic fields as effectors of cellular responses in vitro: Possible immune cell activation. J. Cell Biochem. 2004, 93, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sámano, J.; Torres-Durán, P.V.; Juárez-Oropeza, M.A.; Verdugo-Díaz, L. Effect of acute extremely low frequency electromagnetic field exposure on the antioxidant status and lipid levels in rat brain. Arch Med. Res. 2012, 43, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic lateral sclerosis: An update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef]
- Dharmadasa, T.; Kiernan, M.C. Riluzole, disease stage and survival in ALS. Lancet Neurol. 2018, 17, 385–386. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Traynor, B.J.; Alexander, M.; Corr, B.; Frost, E.; Hardiman, O. An outcome study of riluzole in amyotrophic lateral sclerosis. J. Neurol. 2003, 250, 473–479. [Google Scholar] [CrossRef]
- Zoing, M.C.; Burke, D.; Pamphlett, R.; Kiernan, M.C. Riluzole therapy for motor neurone disease: An early Australian experience (1996–2002). J. Clin Neurosci. 2006, 13, 78–83. [Google Scholar] [CrossRef]
- Riviere, M.; Meininger, V.; Zeisser, P.; Munsat, T. An analysis of extended survival in patients with amyotrophic lateral sclerosis treated with riluzole. Arch Neurol. 1998, 55, 526–528. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Ng, K.; Van Den Bos, M.; Byth, K.; Kiernan, M.C.; Vucic, S. Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in ALS. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 1–9. [Google Scholar] [CrossRef]
- Fang, T.; Al Khleifat, A.; Meurgey, J.H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef]
- Dorst, J.; Ludolph, A.C.; Huebers, A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2018, 11, 1–16. [Google Scholar] [CrossRef]
- Bhandari, R.; Kuhad, A. Edaravone: A new hope for deadly amyotrophic lateral sclerosis. Drugs Today (Barc) 2018, 54, 349–360. [Google Scholar] [CrossRef]
- Yoshino, H. Edaravone for the treatment of amyotrophic lateral sclerosis. Expert Rev. Neurother. 2019, 19, 185–193. [Google Scholar] [CrossRef]
- Kiernan, M.C. Motor neuron disease in 2017: Progress towards therapy in motor neuron disease. Nat. Rev. Neurol. 2018, 14, 65–66. [Google Scholar] [CrossRef]
- Abraham, A.; Nefussy, B.; Fainmesser, Y.; Ebrahimi, Y.; Karni, A.; Drory, V.E. Early post-marketing experience with edaravone in an unselected group of patients with ALS. Amyotroph. Lateral Scler. Front. Degener. 2019, 0, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; van den Berg, L.H.; Shefner, J.M.; Mitsumoto, H.; Mora, J.S.; Ludolph, A.; Hardiman, O.; Bozik, M.E.; Ingersoll, E.W.; Archibald, D.; et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): A randomised, double-blind, phase 3 trial. Lancet Neurol. 2013, 12, 1059–1067. [Google Scholar] [CrossRef]
- Bozik, M.E.; Mitsumoto, H.; Brooks, B.R.; Rudnicki, S.A.; Moore, D.H.; Zhang, B.; Ludolph, A.; Cudkowicz, M.E.; van den Berg, L.H.; Mather, J.; et al. A post hoc analysis of subgroup outcomes and creatinine in the phase III clinical trial (EMPOWER) of dexpramipexole in ALS. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 406–413. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- Vieira, F.G.; LaDow, E.; Moreno, A.; Kidd, J.D.; Levine, B.; Thompson, K.; Gill, A.; Finkbeiner, S.; Perrin, S. Dexpramipexole is ineffective in two models of ALS related neurodegeneration. PLoS ONE 2014, 9, e91608. [Google Scholar] [CrossRef]
- Elia, A.E.; Lalli, S.; Monsurrò, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 45–52. [Google Scholar] [CrossRef]
- Parry, G.J.; Rodrigues, C.M.P.; Low, W.C.; Hilbert, S.J.; Steer, C.J. Ursodeoxycholic Acid May Slow the Progression of Amyotrophic Lateral Sclerosis. Ann. Neurodegener. Dis. 2016, 1, 1013. [Google Scholar]
- Henkel, J.S.; Beers, D.R.; Zhao, W.; Appel, S.H. Microglia in ALS: The good, the bad, and the resting. J. Neuroimmune Pharm. 2009, 4, 389–398. [Google Scholar] [CrossRef]
- Puentes, F.; Malaspina, A.; van Noort, J.M.; Amor, S. Non-neuronal cells in ALS: Role of glial, immune cells and blood-CNS barriers. Brain Pathol. 2016, 26, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H., Jr.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. PNAS 2008, 105, 17913–17918. [Google Scholar] [CrossRef]
- Lauria, G.; Dalla Bella, E.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chiò, A.; Corbo, M.; Eleopra, R.; Fazio, R. Erythropoietin in amyotrophic lateral sclerosis: A multicentre, randomised, double blind, placebo controlled, phase III study. J. Neurol. Neurosurg. Psychiatry 2015, 86, 879–886. [Google Scholar] [CrossRef]
- Miller, R.G.; Block, G.; Katz, J.S.; Barohn, R.J.; Gopalakrishnan, V.; Cudkowicz, M.; Zhang, J.R.; McGrath, M.S.; Ludington, E.; Appel, S.H.; et al. Randomized phase 2 trial of NP001, a novel immune regulator: Safety and early efficacy in ALS. Neurol. -Neuroimmunol. Neuroinflammation 2015, 2, e100. [Google Scholar] [CrossRef]
- Khairoalsindi, O.A.; Abuzinadah, A.R. Maximizing the Survival of Amyotrophic Lateral Sclerosis Patients: Current Perspectives. Neurol. Res. Int. 2018, 2018, 1–29. [Google Scholar] [CrossRef]
- Rosenblum, L.T.; Trotti, D. EAAT2 and the molecular signature of amyotrophic lateral sclerosis. In Glial Amino Acid Transporters; Springer: Cham, Switzerland, 2017; pp. 117–136. [Google Scholar]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Watanabe, K.; Yuki, S.; Akimoto, M.; Sakata, T.; Palumbo, J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 5–10. [Google Scholar] [CrossRef]
- Zoccolella, S.; Santamato, S.; Lamberti, P. Current and emerging treatments for amyotrophic lateral sclerosis. Neuropsychiatr. Dis. Treat. 2009, 5, 577–595. [Google Scholar] [CrossRef]
- Keskin, I.; Forsgren, E.; Lehmann, M.; Andersen, P.M.; Brännström, T.; Lange, D.J.; Synofzik, M.; Nordström, U.; Zetterström, P.; Marklund, S.L. The molecular pathogenesis of superoxide dismutase 1-linked ALS is promoted by low oxygen tension. Acta Neuropathol. 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Benkler, C.; O’Neil, A.L.; Slepian, S.; Qian, F.; Weinreb, P.H.; Rubin, L.L. Aggregated SOD1 causes selective death of cultured human motor neurons. Sci. Rep. 2018, 8, 16393. [Google Scholar] [CrossRef]
- Shvil, N.; Banerjee, V.; Zoltsman, G.; Shani, T.; Kahn, J.; Abu-Hamad, S.; Papo, N.; Engel, S.; Bernhagen, J.; Israelson, A. MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: Implications for familial ALS. Cell Death Dis. 2018, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef]
- Korkmaz, O.T.; Aytan, N.; Carreras, I.; Choi, J.K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7, 8-Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef]
- Tanaka, H.; Shimazawa, M.; Kimura, M.; Takata, M.; Tsuruma, K.; Yamada, M.; Takahashi, H.; Hozumi, I.; Niwa, J.; Iguchi, Y.; et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci. Rep. 2012, 2, 573. [Google Scholar] [CrossRef]
- Nagahara, Y.; Shimazawa, M.; Ohuchi, K.; Ito, J.; Takahashi, H.; Tsuruma, K.; Kakita, A.; Hara, H. GPNMB ameliorates mutant TDP-43-induced motor neuron cell death. J. Neurosci. Res. 2017, 95, 1647–1665. [Google Scholar] [CrossRef]
- Lunn, J.S.; Sakowski, S.A.; Federici, T.; Glass, J.D.; Boulis, N.M.; Feldman, E.L. Stem cell technology for the study and treatment of motor neuron diseases. Regen. Med. 2011, 6, 201–213. [Google Scholar] [CrossRef]
- Changsung, K.; Lee, Ch.H.; Sung, J.J. Amyotrophic Lateral Sclerosis- Cell Based Therapy and Novel Therapeutic Development. Exp. Neurobiol. 2014, 23, 207–214. [Google Scholar]
- Srikanth, M.; Yao, L.; Asmatulu, R. Advances in the Research of Astrocyte Function in Neural Regeneration. In Glial Cell Engineering in Neural Regeneration; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Lepore, A.C.; Rauck, B.; Dejea, C.; Pardo, A.C.; Rao, M.S.; Rothstein, J.D.; Maragakis, N.J. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 2008, 11, 1294–1301. [Google Scholar] [CrossRef]
- Lepore, A.C.; O’Donnell, J.; Kim, A.S.; Williams, T.; Tuteja, A.; Rao, M.S.; Kelley, L.L.; Campanelli, J.T.; Maragakis, N.J. Human glial-restricted progenitor transplantation into cervical spinal cord of the SOD1 mouse model of ALS. PLoS ONE 2011, 6, e25968. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Doreswamy, A.; Gross, S.K.; Tang, X.; Campanelli, J.T.; Maragakis, N.J. Human glial progenitor engraftment and gene expression is independent of the ALS environment. Exp. Neurol. 2015, 264, 188–199. [Google Scholar] [CrossRef]
- Reekmans, K.; Praet, J.; Daans, J.; Reumers, V.; Pauwels, P.; Van der Linden, A.; Berneman, Z.N.; Ponsaerts, P. Current challenges for the advancement of neural stem cell biology and transplantation research. Stem Cell Rev. 2012, 8, 262–278. [Google Scholar] [CrossRef]
- Cossetti, C.; Iraci, N.; Mercer, T.R.; Leonardi, T.; Alpi, E.; Drago, D.; Alfaro-Cervello, C.; Saini, H.K.; Davis, M.P.; Schaeffer, J.; et al. Extracellular vesicles from neural stem cells transfer IFN-γ via Ifngr1 to Activate Stat1 Signaling in Target Cells. Moll. Cell 2014, 56, 193–204. [Google Scholar] [CrossRef]
- Gincberg, G.; Arien-Zakay, H.; Lazarovici, P.; Lelkes, P.I. Neural stem cells: Therapeutic potential for neurodegenerative diseases. Br. Med. Bull. 2012, 104, 7–19. [Google Scholar] [CrossRef]
- Mazzini, L.; Vescovi, A.; Cantello, R.; Gelati, M.; Vercelli, A. Stem cells therapy for ALS. Expert Opin. Biol. 2016, 16, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Skotte, N.H.; Bennett, C.F.; Hayden, M.R. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol. Med. 2012, 18, 634–643. [Google Scholar] [CrossRef]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 2006, 116, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Duque, S.; Joussemet, B.; Riviere, C.; Marais, T.; Dubreil, L.; Douar, A.M.; Fyfe, J.; Moullier, P.; Colle, M.A.; Barkats, M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Molecular 2009, 17, 1187–1196. [Google Scholar] [CrossRef]
- Thomsen, G.M.; Gowing, G.; Latter, J.; Chen, M.; Vit, J.P.; Staggenborg, K.; Avalos, P.; Alkaslasi, M.; Ferraiuolo, L.; Likhite, S.; et al. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J. Neurosci. 2014, 34, 15587–15600. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, G.M.; Alkaslasi, M.; Vit, J.-P.; Lawless, G.; Godoy, M.; Gowing, G.; Shelest, O.; Svendsen, C.N. Systemic injection of AAV9-GDNF provides modest functional improvements in the SOD1 G93A ALS rat but has adverse side effects. Gene Ther. 2017, 24, 245–252. [Google Scholar] [CrossRef]
- Martier, R.; Liefhebber, J.M.; García-Osta, A.; Miniarikova, J.; Cuadrado-Tejedor, M.; Espelosin, M.; Ursua, S.; Petry, H.; van Deventer, S.J.; Evers, M.M.; et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol. Nucleic Acids. 2019, 16, 26–37. [Google Scholar] [CrossRef]
- Harrison, D.; Mehta, P.; van Es, M.A.; Stommel, E.; Drory, V.E.; Nefussy, B.; van den Berg, L.H.; Crayle, J.; Bedlack, R.; the Pooled Resource Open-Access ALS Clinical Trials Consortium. “ALS reversals”: Demographics, disease characteristics, treatments, and co-morbidities. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 495–499. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Phenotypes | Regions | Symptoms | The Prognosis |
|---|---|---|---|
| Limb-onset | UMN | Spasticity, weakness, and brisk deep tendon reflexes; | Overall survival is 5–8 years |
| LMN | fasciculations, wasting, and patients present with gradually ascending distal weakness. | ||
| Bulbar-onset | UMN | Spastic dysarthria, which is characterized by slow, labored, and distorted speech, often with a nasal quality; | 2 to 4 years, and depends on the timing of respiratory and limb involvement. |
| LMN | tongue wasting, weakness, and fasciculations, accompanied by flaccid dysarthria*, and later dysphagia. | ||
| Primary lateral sclerosis | Pure UMN | It is characterized by an ascending spastic tetraparesis with involvement of speech in the majority by 3 years, urinary urgency; | A slowly progressive condition, with survival for decades. |
| Progressive muscular atrophy | Pure LMN | The least well-defined subtype of ALS. Asymmetrical weakness and wasting, often in the legs, which coalesces into four limb lower motor neuron involvement. | About 5 years |
| ALS-frontal lobe dementia syndrome (frontotemporal lobar degeneration, FTLD) | UMN, LMN, and brain cortex | Presentation with frontotemporal dementia. Later developing signs of MND. | An overall survival of less than 3 years. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20, 2616. https://doi.org/10.3390/ijms20112616
Nowicka N, Juranek J, Juranek JK, Wojtkiewicz J. Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2019; 20(11):2616. https://doi.org/10.3390/ijms20112616
Chicago/Turabian StyleNowicka, Natalia, Jakub Juranek, Judyta K. Juranek, and Joanna Wojtkiewicz. 2019. "Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 20, no. 11: 2616. https://doi.org/10.3390/ijms20112616
APA StyleNowicka, N., Juranek, J., Juranek, J. K., & Wojtkiewicz, J. (2019). Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 20(11), 2616. https://doi.org/10.3390/ijms20112616